
Sequencing of individual barcoded cDNAs using Pacific Biosciences and Oxford Nanopore Technologies reveals platform-specific error patterns
- 1Center for Algorithmic Biotechnology, Institute of Translational Biomedicine, St. Petersburg State University, St. Petersburg 199034, Russia;
- 2Brain and Mind Research Institute and Center for Neurogenetics, Weill Cornell Medicine, New York, New York 10065, USA
-
↵3 These authors contributed equally to this work.
Abstract
Long-read transcriptomics require understanding error sources inherent to technologies. Current approaches cannot compare methods for an individual RNA molecule. Here, we present a novel platform-comparison method that combines barcoding strategies and long-read sequencing to sequence cDNA copies representing an individual RNA molecule on both Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT). We compare these long-read pairs in terms of sequence content and isoform patterns. Although individual read pairs show high similarity, we find differences in (1) aligned length, (2) transcription start site (TSS), (3) polyadenylation site (poly(A)-site) assignment, and (4) exon–intron structures. Overall, 25% of read pairs disagree on either TSS, poly(A)-site, or splice site. Intron-chain disagreement typically arises from alignment errors of microexons and complicated splice sites. Our single-molecule technology comparison reveals that inconsistencies are often caused by sequencing error–induced inaccurate ONT alignments, especially to downstream GUNNGU donor motifs. However, annotation-disagreeing upstream shifts in NAGNAG acceptors in ONT are often confirmed by PacBio and are thus likely real. In both barcoded and nonbarcoded ONT reads, we find that intron number and proximity of GU/AGs better predict inconsistencies with the annotation than read quality alone. We summarize these findings in an annotation-based algorithm for spliced alignment correction that improves subsequent transcript construction with ONT reads.
Long-read sequencing is being increasingly used in transcriptomics, particularly for barcoded unique molecules (Gupta et al. 2018; Singh et al. 2019), which yields single-cell and spatially resolved long-read transcriptomes. Various platforms such as Pacific Biosciences (PacBio) (Eid et al. 2009; Koren et al. 2012; Au et al. 2013; Sharon et al. 2013; Tilgner et al. 2014; Weirather et al. 2015), Oxford Nanopore Technologies (ONT) (Oikonomopoulos et al. 2016; Byrne et al. 2017), and linked-read technologies. Linked-read technologies for RNA were originally represented either by synthetic long reads (SLRs) (Tilgner et al. 2015) or usually by more sparsely covered 10x Genomics linked-reads (Tilgner et al. 2018), although more recently other linked-read technologies have emerged (Wu et al. 2019; Chen et al. 2020). Furthermore, for all these platforms a variety of protocols either exists or can be imagined. Comparing the accuracy of these distinct approaches is therefore fundamental in modern transcriptomics just as it has been fundamental for short-read sequence analysis (Engström et al. 2013; Steijger et al. 2013; Li et al. 2014a,b).
A drawback of commonly used strategies is their lack of single-molecule resolution. For example, percent-spliced-in (PSI) values (Wang et al. 2008) of splice sites, or transcripts per million (TPM) (Wagner et al. 2012) values, can easily be compared between multiple strategies. However, these approaches do not allow for the comparison of the accuracy of different strategies for a single molecule. Usually, platforms are compared by the estimated percentages of molecules that behave in a similar way. High concordance of such percentages theoretically suggests that both platforms would behave identically on an individual molecule. However, this theoretical suggestion has so far been impossible to verify because of the impossibility to assess whether two platforms sequence a representation of the same molecule. Our single-RNA-molecule reasoning provides a framework in which an alignment is either correct or false. This is not the case for groups of molecules, in which an alignment can be correct for one molecule and false for another molecule.
Single-cell and spatial barcoding of cDNAs have revolutionized the investigation of complex organs (Macosko et al. 2015; Zeisel et al. 2015). In most single-cell approaches, cDNAs are generated with a polydeoxythymidylic acid (poly(dT)) primer carrying added sequences. In single-cell barcoding, a portion of this added sequence (the barcode, here 16 bases) is identical for all cDNAs from the same individual cell but distinguishes one cell from others. Another portion (the unique molecular identifier [UMI], here 12 bases) is random for each reverse transcription event and thus informs on whether two sequenced reads represent two distinct reverse transcription events or polymerase chain reaction (PCR) duplicates of only one reverse transcription event. Similarly, spatial approaches use barcodes and UMIs to distinguish spatial locations and reverse transcription events. Our advances in long-read sequencing of single-cell (Gupta et al. 2018; Joglekar et al. 2021; Hardwick et al. 2022) and spatially (Joglekar et al. 2021) barcoded cDNAs allow the identification of full-length isoforms for barcoded molecules.
Here, we aim to compare the PacBio and ONT platforms in terms of error profiles, spliced alignments, their discrepancies with the reference, and potential pitfalls in downstream analysis. For this purpose, we use barcoded cDNA copies corresponding to the same RNA molecule sequenced on both platforms.
Results
Identification of RT pairs sequenced on PacBio and ONT platforms
Here we compare cDNAs that are barcoded by their single cell (ScISOr-Seq) of origin or their spatial location (Sl-ISO-Seq), sequenced on both the PacBio Sequel II (circular consensus reads) and ONT systems (Joglekar et al. 2021; Hardwick et al. 2022). A reverse-transcription event is identified by the combination of (1) single-cell/spatial location barcode, (2) a UMI, and (3) the gene to which the molecule is mapped (Fig. 1A). PCR copies of the same reverse-transcription event are sequenced on both platforms (RT read pairs), which enables their comparison for individual RNA molecules. To have the highest-confidence barcodes and ONT-PacBio read pair correspondences, we only use a perfect matching strategy for barcode and UMI detection (Supplemental Note, “Experimental details”). Thus, we compare sequencing errors and intron structure in identified RT read pairs.
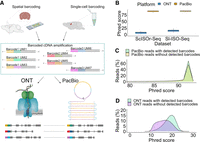
Outline and primary read characteristics. (A) Individual reverse transcription events turn an individual RNA molecule into a barcoded RNA–cDNA hybrid, which is amplified into many cDNA molecules that carry the same barcode and UMI. We previously performed this process in two distinct ways: by single-cell 10x Genomics barcoding and by spatial 10x Genomics Visium barcoding. Aliquots of these cDNAs are then sequenced on PacBio and ONT. Using the identity of barcode and UMI, we can detect individual RNA molecules whose cDNA copies have been sequenced on both ONT and PacBio. We refer to these read pairs as RT read pairs. (B) Comparison between Phred scores of PacBio CCS and ONT reads from both data sets. (C) Phred score distribution for PacBio CCS reads from the Sl-ISO-Seq data set with (light green) and without (yellow) detected barcodes. (D) Phred score distribution for ONT reads from the Sl-ISO-Seq data set with (light blue) and without (purple) detected barcodes.
Molecule identification discards lower-quality ONT reads
We first compare Phred scores of all individual reads sequenced on both platforms independently of molecule detection. The Phred quality score of a base indicates the probability of a base being called correctly. The Phred quality score of a read is computed as the average across Phred scores of all bases in a read. PacBio shows much higher Phred scores than ONT, both in single-cell data and in spatial data (Fig. 1B). Because we conservatively analyze only perfectly matched barcodes, ONT reads with barcodes have significantly higher Phred scores than those without (two-sided Wilcoxon rank-sum P-value = 2.4 × 10−9) (Fig. 1D), whereas no significant difference is detected for PacBio (two-sided Wilcoxon rank-sum P-value = 0.2) (Fig. 1C). Similar observations are made on the single-cell data set, although the difference for ONT reads is even more prominent (Supplemental Fig. S1A,B). Overall, barcode detection in spatial data yields 2,873,455 PacBio reads (of 3,371,331, 85%) and 12,153,599 ONT reads (of 73,181,790, 16%). Although ONT essentially has a deeper sequencing depth than PacBio, the difference between the number of usable reads is not so high when only considering reads with detected barcodes. It is likely that allowing for barcode mismatches could lead to more ONT reads being retained, although that comes at the risk of introducing more inaccurate barcodes.
Sequence comparison using reference-based and reference-free alignments
Our downstream analysis mostly relies on read mappings, so we first aligned reads using different tools: the widely used minimap2 (Li 2018) and specialized transcriptome aligners: deSALT (Liu et al. 2019), GraphMap2 (Marić et al. 2019), and uLTRA (Sahlin and Mäkinen 2021). Of note, we did not use STARlong (Dobin et al. 2013) because it has strong performance for PacBio reads but is not optimized for error-prone ONT data.
Although all three aligners yield largely similar results, GraphMap2 and uLTRA produce slightly shorter alignments than deSALT and minimap2, and uLTRA generates alignments with more prominent differences between aligned lengths of PacBio and ONT reads (Supplemental Table S3). In addition, GraphMap2 produces the least number of RT read pairs because a PacBio read and an ONT read sharing the same barcode and UMI are mapped to different genes more frequently.
Alignments generated by deSALT and especially Graphmap2 show more discordance between PacBio and ONT reads in terms of splice sites than alignments produced by uLTRA and minimap2 (Supplemental Fig. S6). Moreover, minimap2 performs significantly better in terms of isoform detection (see Splice site correction improves transcript discovery precision) (Table 1). To enforce the same strategy for PacBio and ONT, here we use minimap2 for alignment.
Precision, recall, and false-positive rates of StringTie2 results on the simulated ONT data set aligned with different strategies
Mapping RT read pairs (54,752 in the Sl-ISO-Seq and 274,287 in the ScISOr-Seq data) to the genome with minimap2 (Li 2018) showed highly correlated alignment lengths (Fig. 2A; Supplemental Fig. S2A). PacBio reads were significantly longer than their ONT counterparts in ScISOr-Seq and, although less pronounced, in Sl-ISO-Seq data, but differences were small compared with the entire read (Fig. 2B). However, in terms of absolute numbers, the difference is notable: 70% of read pairs have a longer aligned portion (median, 15 bp) in the PacBio; 27%, in the ONT read (median, 9 bp) (Fig. 2C). In comparison to read length, the differences are minor. However, 9- to 15-bp sequences can harbor important elements such as polyadenylation (poly(A)) signals, protein/miRNA-binding sites, microexons, or G-quadruplexes (Lee et al. 2020).
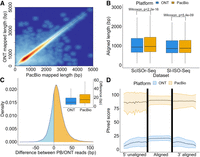
Alignment characteristics of RT read pairs. (A) Heatscatter plot showing aligned lengths of respective PacBio read (x-axis) and ONT read (y-axis) from the RT read pair after mapping to the genome using minimap2 (Sl-ISO-Seq data set, Spearman's Rho: 0.96, p < 2.2×10−16). (B) Comparison between aligned lengths of PacBio and ONT reads for both data sets after mapping to the genome using minimap2. (C) Density plot showing the difference between aligned lengths of PacBio read and ONT read from the RT read pair after mapping to the genome using minimap2 (Sl-ISO-Seq data set) and box plot showing distribution of the differences. In the density plot, cases when PacBio alignment is longer correspond to the yellow area under the curve; the opposite is represented by the blue area. (D) Mean Phred score distribution along the read for aligned (middle) and unaligned (left and right) parts of PacBio (yellow) and ONT (blue) reads based on a (reference-free) pairwise Smith–Waterman alignment of the PacBio and ONT reads from the RT read pair (Sl-ISO-Seq data set). Lower and upper bounds represent the standard deviation of the Phred score distribution.
To delineate common and diverging sequences in reads from RT read pairs, we aligned them to each other using the Smith–Waterman algorithm. Of note, unlike modern mapping algorithms based on heuristics, Smith–Waterman is an exact solution and not subject to future improvements. We divided each alignment pair into an unaligned 5′ part, an aligned portion, and an unaligned 3′ part. In all three compartments within the spatial data, PacBio showed much higher read-wise Phred scores than ONT, but PacBio qualities slightly drop in the nonaligned 5′ portion. However, for the single-cell data, PacBio qualities in the 5′ unaligned portion remained constant and comparable to the aligned portion. In both data sets, ONT qualities deteriorate in the 5′ unaligned portion and gradually decrease in the unaligned 3′ portion (Fig. 2D; Supplemental Fig. S2B).
Sequencing error rates and k-mer identity in alignments to the genome
Deducing sequencing errors from alignments is difficult because in addition to sequencing and alignment errors, PCR errors, single-nucleotide variants (SNVs), and mutations cause a divergence between reads and the genome. We used a three-way comparison between paired PacBio and ONT reads as well as the genome to define a “ground truth” using a majority call among all three sources and to delineate error patterns as a divergence from this ground truth (Methods). ONT has more errors than PacBio (Supplemental Fig. S3A). For PacBio, deletions or mismatches are approximately threefold less abundant than insertions, whose frequency increases toward read ends (Supplemental Fig. S3B). ONT behaves very differently: Deletions dominate over insertions and mismatches, and all error types decrease toward alignment ends (Supplemental Fig. S3C). Forty-one percent of PacBio errors and only 23% of ONT errors occur in homopolymers (Supplemental Fig. S3D). For PacBio, indels within homopolymers are more prevalent than mismatches, and slightly more errors occur in homopolymers toward the 3′end (Supplemental Fig. S3E). For ONT, similar trends were observed, although the insertions are less biased toward homopolymers than in PacBio (Supplemental Fig. S3F). In summary, PacBio has fewer errors than ONT, but a large fraction of PacBio errors occur in homopolymers, whereas ONT errors mostly arise from other areas. Similar observations were obtained for the older ScISOr-Seq data set, although the overall error rate in ONT reads was higher (Supplemental Fig. S4A–D).
Alignments to a genome often use seeding through matching k-mers. We considered 14-mer identity, a commonly used k-mer, for example, in minimap2 (Li 2018) and analyzed each exon alignment separately. As expected, 14-mer identity was lower than single-base identity and specifically affected ONT reads (Supplemental Fig. S3G). For ONT data, we found lower 14-mer identities for slightly longer exons (two-sided Wilcoxon rank-sum test P = 0.005, 1- to 20-bp vs. 21- to 50-bp exons). The most reasonable explanation is that short exons may become unmappable with few sequencing errors, which thus excludes them from this analysis and creates a bias for k-mer identity values. However, for both PacBio and ONT reads, the interquartile range of k-mer identities in short exons is higher compared with longer exons, as a single sequencing error may disrupt all k-mers and lead to 0% k-mer identity (Supplemental Fig. S3G). These effects are noticeably stronger for ONT alignments than for PacBio alignments. After homopolymer compression (Methods), 14-mer identity reached ∼100% for PacBio regardless of exon length. However, for ONT, compression caused high variability in short exons (<21 bp): Although the median increased to 100%, the first quartile decreased to 0%, because a single error in a short exon can affect all 14-mers (Supplemental Fig. S3H). Broadly similar observations were made for ScISOr-Seq data (Supplemental Fig. S4E,F). Thus, homopolymer compression should be applied to ONT reads with care.
Three-way comparison of annotation, ONT, and PacBio shows differences at exon and splice-site calling
Long-read experiments regularly uncover many isoforms that are inconsistent with annotations (Au et al. 2013; Sharon et al. 2013; Tilgner et al. 2014, 2015; Oikonomopoulos et al. 2016; Tardaguila et al. 2018; Kovaka et al. 2019; Tung et al. 2019; Tang et al. 2020; Wyman et al. 2020). Although for short-read experiments, splice site identification and the following splice site quantification has been addressed with large success (Vaquero-Garcia et al. 2016), long-read-based annotation-inconsistent isoforms can be truly novel or simply false. This question can only be conclusively answered for a single molecule as a correct alignment for one molecule can be false for another molecule. Here, we exploit RT read pairs to evaluate inconsistencies between alignments and the annotation using a “three-way comparison” in which the ground truth is defined as a variant supported by the reference and at least one long-read technology for an RT read pair.
We considered 22,600 and 48,993 RT read pairs, in which at least one of the PacBio/ONT reads is assigned to a known transcription start site (TSS) (Lizio et al. 2015) and poly(A) site (Herrmann et al. 2019), respectively (Methods). Indeed, all barcoded reads have a poly(A) tail, which creates a bias toward 3′completeness in RT read pairs, and thus, a known poly(A) site is assigned for a significantly larger portion of reads than a TSS. Moreover, in 95% of RT read pairs, both the PacBio and ONT reads are assigned to the same annotated poly(A) site, whereas agreement on TSS is lower (87%) (Fig. 3A,B). For TSS assignment, a significant portion of disagreeing pairs arises from unassigned ONT reads (8% of all RT read pairs), which suggests that 5′ truncation of PacBio reads is less frequent. We noticed that the results differ for spliced and unspliced reads. Although spliced reads are assigned to TSS more often, the percentage of RT read pairs with both reads spliced and assigned to different TSS is also higher (3.2% for spliced reads vs. 1.7% for unspliced). This may potentially be explained by short starting exons misaligned in ONT reads. For poly(A) sites, however, the difference between spliced and unspliced reads is marginal (Fig. 3C).
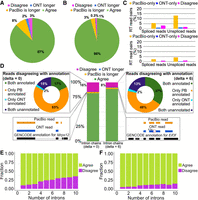
Agreement in RT read pairs of Sl-ISO-Seq data. (A) Fractions of TSS assignments that agree (green), disagree (magenta), or are found only in one read (blue for ONT, yellow for PacBio) from the RT read pair. (B) Same as A, but for poly(A) sites. (C) Percentage of RT read pairs that disagree on the assigned TSS (top) and poly(A) site (bottom): only PacBio read assigned (yellow), only ONT (blue), and both assigned but to different sites (magenta). (D, middle) Percentage of RT read pairs that agree (green), disagree (magenta), or have one chain being longer (blue for ONT, yellow for PacBio [PB]) when splice junctions are compared precisely (left; delta = 0bp) or inexactly (right; delta = 6bp). (Top left) Classification of disagreeing intron chains from RT read pairs with respect to the reference annotation (delta = 0 bp): Both are inconsistent with the annotation (dark blue); both correspond to known (different) transcripts despite the disagreement (green); and PacBio is consistent with the annotation whereas ONT is not (yellow) and vice versa (light blue). (Top right) Classification of disagreeing intron chains with respect to the reference annotation using inexact comparison (delta = 6 bp). (Bottom left) An example of agreeing intron chains from an RT read pair in which PacBio intron chain is longer (Mrps12 gene). (Bottom right) An example of intron chains from an RT read pair that have a 6-bp difference in the donor site of the second intron. Comparing intron chains with delta = 6 bp classifies them as agreeing (Eif3f gene). (E) Fraction of agreeing (green) and disagreeing (magenta) intron chains with respect to intron chain length when compared precisely (delta = 0 bp). (F) Same as E, but with delta = 6 bp.
We then considered for each read mapping the list of all its introns, which we refer to as an intron chain. In 81.8% of RT read pairs (of 28,330 total pairs in which both reads are spliced), the PacBio and the ONT read had identical intron chains. In 2% of cases, the intron chains were not contradictory, but the PacBio read had extra intron(s) at its extremities, whereas ONT reads had longer intron chains only in 0.6% of pairs. In the remaining 15.7%, PacBio and ONT intron chains disagreed with each other. When compared with the annotation, 17% of the disagreeing RT read pairs had both intron chains that corresponded to different known transcripts (green in Fig. 3D, top left), whereas in 15% of cases, both were inconsistent with the annotation (dark blue in Fig. 3D, top left). In these cases, it is not easy to ascertain which mapping is true. However, in a large fraction (65%) of the disagreeing pairs, the PacBio mappings were consistent with the annotation, whereas ONT mappings were not (yellow in Fig. 3D, top left). In this case, assuming that the PacBio intron chain is in fact correct appears more parsimonious than the contrary.
The above statements are based on a single-base interpretation of PacBio and ONT splice sites (delta = 0 bp; Methods). To account for slight shifts in splice-site mapping, we explored inexact intron chain comparison, in which junctions are considered equal if the distance between them does not exceed 6 bp (delta = 6 bp). This reduced disagreements between paired PacBio and ONT reads by 48%. Among the remaining disagreements, in nearly half of the cases the PacBio mapping corresponded to an annotated transcript, whereas the ONT read did not (Fig. 3D, middle and right). Overall, 43% of ONT and 15% of PacBio mappings inconsistent with the annotation at delta = 0 bp were reclassified as annotated with delta = 6 bp. Notably, further increasing delta to 10 bp affects only a small portion of reads: specifically, 78 PacBio and 468 ONT reads (Supplemental Table S6).
In addition, we compared intron chains of PCR duplicated read pairs sequenced on the same platforms for intra-molecular consistency (Supplemental Fig. S8). Indeed, when two reads that correspond to one original RNA molecule disagree in terms of alignment, only one can be correct. As expected, PacBio read pairs originating from PCR duplicates have significantly lower intron chain disagreement (1.8% with delta = 0 bp, 0.4% with delta = 6 bp) compared with ONT (8.3% with delta = 0 bp, 4.7% with delta = 6 bp), thus confirming the observations stated above.
We further hypothesized that the fraction of disagreeing RT read pairs would increase with the number of splice sites per read. Indeed, reads with eight or more introns disagreed with its pair approximately threefold more often than reads with two introns. However, read pairs with eight or more introns still agreed in 70% of cases (Fig. 3E). Using delta = 6 bp reduces disagreements but roughly preserves the trend (Fig. 3F). Broadly similar observations were also made for ScISOr-Seq data (Supplemental Fig. S5). These observations suggest that other factors beyond the intron chain length influence disagreements between PacBio and ONT reads. We therefore investigated sequence characteristics of disagreeing introns.
Sequence characteristics underlying disagreements within RT read pairs
We then analyzed alternative splicing events in 23,356 pairs of PacBio and ONT reads in which both reads in a pair were spliced, uniquely mapped, and unambiguously assigned to an isoform (Methods). Exon skipping with respect to the identified isoform was observed 461 times (1.9%) in ONT data and only 45 times (0.2%) in PacBio data (Fig. 4A). Exon skipping in ONT data is typically observed in exons ≤40 bp; however, in PacBio data, skipping is rarely observed in exons >15 bp (Fig. 4B). Minimap2 uses exact matching of certain k-mers with default k = 15 (Li 2018), followed by dynamic programming. However, sequencing errors, especially in ONT data, may cause the above short exons to be missed.
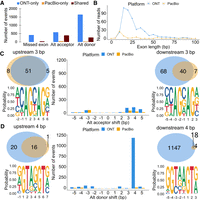
Exon and splice site characteristics underlying disagreements between PacBio and Nanopore in Sl-ISO-Seq data. (A) Number of missed exons (left), alternative acceptors (middle), and donors (right) with respect to the reference annotation that occur only in ONT read (blue), only in PacBio read (yellow), and in both reads from an RT read pair (brown). (B) Length distribution for skipped exons in PacBio reads (yellow) and ONT reads (blue). (C, middle) Number of alternative acceptor sites in PacBio (yellow) and ONT reads (blue) with respect to the distance from the annotated acceptor site. (Top left) Venn diagram for 3-bp upstream alternative acceptor sites in PacBio (yellow) and ONT reads (blue) from an RT read pair. (Bottom left) Nucleotide frequency for loci where 3-bp upstream acceptor sites occur. (Top right) Venn diagram for 3-bp downstream alternative acceptor sites in PacBio (yellow) and ONT reads (blue) from an RT read pair. (Bottom right) Nucleotide frequency for loci where 3-bp downstream acceptor sites occur. (D, middle) Number of alternative donor sites in PacBio (yellow) and ONT reads (blue) with respect to the distance from the annotated donor site. (Top left) Venn diagram for 4-bp upstream alternative donor sites in PacBio (yellow) and ONT reads (blue) from an RT read pair. (Bottom left) Nucleotide frequency for loci where 4-bp upstream donor sites occur. (Top right) Venn diagram for 4-bp downstream alternative donor sites in PacBio (yellow) and ONT reads (blue) from an RT read pair. (Bottom right) Nucleotide frequency for loci where 4-bp downstream donor sites occur.
Alternative acceptors (4.2% of ONT reads; 2% of PacBio reads), and comparatively more often, alternative donors (8.3% for ONT and 1.3% for PacBio) were more commonly observed than exon skipping (Fig. 4A). Thus, overall, 11.6% of ONT reads and 3.7% of PacBio reads showed one or more discrepancies to the annotation. We found similar trends in the single-cell data, albeit with higher inconsistencies for ONT data (Supplemental Fig. S7A,B). Discrepancies between an alignment and annotation found in both PacBio and ONT likely are novel isoforms. Such cases generally exceed discrepancies supported by PacBio-only (Fig. 4A).
From here on, we only consider canonical introns with GU/AG splice sites. We found that inconsistent acceptors were usually shifted by 3–5 bp downstream in ONT-only, whereas 80% upstream 3-bp shift (“NAGNAG”) acceptors were supported by both PacBio and ONT. Thus, downstream ONT acceptor shifts are questionable, whereas upstream NAGNAG shifts appear often true (Fig. 4C). For inconsistent donors, a downstream shift of 4 bp predominantly occurred for ONT data and with a much smaller overlap between both technologies than for the acceptor sites (Fig. 4D). Such shifts are caused by misalignment at the commonly known GUNNGU donor motif (Wang and Ruvinsky 2010). In summary, downstream 4-bp shifts from an annotated donor observed in ONT are doubtful, whereas 3-bp shifts from an annotated acceptor harbor a significant number of trustworthy novel splice sites. Broadly similar observations were made with ScISOr-Seq data (Supplemental Fig. S7C,D).
It is worth noting that modern transcriptome aligners can use annotated splice junctions. This highly reduced the discrepancies between ONT and the annotation but had a marginal effect on PacBio and cases for which ONT and PacBio agreed. Using the annotation makes PacBio seem to have more inconsistencies than ONT, possibly because novel ONT splice sites are overcorrected to the annotation (Supplemental Fig. S7E–H).
Extrapolating characteristics observed in barcoded read pairs to nonbarcoded ONT reads
The observations described above are based on RT read pairs, which require a detected barcode and UMI in both the PacBio read and the ONT read. However, as opposed to PacBio data, ONT reads with barcodes have higher Phred scores than those without (Fig. 1D). To understand quality effects on read characteristics, we analyzed the entire Sl-ISO-seq ONT data, which mimics nonbarcoded transcriptomic experiments. We observed that the aligned length increased from 532 bp for read-wise Phred-score = 10 reads to 815 bp for Phred-score = 20 (Fig. 5A). Similarly, high-quality reads had one more detected intron on average: 2.7 and 3.7 introns for spliced reads with Phred score 10 and 20, respectively (Fig. 5B).
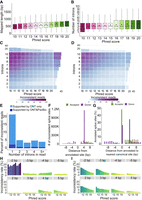
Characteristics of all ONT reads including nonbarcoded ones for Sl-ISO-Seq data. (A) Aligned read length with respect to read Phred score (average across all read bases) for all ONT reads from the Sl-ISO-Seq data set. (B) Read intron chain length with respect to read Phred score for all ONT reads from the Sl-ISO-Seq data set. (C) Heatmap showing average inconsistency rate between read intron chains and annotated intron chains (exact comparison, delta = 0 bp) with respect to read Phred score (x-axis) and intron chain length (y-axis) for all ONT reads from the Sl-ISO-Seq data set. Barplot at the top (on the right side) summarizes the inconsistency rate with respect to only the Phred score (only intron chain length). Purple corresponds to a higher inconsistency rate, and light blue indicates a lower inconsistency. (D) Same as C, but using inexact intron chain comparison (delta = 6 bp). (E) Histogram showing a fraction of inconsistent ONT reads that have at least one intron entirely contained inside an annotated exon. Dark blue represents reads for which the contained intron is supported by at least one PacBio read, and light blue corresponds to the rest of ONT reads. (F) Number of inconsistent donor (purple) and acceptor (green) splice sites in ONT reads from the Sl-ISO-Seq data set with respect to the distance from the annotated splice site. (G) Percentage of annotated canonical donor (green) and acceptor (purple) splice sites with respect to distance to the nearest canonical dinucleotides (GU for donors, AG for acceptors). Zero corresponds to the case when no canonical dinucleotides were detected within 10 bp. (H) Inconsistency rates of individual acceptor splice sites in ONT reads from the Sl-ISO-Seq data set with respect to reads’ Phred scores. Each histogram represents splice sites with a certain distance from the annotated splice site (gray bars on top). (I) Same as H, but for donor splice sites.
Furthermore, we compared each read's intron chain against annotated transcripts (Methods). The inconsistency rate goes down with higher Phred score: 28% of reads are inconsistent with the annotation for Phred-score = 10 (n = 206,675), but only 15% are inconsistent for reads with Phred-score = 20 (n = 2,667,759) (Fig. 5C). Moreover, as found previously (Au et al. 2013; Sharon et al. 2013; Tilgner et al. 2013, 2015), inconsistency increases with longer intron chains; that is, 40% of reads with seven or more introns are inconsistent, which is approximately 2.7-fold more than for reads containing three or fewer introns (∼15%). Similar trends were observed when intron chains were compared using delta = 6 bp, although overall inconsistency rates dropped (Fig. 5D). However, we noticed that the lowest inconsistency rate is observed for reads with two introns (11%), rather than for single-intron reads (17%). Although this observation was previously reported, no explanation was found (Sharon et al. 2013; Tilgner et al. 2013, 2015). We hypothesized that aligners can arbitrarily split a read into two exons, whereas such splits into three or more exons are less likely. Additional analysis (Methods) showed that 19% of inconsistent single-intron alignments have their intron entirely within an annotated exon, approximately fivefold more than for alignments with two introns (3.5%) (Fig. 5E). Moreover, only 12% of such mappings are supported by PacBio reads. Thus, rather than representing true alternative isoforms, some inconsistent single-intron mappings likely occur owing to misalignments.
We further examined individual splice sites by considering all canonical introns in ONT alignments that matched annotated canonical introns with a loose threshold of 10 bp. Similar to barcoded ONT reads, mapped acceptors are rarely shifted upstream and are more consistent with the annotation than donors, which have a dominant 4-bp downstream shift caused by the GUNNGU motif (Fig. 5F). To understand the major source of these shifts, we computed distances from all annotated splice sites to the nearest GU/AG (Fig. 5G). As these distances strongly resemble the distribution of ONT shifts, we hypothesized that shifts may depend on the proximity of GU/AG in general, rather than a particular motif.
Thus, we analyzed the inconsistency rate of annotated splice sites with respect to (1) the nearest GU/AG and (2) read quality (Methods). Donor inconsistency strongly depends on both read quality and distance to the nearest GU (Fig. 5I). For example, the probability of a downstream 4-bp shift caused by a GUNNGU motif is approximately twofold lower than of a downstream 2-bp shift near a GUGU (3.06% and 6.48%, respectively), despite GUNNGU being vastly more frequent than GUGU overall (36,663,585 and 1,066,886 of total detected splice sites near respective motif). Acceptor sites, on the other hand, show visible quality dependency only for downstream shifts, most likely owing to rare occurrences of upstream AGs. The upstream shifts are dominated by NAGNAG acceptors, which show only a 1.5-fold decrease in inconsistency rate between reads with Phred-score 10 and 20 (4.25% vs. 2.86%). As this difference is noticeably smaller than that for all acceptors (approximately 4.4-fold, 3.13% vs. 0.71%), in conjunction with our RT read pair analysis, it suggests that a portion of the upstream NAGNAG shifts may be real.
In summary, because of the elevated number of sequencing errors, ONT read alignments obtained with minimap2 may not provide exact splice site coordinates, especially for the cases when (1) a read has low quality, (2) a read spans multiple introns, and (3) canonical dinucleotides are located near splice sites. To avoid a potential misinterpretation of reads disagreeing with the annotated transcripts, one should treat such alignments with additional care or use inexact intron comparison when matching the annotation.
Splice site correction improves transcript discovery precision
Based on the observations made for ONT data, we implemented an algorithm for correcting splice junctions in individual reads with the aid of the annotation. This algorithm works with aligned reads and is capable of restoring (1) skipped short exons and (2) incorrectly detected splice sites (Methods). To evaluate how the designed algorithm affects transcript discovery, we simulated ONT reads with NanoSim (Hafezqorani et al. 2020) because the ground truth is unknown for the real data used in this study. Although we could use PacBio reads from the same RT read pair for verification, the fraction of ONT reads having an RT read pair is comparably low and not suitable for transcript model construction.
Transcript discovery was performed using StringTie2 (Kovaka et al. 2019). To mimic real-life situations, we removed some transcripts from the annotation before running our correction algorithm and StringTie2. The generated transcripts were matched against the set of “expressed” transcripts (known) and ones that were removed from the annotation (novel) using gffcompare (Pertea and Pertea 2020) to compute precision and recall of different approaches.
To understand the effect of splice site correction, we generated read alignments using (1) deSALT with default options, (2) minimap2 with default options and without correction, (3) minimap2 with the annotation and without additional correction, (4) minimap2 with the annotation and FLAIR (Tang et al. 2020) correction, (5) minimap2 with the annotation and the additional correction by 2passtools (Parker et al. 2021), and (6) minimap2 with the annotation and the additional correction with our algorithm.
Table 1 demonstrates that transcripts generated by StringTie2 using deSALT alignments have significantly lower quality compared with minimap2. Further analysis of deSALT alignments showed that the tool often incorrectly reports the transcript's strand (for 1.3 million out of 2.6 million alignments), which can substantially affect downstream analysis.
Running minimap2 with the annotation greatly improves results for novel transcripts. Using FLAIR for additional correction slightly improved precision but significantly reduced recall (a reduction from 86.4% to 75.7% overall, from 64.2% to 31.0% for novel isoforms). At the same time, the additional correction with our algorithm makes substantial improvements: Overall recall and precision increase by 0.9% and 8.8%, respectively, whereas the precision of novel transcripts improves by ∼50% (from 21.9% to 32.5%) as the number of false positives among novel transcripts decreases 1.6-fold (from 12,135 down to 7368). 2passtools performed similarly well, although it was slightly worse in terms of precision (a reduction from 76.5% to 71.4% overall, from 32.5% to 26.2% for novel isoforms). Although minimap2 and the correction algorithms only used information about known transcripts, this information aids the more precise detection of novel isoforms because they often share one or more exons with known isoforms.
Discussion
Different long-read RNA approaches are being increasingly used for isoform analysis (Koren et al. 2012; Au et al. 2013; Sharon et al. 2013; Tilgner et al. 2014, 2015, 2018; Oikonomopoulos et al. 2016; Garalde et al. 2018; Gupta et al. 2018; Tardaguila et al. 2018; Volden et al. 2018; Depledge et al. 2019; Wang et al. 2019; Tang et al. 2020; Joglekar et al. 2021; Sun et al. 2021; Hardwick et al. 2022). Therefore, understanding how each approach fares in detecting RNA traits is fundamental. Previous and current comparisons (Li et al. 2014a, 2014b; Weirather et al. 2017; Cui et al. 2020; Pardo-Palacios et al. 2021) of long-read technologies focused on such important data properties as overall read mappability, sequencing error rates, quantification analysis, isoform reconstruction, and alternative splicing detection. Indeed, broad conclusions of these studies correlate with our results: ONT does have noticeably higher yield compared with PacBio but also contains significantly more sequencing errors that complicate spliced alignment and consecutive transcript discovery. However, although being highly useful, none of these studies examines individual reads and compares multiple technologies for an individual RNA molecule. Moreover, previous approaches do not analyze spliced alignment error patterns and their dependency on isoform complexity.
Here, we used single-molecule barcoding technologies (Gupta et al. 2018; Joglekar et al. 2021) to sequence cDNA copies of single reverse transcription events on PacBio and ONT. Using perfectly matching barcodes and UMIs, we established the correspondence of a pair of ONT and PacBio reads to an individual RNA molecule. This procedure is highly specific while discarding doubtful PacBio-ONT read pairs but causes a selection for higher-quality reads in ONT but not in PacBio (see Fig. 1C,D). We found important differences that can avoid misinterpretation of data and guide researchers in their choice of technology.
PacBio and ONT reads from an RT read pair frequently differ in length, usually with up to 50 extra nucleotides in the PacBio read, which is small compared with the entire read. However, extra nucleotides in ONT also exist, although less frequently and fewer. These differences are small but can harbor poly(A) signals, Kozak sequences, or splicing factor binding sites. Additionally, we observed that PacBio reads extended more often to a known TSS and poly(A) site than ONT, a criterion important to defining complete isoforms. Of note, despite an overall low error rate, a significant fraction of PacBio errors arises from homopolymers (up to 40%), whereas ONT shows more errors but with less bias toward homopolymers.
With respect to exon–intron structures, PacBio–ONT inconsistencies mostly come from splice site shifts and skipped short exons owing to alignment errors. Such errors appear more often in ONT, although PacBio may also miss exons <15 bp. These inconsistencies increase as intron chains become longer.
The probability of a donor shift in an alignment primarily depends on the distance from the donor site to the nearest GU dinucleotide. However, the GTNNGT donor motif is very common in mammalian genomes, and thus, more donor shifts are explained by this double GT arrangement than with fewer or more separating nucleotides. Donor shifts in ONT reads are usually not confirmed by PacBio reads from the same RNA molecule. For acceptors, the most commonly observed shift is NAGNAG acceptors, and such shifts in ONT are often confirmed in the corresponding PacBio reads. Thus, GUNNGU donors in ONT that diverged from the annotation are most often not real, whereas such diverging NAGNAG acceptors are often likely real.
Because using only barcoded ONT reads creates a bias toward high-quality reads, we also analyzed all ONT reads. Low-quality reads are shorter, cover fewer introns, and disagree with the annotation more frequently than reads with high Phred scores. Other trends detected in RT read pairs comparison, such as higher inconsistency for long intron chains and intron shifts in the proximity of canonical dinucleotides, are generally preserved and therefore useful to nonbarcoded approaches.
We leveraged the above observations in an algorithm for correcting individual read alignments based on gene annotation. Using simulated Nanopore reads, we demonstrate that correcting splice site coordinates and misaligned microexons with our method has a noticeable positive effect on subsequent transcript detection using StringTie2. Moreover, annotation-based correction improves discovery of novel transcripts as they often share exons with known isoforms. Thus, the described findings can be of use for other researchers developing novel algorithms for long-read transcriptome analysis.
Although various sources provide different cost estimates for ONT and PacBio RNA sequencing, it seems that a single PromethION flow cell yield is about three to 10 times larger compared with a single Sequel II SMRT cell. Taking into account significantly higher accuracy of PacBio reads in terms of both per-base quality and the ability to correctly detect splice site positions, we believe that PacBio reads can be especially useful for creating de novo annotations and detecting novel isoforms in annotated genomes. As modern transcript discovery tools such as StringTie2 may generate a significant amount of false-positive isoforms when using ONT data, Nanopore sequencing should be used for automatic annotation with care. However, as Nanopore sequencing generates noticeably more data, it may be applied for estimating expression levels of annotated transcripts and further differential isoforms expression analysis, as well as detecting isoforms with subsequent manual validation. We believe that more studies and benchmarks, such as LRGASP (Pardo-Palacios et al. 2021), will shed additional light on this. Moreover, as the field of long-read transcriptomics continues its rapid expansion, novel protocols and computational methods will ensure more accurate usage of both PacBio and ONT technologies for all kinds of research projects.
Overall, the single-reverse-transcription event approach provides a powerful instrument for platform comparisons. In contrast to the comparisons of distinct molecules, this method offers tertium-non-datur reasoning, in which disagreements are known to be caused by errors of one of the platforms.
Methods
Experimental details
No new samples or sequencing data were generated for this study; however, we provide a brief description of the samples used and experimental protocols followed in the Supplemental Material. The description is taken from Joglekar et al. (2021). Sequenced data were previously submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession numbers GSE158450 and GSE178175. Read statistics are shown in Supplemental Tables S1 and S2.
Alignment to the reference genome using minimap2
PacBio reads were mapped to GRCm38 mouse reference genome with minimap2 v2.17 (Li 2018) using -t 16 -a -x splice:hq ‐‐secondary = no options. ONT reads were mapped with the -t 16 -a -x splice -k 14 ‐‐secondary = no options. When GENCODE M21 mouse annotation was used for read mapping, it was converted to BED format using paftools.js gff2bed command (included in minimap2 package) and provided to minimap2 using the ‐‐junc-bed option.
Alignment to the genome using deSALT
PacBio reads were mapped to GRCm38 mouse reference genome with deSALT v1.5.6 (Liu et al. 2019) using the -t 16 -x ccs options. ONT reads were mapped with the -t 16 -x ont1d options.
Alignment to the genome using GraphMap2
Reads were mapped to GRCm38 mouse genome with GraphMap2 v0.6.5 (Marić et al. 2019) using the -t 16 -x rnaseq options. In addition, GraphMap2 was run with GENCODE M21 mouse annotation provided using the ‐‐gtf option; however, the run failed with an error.
Alignment to the genome using uLTRA
PacBio reads were mapped to GRCm38 mouse reference genome with uLTRA v0.0.4.1 (Sahlin and Mäkinen 2021) using the ‐‐isoseq ‐‐t 24 options. ONT reads were mapped with the ‐‐ont ‐‐t 24 options.
Pairwise read alignment
Pairwise read alignment was performed using the Smith–Waterman local alignment algorithm implemented in the SSW Python library (Zhao et al. 2013) with default options.
Sequencing error rate
Sequencing error rates were computed based on minimap2 alignments using a three-way comparison between the reference genome and RT read pairs. An error at a certain position in a read from RT read pair was reported only when the alignment shows a difference from the genome (i.e., insertion, deletion, or substitution), whereas the second read from the pair either matches the genome or contains an alternative discrepancy at this position (e.g., another base is inserted). Identical differences from the reference genome (same position and nucleotide) detected in both PacBio and ONT reads from an RT read pair were not classified as sequencing errors. An error is deemed to occur within a homopolymer region if any 3-bp window in the genome that contains an error position consists of the same nucleotides.
k-mer identity and homopolymer compression
The k-mer identity (k = 14bp) with the reference genome was calculated using minimap2 alignments for each exon individually. We first extracted all genomic k-mers from the respective mapped region (of the exon) and then calculated the fraction of the k-mers that occurred within this exon in the read. Homopolymer compression (Au et al. 2012) was performed by substituting all stretches of identical nucleotides (≥2 bp) with a single nucleotide of the same kind in both read sequence and reference sequence from the respective mapping region. The k-mer identity was then computed in the same way as for noncompressed sequences.
TSS/poly(A) analysis
TSS and poly(A) sites were assigned to each read as previously performed (Joglekar et al. 2021). For published TSS, we used high-quality calls from the FANTOM5 Consortium (Lizio et al. 2015). Among all published TSS calls, within 100 bp of the 5′ end of the read mapping, we assigned the closest TSS to the read and none if there are no such calls within 100 bp. Using very recent poly(A) site calls (Herrmann et al. 2019), we applied a similar procedure to assign poly(A) sites to the read (within 100 bp of the 3′ end of the read mapping).
An RT read pair is considered as “agreeing” on TSS/poly(A) site assignment if both reads have an assigned TSS/poly(A) site and the two assigned sites are identical.
Intron chain comparison and inconsistency detection
Intron chains were compared against each other as ordered lists of coordinate pairs. In the precise intron chain comparison, two introns are considered equal if their splice site coordinates are identical (delta = 0 bp), whereas in the inexact comparison, each splice site is allowed to differ by delta = 6 bp at most. Intron chains are considered as agreeing if they are equal or if one chain is a subchain of another with respect to the given delta value and disagreeing otherwise.
To detect inconsistencies between reads in RT read pairs, intron chains for both reads were extracted from the BAM files obtained with minimap2 and compared against each other as described above. Similarly, to detect agreement between a read and the annotation, a read intron chain extracted from the BAM file was compared against intron chains of known transcripts from GENCODE M21 comprehensive mouse annotation. Read is deemed to be consistent if its intron chain agrees with at least one annotated transcript and is deemed inconsistent otherwise. Reads that do not overlap with annotated exons (i.e., entirely map to intergenic or intronic regions) are considered as uninformative and are ignored in the analysis.
Classification of splicing modifications
To classify splice site inconsistencies with the gene annotation, reads were assigned to known transcripts using a custom script, assign_reads.py available at GitHub (see Data access), which assigns mapped reads to known isoforms based on intron chains and nucleotide identity. For PacBio reads, the script was run with the ‐‐data_type pacbio_ccs option; for ONT reads, the ‐‐data_type nanopore was used. Benchmarking of the method on the simulated data is presented in the Supplemental Note, “Benchmarking of the read-to-isoform assignment algorithm” (Supplemental Table S4).
For further analysis, we selected unambiguous assignments with respective reported splicing modifications (skipped exons, alternative donor, or acceptor site). To investigate alternative donors and acceptors (i.e., shift frequency, nucleotide content), only introns with canonical splice sites were used (GT-AG).
The output of the script was also used to track the origin of inconsistent nonbarcoded reads. To detect reads having at least one intron entirely located within an annotated exon, we selected uniquely assigned reads having this specific type of inconsistency (additional novel intron according to our categories).
Splice site analysis for nonbarcoded reads
To analyze splice site consistency in nonbarcoded reads, we assigned each read intron separately to an annotated intron (rather than the entire intron chain) with a loose threshold delta = 10 bp. Such an approach allows us to maximize the number of investigated splice sites and consider individual introns even from inconsistent chains. For the analysis, we selected only cases when both read and annotation intron have canonical splice sites (GU/AG). We say that an assigned read intron correctly detects a splice site if its position is equal to the annotated splice site (0-bp difference), it detects incorrectly otherwise. The inconsistency rate of an annotated splice site is defined as the number of incorrect calls divided by the total number of read introns assigned. Each annotated splice site was classified according to the distance to the nearest GU (for donors) or AG (for acceptors) in the vicinity of 10 bp. It allowed us to compute overall inconsistency rates for splice sites with respect to this distance.
Splice site correction algorithm
The correction algorithm takes aligned reads and genome annotation as input. Each read is processed individually, as opposed to the classic transcript construction method that relies on clustering and splice site consensus (Kovaka et al. 2019; Tang et al. 2020; Wyman et al. 2020; Sahlin and Medvedev 2021). An aligned read is first assigned to a reference isoform based on inexact intron chain matching and exon similarity as described above. Further, each read is examined with respect to the accuracy of the detected intron structure. Coordinates of corrected alignments are output in BED12 format. The algorithm is available at GitHub (correct_splice_sites.py; see Data access).
Splice site correction algorithm: restoring skipped exons from neighboring splice sites
A reference exon is considered to be skipped during the alignment if (1) it is <50 bp, (2) it is spanned by a read intron, and (3) adjacent exons in the alignment contain extra sequences reaching into the annotated introns surrounding the reference exon and these two extra sequences are of a similar total length as the reference exon (Supplemental Fig. S9).
Splice site correction algorithm: correcting individual splice sites
With the same considerations as above, an individual spice site in the read is to be corrected if (1) it is no further than delta = 6 bp apart from a known splice site and (2) the read alignment has indels close to this position.
Simulating ONT data
To simulate ONT data we used the NanoSim software in transcriptome mode (Hafezqorani et al. 2020) using the pretrained ONT cDNA error model. However, examining the code, we found that NanoSim randomly selects a starting position of a read in an mRNA to simulate truncation. This is performed using a uniform distribution, thus assuming that 5′ and 3′ are identical, which is not the case for the real data. To avoid this pitfall, we mapped raw ONT reads to the reference transcripts using minimap2 (with -x map-ont option) and estimated the probabilities of the initial sequence being truncated on each side by N% of its length. We thus modified the NanoSim truncation procedure so that reference sequences are clipped according to empirically derived probabilities (Supplemental Fig. S10). In addition, we turned off the simulation of random decoy reads, which represent the background noise of the sequencing experiment. We simulated 30 million ONT reads using transcripts from the Mouse GENCODE v26 basic annotation (Frankish et al. 2021). In addition, a 30-bp poly(A) tail was attached to every transcript before simulation. Each transcript with at least one generated read was considered as “expressed” and then represented the ground truth.
Evaluating transcript model construction
We first generated a reduced genome annotation by removing 20% of expressed spliced transcripts. Removed transcripts are considered novel, whereas expressed transcripts kept in the annotation represent the set of known models. Using these two sets allowed to independently evaluate the ability of the algorithm to report known and discover novel isoforms. StringTie2 results were similarly split into novel and known models based on the information provided in the GTF file, and gffcompare (Pertea and Pertea 2020) was further launched to estimate precision and recall.
In addition, we performed the series of experiments with different fractions of excluded expressed isoforms to analyze how this parameter affects the results (Supplemental Note, “Novel isoform discovery with different fractions of unknown transcripts,” Supplemental Table S5).
Running StringTie2
StringTie2 (Kovaka et al. 2019) was run with -L option for long reads and the reduced genome annotation.
Running FLAIR
Correction module of FLAIR (Tang et al. 2020) was run with minimap2′ alignments (converted to BED file using bam2Bed12.py) and the reduced genome annotation as an input.
Running 2passtools
First, 2passtools (Parker et al. 2021) “score” command was run on minimap2′ alignments. Results were filtered with the “filter ‐‐exprs “decision_tree_2_pred'” command and provided to minimap2 using the ‐‐junc-bed option.
Data access
Simulated data generated in this study are available at Zenodo (https://doi.org/10.5281/zenodo.6325107). The barcode detection tool is available as the “GetBarcodes” function in the scicorseqr R-package (https://github.com/noush-joglekar/scisorseqr). All scripts used for data analysis and spliced alignment correction are available as Supplemental Code and at GitHub (https://github.com/ablab/platform_comparison).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Alyona Sidorova and Alexandra Bazarova for their aid with the analysis. This work was supported by the National Institute of General Medical Sciences (grant 1R01GM135247-01 to H.U.T.), St. Petersburg State University, Russia (grant ID PURE 93023187 to A.M. and A.D.P.), and Brain Initiative (grant 1RF1MH121267-01). Scientific research was performed at the Research Park of St. Petersburg State University Computing Center.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276405.121.
-
Freely available online through the Genome Research Open Access option.
- Received November 18, 2021.
- Accepted March 5, 2022.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








