
Extensive sampling of Saccharomyces cerevisiae in Taiwan reveals ecology and evolution of predomesticated lineages
- Tracy Jiaye Lee1,2,3,
- Yu-Ching Liu1,
- Wei-An Liu1,
- Yu-Fei Lin1,
- Hsin-Han Lee1,4,5,
- Huei-Mien Ke1,
- Jen-Pan Huang1,
- Mei-Yeh Jade Lu1,
- Chia-Lun Hsieh1,
- Kuo-Fang Chung1,
- Gianni Liti6 and
- Isheng Jason Tsai1,2,3,4,5
- 1Biodiversity Research Center, Academia Sinica, 115 Taipei, Taiwan;
- 2Biodiversity Program, Taiwan International Graduate Program, Academia Sinica and National Taiwan Normal University, 115 Taipei, Taiwan;
- 3Department of Life Science, National Taiwan Normal University, 116 Taipei, Taiwan;
- 4Bioinformatics Program, Taiwan International Graduate Program, National Taiwan University, 106 Taipei, Taiwan;
- 5Bioinformatics Program, Institute of Information Science, Taiwan International Graduate Program, Academia Sinica, 115 Taipei, Taiwan;
- 6Université Côte d'Azur, CNRS, INSERM, IRCAN, 06107 Nice, France
Abstract
The ecology and genetic diversity of the model yeast Saccharomyces cerevisiae before human domestication remain poorly understood. Taiwan is regarded as part of this yeast's geographic birthplace, where the most divergent natural lineage was discovered. Here, we extensively sampled the broadleaf forests across this continental island to probe the ancestral species’ diversity. We found that S. cerevisiae is distributed ubiquitously at low abundance in the forests. Whole-genome sequencing of 121 isolates revealed nine distinct lineages that diverged from Asian lineages during the Pleistocene, when a transient continental shelf land bridge connected Taiwan to other major landmasses. Three lineages are endemic to Taiwan and six are widespread in Asia, making this region a focal biodiversity hotspot. Both ancient and recent admixture events were detected between the natural lineages, and a genetic ancestry component associated with isolates from fruits was detected in most admixed isolates. Collectively, Taiwanese isolates harbor genetic diversity comparable to that of the whole Asia continent, and different lineages have coexisted at a fine spatial scale even on the same tree. Patterns of variations within each lineage revealed that S. cerevisiae is highly clonal and predominantly reproduces asexually in nature. We identified different selection patterns shaping the coding sequences of natural lineages and found fewer gene family expansion and contractions that contrast with domesticated lineages. This study establishes that S. cerevisiae has rich natural diversity sheltered from human influences, making it a powerful model system in microbial ecology.
The yeast genus Saccharomyces, which includes S. cerevisiae, is a powerful model system for revealing patterns of genomic variation underlying reproductive isolation and adaptation in eukaryotic microorganisms. Surveys of population genetic data have been used in S. cerevisiae to date the origin of key domestication events (Gallone et al. 2016; Duan et al. 2018; Peter et al. 2018), to determine life cycle frequencies in nature (Tsai et al. 2008), to determine the genomic basis of adaptation at continental scale (Duan et al. 2018; Peter et al. 2018), and, more recently, to establish its geographical origin and dispersal history (Xia et al. 2017). Phylogenomic analyses of the Saccharomyces sensu stricto complex and extensive sequencing of collections across the world suggest that S. cerevisiae originated in East Asia (Duan et al. 2018; Peter et al. 2018). The 1011 Genome Project—the most broad large-scale yeast population genomic study—discovered that three wild isolates from Taiwan showed an unprecedented high genetic diversity compared with populations from the rest of the world (Peter et al. 2018). Population genomics of 266 domestic and wild isolates in China revealed six wild lineages from primeval forests. The newly identified CHN-IX group represents the most diverged lineage (Duan et al. 2018). Isolates from this group and the three Taiwanese isolates were grouped into a single lineage that showed a disjunct geographic distribution (Bendixsen et al. 2021). Although considerable knowledge is available on the biogeography and population genetics of plants and animals across continents (Whittaker et al. 2017), little is known about how eukaryotic microorganisms such as S. cerevisiae disperse, establish, reproduce, and persist in nature (Liti 2015).
Most S. cerevisiae biology has been based on experiments on a handful of laboratory domesticated strains, but comprehensive analyses of the ecology and evolutionary biology of S. cerevisiae in the wild are still unavailable. In nature, S. cerevisiae have been isolated from the bark, fruits, surrounding soil, and leaves of plants belonging to several different families (Naumov et al. 2013), with early reports suggesting that the yeast is most successfully isolated from the oak family Fagaceae (Sniegowski et al. 2002; Sampaio and Gonçalves 2008; Wang et al. 2012). S. cerevisiae contains high genetic diversity in certain populations, including lineage-specific variants that display clear population structures (Barnett 1992; Wang et al. 2012; Cromie et al. 2013; Strope et al. 2015; Gallone et al. 2016; Gonçalves et al. 2016; Zhu et al. 2016; Duan et al. 2018; Legras et al. 2018; Peter et al. 2018) and explain phenotypic variance similar to common variants (Fournier et al. 2019). Samples from natural habitats tend to be homozygous diploids forming unique populations with minimal genetic admixture, whereas lineages associated with human activities were likely heterozygous, containing higher ploidy and greater genetic admixture leading to a mosaic genome makeup (Diezmann and Dietrich 2009; Liti et al. 2009; Wang et al. 2012; Almeida et al. 2015). The diverse natural lineages of S. cerevisiae present in East Asia provide an excellent opportunity to study the natural diversity of this species, which was previously believed to be fully domesticated (Fay and Benavides 2005).
Taiwan is a continental shelf island with the fifth highest tree density in the world (Crowther et al. 2015). Among the 13 climate-related forests types in Taiwan, five are Fagaceae-dominated natural forests on low- and mid-elevation mountains (Li et al. 2013), thus a potentially ideal natural habitat for S. cerevisiae. Taiwan also harbors a high phylogenetic diversity of flowering plants (53 out of 64 angiosperm orders present under the APG IV classification system) (Lin and Chung 2017) and endemism compared with other oceanic islands (Hsieh 2002), raising the possibility that the associated microbial populations are genetically different from their continental counterparts. Here, we set out to characterize the intra-genetic diversity, relative abundance, and distribution of S. cerevisiae in Taiwanese forests over 4 yr of broad sampling. Our study provides novel insights of the predomestication phase of S. cerevisiae and broadens our understanding of the ecological and biogeographic implications before anthropogenic impacts.
Results
Deep sampling of natural S. cerevisiae from Taiwanese forests
From July 2016 to October 2020, our sampling strategy consisted of maximizing the number of localities associated with Fagaceae hosts and sampling a broad range of plant families present in Taiwanese broad-leaved forests (Fig. 1A; Supplemental Table S1). We surveyed 693 plant hosts belonging to 43 orders, 86 families, and 156 genera (Supplemental Table S2) collected over 113 nonoverlapping 1-km2 grids. Various substrates (twigs, bark, leaves, flowers, fruits, and topsoil around trees) were collected from each tree and subject to selective media enrichments, resulting in 5526 independent incubations (Supplemental Table S3). The successful isolation rates of S. cerevisiae per sample and per tree host were 1.9% and 10.8%, respectively, higher than from Brazilian forests (Barbosa et al. 2016) and Slovenia oak forests (Dashko et al. 2016) but lower than from North American oaks (Sniegowski et al. 2002) and Chinese wild niches (Wang et al. 2012). These isolates were recovered across altitudes of 0–2100 m from 18 plant families (Fig. 1B), with a majority from Fagaceae including four genera (27 Quercus, nine Lithocarpus, eight Castanopsis, and one Fagus species). Ten plant genera had higher isolation rates than Quercus, ranging from 40% to 100% per plant, albeit this recovery rate applied for as few as one tree (Supplemental Table S2). Among Fagaceae, Quercus pachyloma showed the highest isolation rate (75%; three out of four trees). Of the 339 lichen samples, four yielded successful isolations. Among the types of substrates, litter had the highest isolation rate (8.1%), providing the majority of recovered S. cerevisiae isolates (26.2%), followed by fruit, soil, bark, and leaves (∼4%–5% each). In general, the majority of samples were collected from July to December, and we found the isolation rate to be highest in July (18.9% per host tree), followed by September and October (17.5% and 11.3%, respectively). Isolation rates in other months remained around 0%–11% (Supplemental Table S3).
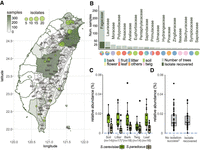
Sampling and isolation of S. cerevisiae in Taiwan. (A) Map of Taiwan showing sampling efforts in each county, with darker shades representing areas with higher numbers of samples collected and circles denoting the locations where S. cerevisiae was successfully isolated. One isolate found on Dongsha Island is not shown on this map. (B) Eighteen plant families from which S. cerevisiae was isolated. The darker color on each bar corresponds to the number of plants that yielded a successful isolation. Another 73 plant families from which we did not obtain any S. cerevisiae isolates are not shown. Pie charts below each bar represent the substrate surrounding plants from which samples were recovered. (C,D) Pairwise comparisons found no differences in the relative abundances of S. cerevisiae among bark, leaf, or twig (C; Wilcoxon-rank with Bonferroni correction: bark–leaf, P = 1.0; bark–twig, P = 0.118, leaf–twig, P = 0.461) and between samples with or without isolation success (D).
Recurrent sampling of eight trees over 2 yr showed differential isolation successes (Supplemental Table S4), suggesting that S. cerevisiae had different abundances in different parts or trees. Focusing on a total of five substrates from 18 trees within ∼100 m2 of this forest (Supplemental Fig. S1; Supplemental Table S4), ITS amplicon sequencing succeeded in detecting just two amplicon sequence variants (ASVs) belonging to the Saccharomyces genus: S. cerevisiae and Saccharomyces paradoxus. In contrast to surveys in temperate and boreal forests (Charron et al. 2014; Kowallik and Greig 2016; Brysch-Herzberg and Seidel 2017), S. cerevisiae had a higher relative abundance calculated as the percentage of the total taxa-classified reads than did S. paradoxus in the subtropics (Fig. 1C). The sequence relative abundance of S. cerevisiae was on average 0.012% in these trees belonging to seven families regardless of substrates sampled; this suggested that, despite being ubiquitous in nature, S. cerevisiae lives in small populations. The relative abundances of S. cerevisiae were found to be constant between pairwise comparisons of bark, leaves, and twigs (Wilcoxon-rank with Bonferroni correction: bark–leaf, P = 1.0; bark–twig, P = 0.118; leaf–twig, P = 0.461) (Fig. 1C), among tree families (Supplemental Fig. S2, P = 1.0), and on whether a S. cerevisiae isolate was recovered (P = 0.89) (Fig. 1D). In addition, bioclimatic variables extracted from GPS coordinates also showed no difference between sites at which isolates were and were not recovered (Supplemental Information; Supplemental Table S5). Together, these results imply that the primary habitat of S. cerevisiae is unlikely associated with a single tree host.
Multiple natural S. cerevisiae lineages in Taiwan
We sequenced the genomes of 121 isolates with a median coverage of 91× depth (Supplemental Table S6). All isolates were primarily homozygous (average heterozygosity: 0.01%) diploids, with the exception of isolate PD36A, which was a triploid (Supplemental Fig. S3) estimated by flow cytometry (Supplemental Information). We constructed a maximum likelihood phylogeny based on 765,169 SNPs segregating in 340 isolates (Fig. 2A) by including 219 representative isolates previously studied from multiple habitats (Barbosa et al. 2016; Duan et al. 2018; Peter et al. 2018; Pontes et al. 2019) that sampled all the major worldwide wild and domesticated lineages. The topology of the isolate phylogeny is largely consistent with a previous neighbor joining tree from the 1011 S. cerevisiae Genome Project (Peter et al. 2018): The natural isolates were mostly grouped according to sampling locations, whereas industrial isolates were grouped according to fermentation sources. In particular, the wine/European lineage and Asian fermentation lineage were separated by a suite of natural isolates, suggesting independent domestication events (Fay and Benavides 2005; Liti et al. 2009; Gonçalves et al. 2016; Gallone et al. 2018). The African palm wine lineage was separated from the West African cocoa lineage and placed near the branch leading to the Asian fermentation lineage. Furthermore, the CHN-VI/VII lineage, which was collected from fruits, was further separated into two lineages consistently with geographical proximity of its members (designated as CHN-VI/VII.1 and CHN-VI/VII.2 in Fig. 2A,C; Supplemental Table S6).
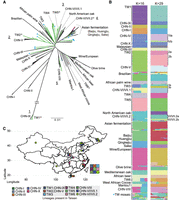
Phylogeny and population structures of 340 S. cerevisiae isolates. (A) Unrooted phylogeny based on 765,169 genome-wide SNPs. Bootstrap support was >90% in all major lineages except inner nodes within some lineages, as indicated by asterisks. Natural, industrial, and fermentation-related isolates discovered in Taiwan are colored in green, blue, and magenta, respectively. Mosaic Taiwanese isolates from ADMIXTURE analyses are labeled with blue dots on branch tips. Five cases in which Taiwanese and Chinese isolates were found to be monophyletic are indicated with underscored numbers. The Asian fermentation lineage includes Baijiu-, Huangjiu-, Qingke jiu-, sake-, and fermentation-related isolates from Taiwan, as shown in B. (B) Population structure from ADMIXTURE analysis at K = 16 and 29. Labels on the left side of the bars indicate each group from K = 16, and some were further separated in K = 29, which is annotated on the right side. Natural Taiwanese isolates with admixed genome makeup are shown together in the TW mosaic group. (C) Map of China and Taiwan indicating where the S. cerevisiae natural lineages were found (colored squares and circle). CHN-IV isolates that were sampled from Japan are not shown on this map.
Previous studies of natural S. cerevisiae revealed that most lineages comprise isolates from neighboring geographic origins (Duan et al. 2018; Peter et al. 2018); however, natural Taiwanese isolates are found throughout the phylogeny despite the small size of the island (Fig. 2A). The population structure of the 340 isolates used for the phylogeny was analyzed using ADMIXTURE (Alexander et al. 2009) with K from two to 30. The cross-validation (CV) error was lowest at K = 29 (CV error = 0.09025), although it only differed <1% between K = 16 and 30 (Fig. 2B; Supplemental Fig. S4). ADMIXTURE at K = 16 was largely consistent with the phylogenetic lineages such as placing CHN-VI/VII into two genetic groups. ADMIXTURE at K = 29 further separated two instances in which a group was split into solely either Chinese or Taiwanese isolates, suggesting the presence of lineage-specific segregating sites as a result of geographical isolation (Fig. 2B; Supplemental Table S7). Some groups comprising isolates from a proximate geographical origin were further split into smaller groups, suggesting ongoing genetic differentiation. Based on ADMIXTURE K = 29, we reused previously assigned group names (Duan et al. 2018; Peter et al. 2018) and designated these differentiated groups and new lineages exclusively found in Taiwan TW1 to TW6 (the most diverged lineage was TW1, and they were progressively labeled clockwise) (Fig. 2). Examples include the recovery of 28 TW1 isolates clustered with CHN-IX (Duan et al. 2018; Bendixsen et al. 2021), together representing the most divergent lineage to date, and a new TW4 lineage that did not contain any Chinese strains (Fig. 2). This new lineage included isolates sampled from lichens and four isolates sampled from mushrooms that were previously placed in an undefined lineage (Peter et al. 2018), suggesting a possible association with other fungi (Spribille et al. 2016). In other instances, Taiwanese isolates were found in three previously assigned groups such as CHN-VI/VII.1, CHN-VI/VII.2, and CHN-VIII. Isolates of the most diverged TW1/CHN-IX lineage were separated by ∼1400 km, with four other natural lineages (CHN-I, -V, -VI/VII, and -X) in between. Twenty-three isolates from northern Taiwan (TW2) clustered with the CHN-V population sampled as far as 1500 km apart. Together, these results suggest that Taiwan harbors the highest number of lineages that show disjunct distributions followed by the Hubei–Shanxi region (nine and five, respectively) (Fig. 2C).
Evidence of admixture in natural lineages
Both inter- and intra-species spontaneous hybridizations have been documented in Saccharomyces species. For instance, the wild S. paradoxus SpC* lineage present in North America (Eberlein et al. 2019) and the domesticated S. cerevisiae Alpechin lineage (D'Angiolo et al. 2020) are classic examples of past hybridizations that played genomic and phenotypic diversities (Barbosa et al. 2016; Duan et al. 2018; Peter et al. 2018; Eberlein et al. 2019). Most Taiwanese isolates tend to have little admixture, with 20% and 5% (27/137, 7/137) of isolates containing at least 10% of the genetic component from two and at least three genetic ancestries (Fig. 2B; Supplemental Table S7), respectively. We confirmed the genetic components of domesticated strains’ origins in wild isolates from African cocoa (Peter et al. 2018), olive brines, and Brazilian forests (Barbosa et al. 2016) and identified an additional TW4 group sharing major genetic components with the steamed buns (Mantou) and wine/European lineages, albeit recovered from nature. Other Taiwanese admixed isolates were apparent on the phylogenetic tree as isolated branches and had different levels of admixture from domesticated lineages (Fig. 2A). Additionally, all Taiwan isolates recovered from fruits contain the CHNVI/VII-2a genetic component (Supplemental Fig. S5); this coincides with the nonadmixed CHNVI/VII-2a isolates, which have the widest geographically distribution in Asia (Fig. 2C).
To confirm that gene flow occurred between genetic groups, we applied TreeMix (Pickrell and Pritchard 2012) to designated groups from ADMIXTURE K = 16 (Fig. 3A; Supplemental Information; Supplemental Fig. S6). The TreeMix phylogeny first indicated extensive gene flow among domesticated lineages such as solid- and liquid-state fermentation products and between natural lineages sister to domesticated lineages. Examples include isolates from steamed buns (Mantou) and Asian alcoholic beverages (sake and Qingke jiu), as well as TW6 forest isolates. Second, the phylogeny also identified gene flow between natural lineages sister to the wine/European and Asian fermentation lineages. The CHN-VIII group emerged from both the wine/European and fruit-enriched CHN-VI/VII-2 lineages, which contain isolates from fruits and the natural environment across the Asian continent, including Taiwan. We also recovered hybrids between natural lineages that coexisted in proximity. Two isolates, each belonging to a TW4 or TW2 lineage, came from fallen fruit, whereas PD38A was isolated from fruit growing on a Castanopsis fargesii tree (Fig. 3A). This PD38A hybridization timing was likely to be recent given the presence of large haplotype blocks not extensively broken down by recombination-containing variants identical to each parental lineage (Supplemental Fig. S7). Overall, these results suggest that hybridizations were common in S. cerevisiae and that some admixed lineages have persisted in nature. Reanalysis of the TreeMix phylogeny based on ADMIXTURE group K = 29 shows consistent results: Recurrent migrations occurred between lineages, leading to the wine/European and Asian fermentation lineages (Supplemental Information; Supplemental Figs. S8, S9). To incorporate these findings into a comparative resource, we further sequenced the genomes of 24 Taiwanese isolates representing all the natural lineages discovered in Taiwan using Oxford Nanopore reads (Supplemental Table S8).
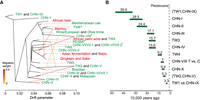
Migration and divergence time between lineages. (A) Migration edges (yellow to red colored lines) estimated by TreeMix showing seven migration edges on the phylogeny. Different edge colors indicate the strength of migration. Lineages were colored according to isolation sources (red and green denote domesticated and wild environments, respectively). Asterisks denote lineages that contain multiple genetic components from different K from the ADMIXTURE analyses. (B) Molecular estimate of time to the most recent common ancestor in different S. cerevisiae lineages. The estimates are shown in Supplemental Table S9A.
Using molecular calibrations, the divergence between different natural lineages as well as the Chinese/Taiwanese split was inferred using either pairwise divergence or a phylogenomic approach (Supplemental Information; Supplemental Fig. S10; Supplemental Table S9). A more recent divergence was estimated from the former approach, in which the lineages were on average diverged 0.03–0.07 million years ago (Ma) (Fig. 3B) compared with 0.54–1.11 Ma inferred from the phylogeny (Supplemental Table S9). Together, these estimates fell during the Pleistocene epoch, suggesting that the split may represent a vicariant event resulting from the submergence of the Taiwan Strait land bridge during interglacial periods and/or the uplift of Taiwanese mountains (Teng 1990) during this period.
Biogeography of wild S. cerevisiae lineages
In nature, single genetically homogenous fungal populations are generally found in distinct geographical regions as a result of isolation by distance (IBD) (Branco et al. 2017; Chung et al. 2017; He et al. 2022). In contrast, the presence of multiple S. cerevisiae lineages at the same locality in Taiwan, even on the same tree, is striking (Fig. 4A; Supplemental Fig. S11; Supplemental Table S6). In one sampling area, four lineages were recovered <35 km apart in central Taiwan (TW1–TW4 and mosaics, n = 10) (Supplemental Fig. S11). In another sampling site, the Fushan Botanical Garden, we obtained 23 isolates comprising three lineages, and admixed isolates were recovered (Fig. 4A). Both significant negative and positive correlations between genetic and geographical distance were observed in isolate pairwise comparisons in close distances (P < 0.05 with 1000 permutations) (Supplemental Fig. S12). However, no such association was found of the whole region (Mantel's r = 0.07, P = 0.23) (Fig. 4B), suggesting that in a given region, the relationships between isolates were less determined by the population structure of single lineages but were dictated by the heterogeneity of multiple lineages coexisting at small spatial scale. The admixed isolates did not contain genetic components from adjacent isolates but instead from CHN-VI/VIII.2a and others (Supplemental Fig. S13). In addition, these combinations of coexisting lineages were not present in a similar locality range in China (Fig. 2C), suggesting that the coexisting of lineages was established by independent dispersal events.
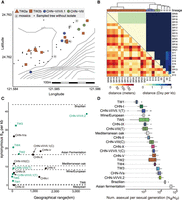
Patterns of genetic variation and geographical distribution. (A) Fine-scale geographic sampling at Fushan Botanical Garden in Taiwan. A total of 106 tree sites constituting 286 substrates were sampled in this region. Different colors represent different lineages, and filled circles denote sampled trees from which S. cerevisiae was not successfully isolated. (B) Genetic and geographic distance of isolate pairs identified in A. (C) Lack of correlation between genetic diversity θW at the synonymous site and geographical range across lineages. Diversity for lineages in which the geographical range is unavailable is indicated with dashed lines. (D) Frequency of asexual per sexual generations across lineages.
The overall genetic diversity of Taiwanese isolates was comparable to that of Chinese isolates (Taiwan θπ = 5 × 10−3 vs. China θπ = 6 × 10−3), even though the samples were only meters to tens of kilometers apart (Supplemental Fig. S14). This reinforced that the pattern of S. cerevisiae diversity in a geographical region was shaped by the presence of multiple lineages and heterogeneity of metapopulations in the same habitat. Up to a twofold difference was observed in genetic diversity between lineages, with the aforementioned most-widespread CHN-VI/VII.2a group harboring the greatest diversity (Fig. 4C; Supplemental Table S10). In contrast, when comparing isolates on the same tree at an extreme microgeographic scale, we found instances of all isolates being clonal or from different lineages with pairwise differences differing by approximately 35,000-fold (one to 35,922 maximum number of pairwise mismatches of isolates recovered on the same tree; θπ = 8.3 × 10−8–2.9 × 10−3) (Supplemental Table S11). Three out of seven lineages have shown a linear IBD (Meirmans 2012) signature, including the aforementioned TW2 lineage (P < 0.05) (Supplemental Fig. S15). The TW2 lineage showed a central-southern Taiwan discontinuous distribution, where isolates are found as much as 194 km apart. This suggests that the greater the geographical range, the higher the likelihood of genetic differentiation. Indeed, greater sequence divergence was shown when intra-lineage isolates between lineages were >10 km apart (P < 0.001, Wilcoxon rank-sum test) (Supplemental Fig. S16), which supported genetic differentiation as a result of geographical isolation (Liti et al. 2006).
Population genomics across lineages
Patterns of segregating sites can be used to infer the relative contributions and frequencies of reproduction modes in nature (Tsai et al. 2008). Wild S. cerevisiae isolates were highly inbred: Wright's inbreeding coefficient F was an average of 0.99, and clones made up 16%–100% of each lineage (Supplemental Table S6), suggesting that most generations were mitotic regardless of lineage. We estimated that the effective population size of mutational (Ne) and recombinational (Nρ) diversity for all chromosomes was 4.1 × 106–7.7 × 107 and 197–12,821, respectively, averaging across chromosomes (Supplemental Table S12) of selected lineages (Supplemental Table S13). The differences between both Ne estimates equates to approximately 382–61,264 mitotic cell divisions for every meiosis event (Fig. 4D). Such estimates overlap with previous estimates of 12,500–62,500 clonal generations based on the decay of heterozygosity during mitosis (Magwene et al. 2011), 1000–3000 in two genealogically independent populations of S. paradoxus (Tsai et al. 2008), and fewer than 800,000 generations in the fission yeast Schizosaccharomyces pombe (Farlow et al. 2015).
We calculated the mean neutrality index (NI) NITG (Stoletzki and Eyre-Walker 2011) for each lineage using polymorphism data from each lineage and S. paradoxus as an outgroup (Fig. 5A). NITG was higher in the domesticated lineages such as wine/European as well as the most diverged TW1/CHN-IX among the natural lineages, suggesting more selection in purging the deleterious alleles in these lineages. We found that variations in NITG in natural lineages were not due to the differences in effective population size inferred from mutational diversity (Kendall's τ = −0.26, P = 0.11) (Supplemental Fig. S17) but from recombinational size (Kendall's τ = 0.33, P = 0.047) (Fig. 5B), suggesting that the selection efficacy was greater when recombination occurred during sexual reproduction, consistent with the results of experimental evolution in a laboratory setting (Goddard et al. 2005). Such a relationship was more significant when lineages with low recombination were removed (Brazilian and the Asian fermentation lineage was excluded, Kendall's τ = 0.52, P = 0.002) (Fig. 5B), indicating similar efficacy of selection in the absence or low recombination.
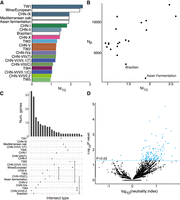
Population genomics across lineages. (A) NITG estimates in natural and two domesticated lineages. (B) Relationship between NITG and Nρ across natural lineages. (C) Lineage-specific and shared genes with NI < 1. (D) ΝΙ from the McDonald–Kreitman test for each gene in the TW3 lineage with S. paradoxus as the outgroup. Genes that were significantly different from NI = 0 were highlighted in blue.
We next investigated the extent of selection at the gene level within each lineage by conducting the McDonald–Kreitman test (McDonald and Kreitman 1991). Overall, we found 18–503 genes with a NI > 1 in each lineage (Fisher's exact test, P < 0.05) (Supplemental Table S14) compared with one to 38 genes with NI < 1 (Fisher's exact test, P < 0.05) (Fig. 5C; Supplemental Table S15), indicating that more genes had an excess of amino acid polymorphisms than were under positive selection (Fig. 5C; Supplemental Fig. S18). The most genes with NI > 1 in the Asia lineages belonged to the most diverse TW3 (144 genes) (Fig. 5D). The majority of these genes indicative of departure from neutrality were observed in only one lineage, emphasizing the lineages’ independent evolutionary history (Supplemental Fig. S19). These negatively selected genes together were found to be enriched in biological processes such as response to stimulus, cell communication, and intracellular signal transduction (Supplemental Table S16). Within each lineage, no genes under either purifying or positive selection were enriched in any particular biological processes, except for the Brazilian lineage, which contained a sufficient number of genes showing NI > 1. Three genes (CDC10, CIT2, and SAT4) in both the wine/European and Asian fermentation lineages showed the largest overlap of genes with NI < 1 among the lineages (Fig. 5C). CIT2 encodes a citrate synthase that was involved in ethanol tolerance (Kasavi et al. 2014). Similarly, 25 genes under negative selection in these two lineages were the only overlap category found to be enriched in biological processes, including RAS protein signal transduction (Supplemental Table S17), which were also targets of adaptation across different experimental evolution experiments (Long et al. 2015). Together these results suggest that the common selective pressure from domestication may have driven the adaptations of these genes. It is unlikely that this overlap was the result of stronger divergent selection with the S. paradoxus outgroup because the pattern was consistent when we used the McDonald–Kreitman with TW3 and CHN-VIII as outgroups, as they were sister to each of the domesticated lineages (Supplemental Fig. S20).
Population differentiation dynamics between lineages
The presence of different levels of shared genetic components observed between the Chinese and Taiwanese isolates among the five shared lineages suggested a distinct differentiation between the disjunct populations. The average ratio of nonsynonymous to synonymous substitution rates (dN/dS) between the China and Taiwan isolates across lineages was 0.21 (Fig. 6A), suggesting that there was pervasive negative selection acting on the coding sequences of S. cerevisiae, with only 40–303 out of 6572 genes showing signals of positive or balancing selection (dN/dS > 1) across the Taiwanese lineages. Consistent with observations from NITG, most of these genes were lineage specific, with only AIM21, involved in mitochondrial inheritance, detected in four out of five lineages (Supplemental Fig. S21; Supplemental Table S18), suggesting that selection acted independently in these lineages.
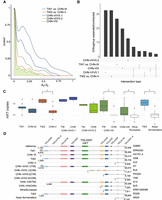
Dynamics between lineages. (A) Density plot of dN/dS showing the majority of genes with dN/dS < 1. (B) Number of specific and shared orthogroups showing significant difference between pairwise lineage comparisons. (C) Distribution of HXT genes in each lineage. (D) Synteny of HXT and adjacent genes on Chr IV 5′ subtelomere. One representative S. cerevisiae isolate in each lineage was chosen. Numbers denote genome coordinates. Numbers in brackets were annotated genes until chromosome end.
Gene duplication played an important role in the evolution of domesticated S. cerevisiae strains showing more rapid copy number variation than wild strains (Bergström et al. 2014; Yue et al. 2017; Duan et al. 2018). To investigate the extent to which gene families differed between sister natural lineages, we de novo assembled, annotated, and inferred the orthogroup (OG) of nonclonal isolates using OrthoFinder (Emms and Kelly 2015). Compared with domesticated lineages (116 CHN-VIII vs. wine/European and 111 TW3 vs. Asian fermentation) (Supplemental Fig. S22), only 17–49 OGs were found to differ between the Chinese and Taiwanese lineages since their split (Wilcoxon rank-sum test, P < 0.05) (Fig. 6B; Supplemental Fig. S23). A large fraction (36.7%–94.7%) were single-copy expansion or contractions (Supplemental Table S19), lineage specific, and enriched in subtelomeres (Supplemental Table S20). The category that overlapped the most comprised seven OGs that were significantly different in two coexisting lineages: CHN-VIII and TW2 (Fig. 4A; Supplemental Fig. S24). In addition, the largest OG inferred was made up of hexose transporter genes (HXT), which are involved in polyol transport; this OG was significant in four out of seven lineage comparisons (Fig. 6C). Copy numbers differed both between domesticated and natural lineages and among the natural lineages (Supplemental Fig. S25). The Taiwanese lineages typically showed expanded HXT copies compared with Chinese or domesticated lineages, and inspecting isolates with long-read assemblies revealed these copies were colinear regardless of lineage (Fig. 6D; Supplemental Fig. S26). Together these results suggest that the Taiwanese isolates may have maintained a larger HXT repertoire, perhaps allowing them to use different sugar types or concentrations.
Discussion
A comprehensive understanding of the natural history of the budding yeast S. cerevisiae is key to further using one of the most human-exploited microorganisms. In this study, we leveraged a 4-yr extensive sampling in Taiwan and combined metabarcoding approach to uncover S. cerevisiae’s ubiquitous presence but low abundance in broadleaf forests. We isolated and whole-genome-sequenced 121 isolates to confirm the presence of the most diverged lineage, TW1 (Bendixsen et al. 2021) and uncover five additional lineages that shared ancestries with lineages found in China as well as four new lineages exclusively found in Taiwan. We show that sympatric lineages coexist in different parts of Taiwan and identified introgressions between lineages. We found that the population structure of S. cerevisiae can be explained by a markup of different lineages that each outcrossed, on average, once in every 382–61,264 mitotic generations. These differences resulted in different selection efficacies across the lineages. The availability of high-quality S. cerevisiae assemblies presented here, in addition to genetic resources, molecular tools, and genome resources such as the 1011 genomes collection (Peter et al. 2018) already available in this model organism provides an exciting new platform to study microbial ecology.
Although S. cerevisiae has repeatedly been recovered from oak bark in the Northern Hemisphere (Sniegowski et al. 2002; Robinson et al. 2016) and is the only substrate of isolation in recent studies (Goddard and Greig 2015), our findings shows that S. cerevisiae is present as a generalist occurring at low abundance in a variety of broadleaf forest substrates. In addition to temperature, we speculate that isolation success for S. cerevisiae was shaped by coexisting microbial communities (Kowallik et al. 2015) competing with S. cerevisiae in the enrichment media. In addition, at a lineage level, S. cerevisiae was found to be associated with particular environments, suggesting that it may have had an ecological niche (Goddard and Greig 2015): TW4 was isolated only from fungal fruiting bodies and lichens, although further work is needed to conclude a possible symbiotic relationship, and a CHN-VI/VII.2 genetic component was present in many lineages and enriched in isolates recovered from the tree fruit substrate. Higher frequencies of admixed isolates observed in fruits may simply be a result of increased contacts with other lineages. Alternatively, fruits and organisms associated with those fruits such as frugivorous animals and vectors may represent niches that promote hybridization; for instance, sporulation has been suggested to be an adaptation that allows cells to survive in nutrient-depleted conditions such as insects’ intestines during experimental passaging (Thomasson et al. 2021). Notably, the presence of CHN-VI/VII.2 genetic components in many natural lineages across the world, as well as in admixed isolates found in fruits, raises the possibility that the common ancestors dispersed from East Asia were from this lineage. In addition to abiotic factors, we speculate that such dispersal events of fruits may be aided by insects and human foraging.
We found that, unlike the general expectation in biogeographic studies that an island only contains a subset of genetic diversity from the mainland population, the genetic diversity of S. cerevisiae populations from Taiwan can be as diverse as those found in the Asia continent. The persistence of ancestral lineages may be a result of Taiwan being a high environmentally heterogeneous region (Ali 2018; Lin et al. 2020) and its prolonged bioclimatic stability (Tsukada 1966) than that of nearby eastern China. Alternatively, the geographic scale for distinguishing island and mainland populations and the importance of habitat diversity may differ between microorganisms (Davison et al. 2018) and other macro-organisms, such as animals and plants. The biogeography of S. cerevisiae appears to be similar to that of its associated flora in East Asia. Disjunct distributions of plants between Taiwan and different parts of China are common (Jianfei et al. 2012). The phylogeography of representative herbaceous and woody plants indicates that these representatives originated in mainland China and then migrated to Taiwan and the Ryukyu Archipelago during the Pleistocene as sea-level fluctuations yielded recurring land bridges (Chiang and Schaal 2006; Niu et al. 2018; Jiang et al. 2019). We note that the Pleistocene was also the period when several tree species extinctions first took place across both the Americans (Seersholm et al. 2020) and Europe (Magri et al. 2017); this was followed by a rapid migration of Quercus that made it the dominant tree genus (Magri et al. 2017), which may have played a role in the restricted S. cerevisiae lineages observed outside East Asia. A systematic sampling of S. cerevisiae in the mainland continent—especially regions containing flora records showing a disjunct distribution like in Taiwan, for example, the Himalaya–Hengduan mountains (Niu et al. 2018), as well as plate boundaries—may help us better understand the biogeography of S. cerevisiae.
Our findings of rampant hybridization events between wild, wild with domesticated, and domesticated lineages bring new perspectives to the ongoing debate over whether S. cerevisiae domestication happened once (Duan et al. 2018; Han et al. 2021) or multiple times (Almeida et al. 2015; Peter et al. 2018). By revealing frequent hybridizations between natural lineages, we show that isolates used in Asian and European fermentations may have been domesticated independently from the lineage CHN-VI/VII.2, and the single-domestication-event notion may be confounded by admixed isolates. Isolates from Asian fermentations were sister to the CHN-VI/VII.2 lineage, and subsequent genetic differentiations of this group have led to independent lineages such as the North American oak group or the Mediterranean oaks group, which is sister to the European/wine isolates (Figs. 2A, 3A). Isolates outside of East Asia likely bear genetic components of this group. This may result in the placement of these isolates in or close to this group in a phylogeny. Ongoing hybridizations also complicate the inference; for instance, the Brazilian rum population is a result of hybridization between European/wine and North American groups (Almeida et al. 2015). Efforts to identify signatures of domestication environments (Han et al. 2021) may also be challenging when admixture is detected between these lineages. Isolation and recording the frequencies of these admixed isolates in nature could provide further insights into the conditions in which new lineages emerge.
Inferring population history in S. cerevisiae with different frequencies of asexual and sexual generations (Tsai et al. 2008) is challenging when using population genetics methods designed around human heterozygosity and recombination rates (Li and Durbin 2011). Disagreement in the divergences estimated from the phylogeny and pairwise divergence between isolates was observed. The phylogenomic method assumes no gene flow and recombination with different lineages, and although S. cerevisiae is a predominantly asexual organism, recombination ρ was still detected and thus inflates divergence (Schierup and Hein 2000; Li et al. 2019). Conversely, estimates from pairwise divergences were more consistent to other reports (Leducq et al. 2016) but may underestimate the true divergence as we do not know the extent of quiescence of different S. cerevisiae lineages (Gray et al. 2004). Recent advances in directly tracking genotype evolution across natural habitats (Xia et al. 2017; Rudman et al. 2022) may lead to more accurate inferences once some of the fundamental parameters such as average generation time can be obtained in nature.
To conclude, we combined deep sampling, metabarcoding, isolate collection, and whole-genome resequencing to illuminate the predomestication phase of Saccharomyces cerevisiae at an unprecedented resolution. The roles of S. cerevisiae in the temperate forest environment have been studied in detail (Mozzachiodi et al. 2022), and we reveal that multiple natural lineages of S. cerevisiae persist in the subtropic and tropical broadleaf forests in Taiwan, indicating that the species is found everywhere but that some genetically differentiated lineages prefer certain substrates. These observations help us to revisit our understanding of eukaryotic microorganism evolution; for instance, an alternating life cycle seems to be a convenient life history trait when genetically diverged partners are around. As more and more ecosystems, for example, tropical cloud forests (Karger et al. 2021), and biodiversity are lost, actions should be taken to conserve and reveal the ecology and evolution of not just S. cerevisiae but also species with a proposed geographical origin. The availability and gene flow between these lineages also allow future experiments, such as on hybrid fitness, to be designed to resemble the subject's natural scenarios rather than relying on domesticated strains.
Methods
Sampling and isolating Saccharomyces cerevisiae
From September 2016 to October 2020, we collected a total of 2461 environmental samples from various substrates (bark n = 340, twigs n = 328, leaf n = 528, litter n = 320, and fruit n = 78) surrounding 693 plant hosts (Supplemental Table S2). A total of 339 lichen samples, aliquots from six fermentation practices, and 68 from other sources (insect corpse n = 43, fruiting body n = 14, industrial strains n = 5, and others in which biomaterial was sampled only once n = 6) were also collected. Collection time and GPS coordinates in GPX format of host plants were recorded on the day of collection. Leaves and flowers of host plants were photographed. Bioclimatic variables of sampling sites were retrieved from the CHELSA (Karger et al. 2017) database (v. 1.2) using recorded GPS coordinates. Digital terrain models (DTMs) of sampling sites were retrieved from Taiwan's Open Government Data website (https://data.gov.tw/dataset/35430). Environmental samples were collected using alcohol-sterilized tweezers or spoons and stored in zip bags. Whenever possible during the sampling trips, metadata such as the identity of the host plant, lichens, and altitude were recorded. Samples were redistributed into 50-mL falcon tubes and stored at room temperature. Each sample was divided into two proportions and immersed in two enrichment media: a liquid medium made up of either (1) 3 g/L yeast extract, 3 g/L malt extract, 5 g/L peptone, 10 g/L sucrose, 7.6% EtOH, 1 mg/L chloramphenicol, and 0.1% of 1-M HCl as used previously (Sniegowski et al. 2002) or (2) YPD containing 10% dextrose and 5% ethanol adjusted to pH 5.3 as used previously (Hyma and Fay 2013). Samples were incubated at 30°C until signs of microbial growth and fermentation were detected, such as white sediment and effervescence. Sediments were then streaked onto YPD agar plates. Single colonies were picked out and incubated in potassium acetate medium for 7–10 d at 23°C (Liti et al. 2017). Single colonies with ascus-like (four spores) structures under microscope were picked out and streaked onto YPD agar plates. Sanger sequencing and gel electrophoresis of the ITS1-5.8S-ITS2 region PCR-amplified with the ITS1F/ITS4 primer set were performed to identify the species of isolates (White et al. 1990; Gardes and Bruns 1993). Pilot sampling, modification, and rationale during the course of sampling strategies are further provided in the Supplemental Information. Sampling efforts were visualized using the R's package ggplot2 (v. 3.3.5) and annotated with metR (v. 0.10.0; https://github.com/eliocamp/metR) and ggspatial (v. 1.1.5; https://paleolimbot.github.io/ggspatial/). To determine ploidy levels for our isolates, we performed flow cytometry analysis for the 105 Taiwanese isolates from this study using propidium iodide (PI) staining assay using previously established protocols (Supplemental Information; Todd et al. 2018).
DNA extraction
Field-collected environmental samples can vary, so we preprocessed these samples and extracted their DNA differently (for details, see Supplemental Information). For whole-genome sequencing of S. cerevisiae, isolates taken from frozen stocks were streaked out onto YPD plates and incubated at 30°C until colonies became visible. Single colonies were then incubated in 5 mL YPD liquid medium overnight at 30°C in a shaker at 200 rpm. High-molecular-weight genomic DNA was extracted using protocol previously described (Denis et al. 2018). DNA quality was determined by Qubit readings; A260, A280, and A260/280 ratios on NanoDrop; and gel electrophoresis.
Library construction and whole-genome sequencing
For Illumina sequencing, paired-end libraries were constructed using the Illumina Nextera or NEB Next Ultra DNA library preparation kit with the manufacturer's protocol. The first 91 isolates were sequenced by Illumina HiSeq 2500, and the remaining 30 were sequenced by NovaSeq to produce 125- and 150-bp paired-end reads, respectively. Oxford Nanopore libraries were prepared using SQK-LSK109 with 12 isolates multiplexed by a EXP-NBD104 and EXP-NBD114 barcoding kit (v. NBE_9065_v109_revV_14Aug2019) and sequenced by a R9.4.1 flow cell on a GridION instrument. A total of 24 isolates were run on two flow cells. Nanopore FAST5 files were base-called using Guppy (v. 4.0.11).
Amplicon sequencing and analysis
Amplicon libraries were constructed as previously described (Tedersoo et al. 2014) from 89 environmental samples (18 bark, 18 twig, 18 leaf, 18 litter, 17 soil), three positive controls (S. cerevisiae S288C, S. paradoxus YDG197, and laboratory isolate Pseudocercospora fraxinii), and DNA from two Escherichia coli as a template to confirm primer specificity toward only fungal species. The ITS3ngs (5-CANCGATGAAGAACGYRG-3′) and ITS4ngsUni (5′-CCTSCSCTTANTDATATGC-3′) primer pair (Tedersoo et al. 2015) was used. Two no template controls were included during the PCR step to confirm that amplicon generation was free of contaminating DNA. To determine the background amplicon noise from experimental pipeline, a sterile filter was treated and processed as one of the field samples. Amplicons were normalized using the SequalPrep normalization plate kit (Thermo Fisher Scientific A1051001) and then pooled and concentrated using AMPure XP (Beckman Coulter A63881). Finished DNA libraries were sequenced on the Illumina MiSeq platform using 2 × 300-bp pair-end sequencing chemistry.
Raw sequencing reads containing the Illumina sequencing index were demultiplexed using sabre (v. 1.0; https://github.com/najoshi/sabre). Sequencing quality was determined using FastQC (v. 0.11.7; https://github.com/s-andrews/FastQC). Reads were quality filtered based on a Qscore > 20, and 50 bp was trimmed from the 3′ end using usearch (v. 11.0.667) (Edgar 2010). Filtered reads were processed following the UPARSE (Edgar 2013) pipeline. In brief, paired reads were merged and dereplicated into unique sequences. Unique sequences were filtered using usearch default settings. Filtered sequences were denoised into zero-radius operational taxonomic units (zOTUs) using the unoise2 (Edgar 2016b) algorithm. The taxonomy of zOTUs was classified using the SINTAX (Edgar 2016a) algorithm (Edgar 2016) against the UNITE (Nilsson et al. 2018) Fungal database (v. 8.2). Merged reads were assigned into zOTUs with 100% sequence identity and tabulated using the usearch_global function. Processed reads were analyzed in the RStudio environment (v. 1.2.5033). Sequencing data were analyzed with phyloseq (v. 1.34) (McMurdie and Holmes 2013). Statistical significance was tested for using kruskal.test from the stats package in R (R Core Team 2021).
Variant calling
To determine the evolutionary history of new Taiwanese isolates, we collected a total of 219 published genomes representing established S. cerevisiae industrial and natural populations: 102 isolates from the 1011 Genome Project (31 wine/European, eight Mediterranean oak, six African beer, six African palm wine, four West African cocoa, four Malaysian bertam palm nectar, six North American oak, six sake, 11 Asian fermentation, one CHN-I, one CHN-III, four CHN-IV, one CHN-V, six mixed origin groups, and seven other isolates of Taiwanese origin) (Peter et al. 2018), 93 isolates from the Chinese population (69 CHN-I to CHN-X isolates excluding those previously sequenced in the 1011 Genome Project, five isolates from Mantou1, six Huaugjiu, seven Baijiu, and six Qingke jiu) (Duan et al. 2018), 16 isolates from the Brazilian wild lineage (Barbosa et al. 2016), and eight isolates from olive brine (Pontes et al. 2019). This combined with the 121 isolates from this study yielded a total of 340 individuals, 30% of which originated from industrial sources and 70% from the natural environment (Supplemental Table S6). Read quality was examined with FastQC (v.0.11.9; https://github.com/s-andrews/FastQC). Read quality and adaptor trimming were performed using Trimmomatic (v0.36; pair end mode, ILLUMINACLIP; LEADING:20; TRAILING:20; SLIDINGWINDOW:4:20; MINLEN:150) (Bolger et al. 2014). For the 340 samples, 64%–95% of the raw paired reads from the 340 samples was kept after trimming. Trimmed reads were each mapped to the S288C reference genome version R64-2-1 using the Burrows–Wheeler aligner (v. 0.7.17-r1188) (Li and Durbin 2009), and the mapping rate was 91%–99%. Duplicate reads were marked using GATK MarkDuplicates (v. 4.1.9.0) (McKenna et al. 2010). Variants were first called in a multisample manner and filtered using BCFtools v. 1.8 (-d 1332; QUAL 30, MQ 30, AC ≥ 2 and 50% missingness; genotype-filtered with minDP 3) (Danecek et al. 2021). Eighty-eight percent (1,150,658/1,306,082) of variants were retained. Second, variants were also called and filtered with FreeBayes (Garrison and Marth 2012) and VCFtools (v. 1.3.2 and v. 0.1.15, respectively; minDP 3, QUAL 30, MQ 30, AC ≥ 2, and 50% missingness; sites with 0.25 < AB < 0.75 and 0.9 < MQM/MQMR < 1.05 were retained) (Danecek et al. 2011). Fifty-six percent of sites were retained based on these criteria (818,025/1,443,685). Finally, 808,864 intersecting variants discovered from both callers were used for further analysis. The functional effects of variants were annotated with SnpEff (v. 4.3t) (Cingolani et al. 2012).
Assembly, annotation, and ortholog identification
Nanopore reads of each isolate were assembled using Canu (v. 1.9) (Koren et al. 2017). For isolates without long reads, Illumina paired-end reads were assembled using SPAdes (v. 3.14.1, options k-mer size 21, 33, 55, 77, and ‐‐careful) (Bankevich et al. 2012). Consensus sequences of the assemblies were polished with four rounds of Racon (v. 1.4.11) (Vaser et al. 2017), one round of Medaka (v. 1.0.1) using nanopore raw reads, and five rounds of Pilon using Illumina reads. The assemblies were further scaffolded using RagTag (Alonge et al. 2019) against the S288C genome reference. Annotations were then transferred using Liftoff (Shumate et al. 2021), with additional de novo annotations using AUGUSTUS (Stanke et al. 2006) on regions without any transferred annotations. OG was inferred using OrthoFinder (v. 2.5.4) (Emms and Kelly 2015). OGs that were differentially abundant between assemblies produced using different sequencing technologies were excluded from further analyses. The assembly metrics and description of the nanopore assemblies are shown in Supplemental Table S8.
Phylogenomic analyses
After removing 43,695 invariant sites resulting from ambiguous nucleotide codes among all isolates, the remaining 765,169 variable sites were used to construct a phylogeny for the 340 isolates. The resulting best-fit model was indicated by BIC to be TVMe + R3 first with IQ-TREE. In addition, a maximum-likelihood phylogeny was inferred using IQ-TREE with the TVMe + R3 + ASC model and a 1000 ultrafast bootstrap approximation (Hoang et al. 2018; Minh et al. 2020). A separate S. cerevisiae lineage phylogeny was inferred and used in MCMCtree method of the PAML (Yang 2007) package to estimate the divergence time among the S. cerevisiae lineages (Supplemental Information).
Diversity, population structure, and demography estimates
For the population structure estimate, biallelic SNPs were kept and filtered based on linkage disequilibrium. Sites that are linked were filtered out using PLINK (v1.90b4) (Chang et al. 2015), excluding pairs of loci with r2 > 0.5 (‐‐indep-pairwise 50 10 0.5 ‐‐r2). The remaining 482,161 sites were used for ancestry estimation by ADMIXTURE (Alexander et al. 2009) using K = 2 to K = 30 with fivefold CV from five runs of different seed numbers. CV errors for each K-value in five runs were compared to choose the representative number of clusters. Migration signals on the phylogeny were estimated with TreeMix using 1000 bootstraps for natural populations according to clusters in K = 16. The numbers of migration edges were estimated, aided by the optM (v. 0.1.5) package (Fitak 2021), and presented in Supplemental Information.
A consensus genome sequence containing variants for each isolate was generated from the SNPs matrix using BCFtools (Danecek et al. 2021) consensus (v. R64-2-1) with the S288C reference genome sequence. For the IBD analysis, the geographical distance between isolates was measured using the sf package in R for Taiwanese isolates with GPS records. For Chinese isolates, because GPS records were not available, we used approximate coordinates for each sample site (Duan et al. 2018) as recommended by the investigators. To estimate the maximum geographical distance within the Chinese lineage, we chose sample sites that were the furthest apart. For instance, for CHN-V, the distance between Shanxi and Hainan was used. For lineages sampled from only one site (CHN-II, CHN-IX), the largest range of the site was used. Diversity estimates for 16 nuclear chromosomes and the corresponding coding/noncoding regions were examined by VariScan (Vilella et al. 2005) with RunMode 11 (n < 4) and 12 (n ≥ 4). These diversity estimates were used to infer frequency of sex according to the method of Tsai et al. (2008) and are detailed in Supplemental Information.
Data access
The sequencing data of the 121 S. cerevisiae isolates and ITS amplicon sequences of 89 samples generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA755173. The accession numbers of the isolates are also shown in Supplemental Table S6. The zOTU table for the amplicon data and all of the scripts written to perform this study were deposited at GitHub (https://github.com/tjleez/popgen.methods) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Cheng-Ruei Lee for the insightful comments on the ADMIXTURE analyses. We thank Mao-Ning Tuaumu for the helpful suggestions on how to deal with bioclimatic variable data. We thank Nguyen Huu-Vang, Cheng-Ruei Lee, Jun-Yi Leu, Ben-Yang Liao, Dang Liu, and John Wang for commenting on earlier versions of the manuscript. We thank Jun-Yi Leu for experimental advice. We thank Bo-Fei Chen and Ling-Tin Kao for helping with the initial sampling trips. We thank Tze-Fu Hsu, Yi-Hsiu Kuan, H. Thorsten Lumbsch, and Matthew Nelsen for collecting/providing some of the biomaterials. We thank Shou-Fu Duan and Feng-Yan Bai for providing early access to sequencing data and recommendations on how to approximate the geographical positions of the Chinese isolates. We thank the National Center for High-Performance Computing for its computer time and for letting us use its facilities. I.J.T. was supported by the Ministry of Science and Technology, Taiwan under grant 110-2628-B-001-027 and Career Development Award AS-CDA-107-L01, Academia Sinica.
Authors contributions: I.J.T. conceived and led the study. T.J.L., Y.-C.L., and W.-A.L. performed the sampling and isolation of Saccharomyces cerevisiae. J.-P.H., C.-L.H., and K.-F.C. helped with the sampling and identified the lichen and plant samples. T.J.L., W.-A.L., Y.-F.L., and H.-M.K. conducted the experiments. Y.-F.L. performed the amplicon analyses. H.-H.L., H.-M.K., and I.J.T. performed the sequencing and assemblies of the S. cerevisiae genomes. T.J.L., Y.-C.L., H.-H.L., and I.J.T. performed the population genomic analyses. Y.-C.L., H.-H.L., and I.J.T. performed the comparative genomics, phylogenomic analyses, and the divergence time estimation. M.-Y.J.L. carried out Illumina sequencing of the isolates. T.J.L. and I.J.T. wrote the manuscript with substantial input from J.-P.H., K.-F.C., and G.L.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276286.121.
-
Freely available online through the Genome Research Open Access option.
- Received October 13, 2021.
- Accepted March 25, 2022.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








