
GC content, but not nucleosome positioning, directly contributes to intron splicing efficiency in Paramecium
- Stefano Gnan1,4,
- Mélody Matelot2,4,5,
- Marion Weiman3,6,
- Olivier Arnaiz3,
- Frédéric Guérin2,7,
- Linda Sperling3,
- Mireille Bétermier3,
- Claude Thermes3,
- Chun-Long Chen1 and
- Sandra Duharcourt2
- 1Institut Curie, Université PSL, Sorbonne Université, CNRS UMR3244, Dynamics of Genetic Information, Paris, 75005 France;
- 2Université Paris Cité, CNRS, Institut Jacques Monod, F-75013 Paris, France;
- 3Université Paris-Saclay, CEA, CNRS, Institute for Integrative Biology of the Cell (I2BC), 91198 Gif-sur-Yvette, France
-
↵4 These authors contributed equally to this work.
Abstract
Eukaryotic genes are interrupted by introns that must be accurately spliced from mRNA precursors. With an average length of 25 nt, the more than 90,000 introns of Paramecium tetraurelia stand among the shortest introns reported in eukaryotes. The mechanisms specifying the correct recognition of these tiny introns remain poorly understood. Splicing can occur cotranscriptionally, and it has been proposed that chromatin structure might influence splice site recognition. To investigate the roles of nucleosome positioning in intron recognition, we determined the nucleosome occupancy along the P. tetraurelia genome. We show that P. tetraurelia displays a regular nucleosome array with a nucleosome repeat length of ∼151 bp, among the smallest periodicities reported. Our analysis has revealed that introns are frequently associated with inter-nucleosomal DNA, pointing to an evolutionary constraint favoring introns at the AT-rich nucleosome edge sequences. Using accurate splicing efficiency data from cells depleted for nonsense-mediated decay effectors, we show that introns located at the edge of nucleosomes display higher splicing efficiency than those at the center. However, multiple regression analysis indicates that the low GC content of introns, rather than nucleosome positioning, is associated with high splicing efficiency. Our data reveal a complex link between GC content, nucleosome positioning, and intron evolution in Paramecium.
In eukaryotes, genomic DNA is compacted by histones into chromatin. The basic unit of chromatin is the nucleosome, which comprises a histone octamer made of the four core histones (H2A, H2B, H3, and H4) and 146–147 bp of DNA wrapped around it (Luger et al. 1997; Parmar and Padinhateeri 2020). Nucleosomes are not randomly located along the genome but are positioned with respect to DNA sequence. The affinity of DNA for histone octamers and the energy needed to bend different DNA fragments around the histone octamer are influenced by the primary DNA sequence, which is therefore an important determinant of nucleosome positioning along the genome (Iyer and Struhl 1995; Segal et al. 2006; Peckham et al. 2007; Tillo and Hughes 2009; Tillo et al. 2010; Vaillant et al. 2010; Lorch et al. 2014). Nucleosome positioning is highly dynamic, and its regulation is crucial to control chromatin accessibility, recruitment of chromatin modifiers, and transcription factors (Jiang and Pugh 2009; Prendergast and Semple 2011; Bartholomew 2014; Kornberg and Lorch 2020). In most genomes, genes display a nucleosome-free region (NFR) at the 5′ of their transcription start site (TSS) owing to the formation of complexes made by transcription factors around promoter regions (Bernstein et al. 2004; Yuan et al. 2005; Jiang and Pugh 2009). A second NFR is present at transcription termination sites (TTSs), likely owing to the adverse nucleotide composition of the poly(A) signal (Fan et al. 2010; Chereji et al. 2016). Nucleosomes are organized in regular arrays with a periodic distance called the nucleosome repeat length (NRL). Such periodicity is especially evident over gene bodies, and it is species and cell type specific (Allan et al. 2013; Beshnova et al. 2014). Genome-wide studies have shown that nucleosomes are preferentially positioned in exons compared with introns in diverse organisms, including Schizosaccharomyces pombe, Drosophila, worms, and humans (Andersson et al. 2009; Nahkuri et al. 2009; Schwartz et al. 2009; Spies et al. 2009; Tilgner et al. 2009; Iannone et al. 2015). Several lines of evidence indicated that a well-positioned nucleosome might slow down RNA polymerase II and favor exon inclusion and alternative splicing (Wilhelm et al. 2011; Jonkers et al. 2014), suggesting a functional role of nucleosome arrays during mRNA maturation. This is in agreement with recent studies showing that intron splicing can occur in a cotranscriptional manner (Brody et al. 2011; Herzel et al. 2017). Some studies have suggested that GC richness at exons, and not nucleosome positioning per se, is important for intron splicing (Amit et al. 2012; Gelfman et al. 2013). Yet, the contribution of nucleosome positioning to intron splicing efficiency has not been investigated thoroughly. The nonsense-mediated decay (NMD) machinery recognizes and degrades transcripts containing premature termination codons (Lykke-Andersen and Jensen 2015; Kurosaki et al. 2019). Therefore, most of the missplicing or unsplicing events are removed rapidly by this powerful surveillance mechanism to avoid the production of erroneous proteins. To date, most studies estimated splicing efficiency from NMD-proficient cells, which eliminate most missplicing or unsplicing events and therefore cannot provide a solid evaluation of the intrinsic efficiency of intron splicing.
The ciliate Paramecium tetraurelia is a unicellular eukaryotic model organism. Like all ciliates, two distinct types of nuclei coexist within the same cytoplasm in P. tetraurelia (Aury et al. 2006). The diploid germline micronucleus (MIC) is transcriptionally silent during vegetative growth and transmits the germline genome to sexual progeny through meiosis, whereas the highly polyploid somatic macronucleus (MAC) is responsible for gene expression (Bétermier and Duharcourt 2014). The more than 90,000 introns annotated in the MAC genome are among the shortest reported in eukaryotes (18–33 nt, 25 nt on average) (Jaillon et al. 2008). How such a large number of tiny introns can be efficiently spliced is not known. In P. tetraurelia, no exon skipping has been reported so far (Jaillon et al. 2008; Saudemont et al. 2017). Introns are associated with weak splice signals, as shown by the very low information content of 5′ and 3′ splice sites, with only the first and last three bases of introns being highly constrained (Jaillon et al. 2008). A strong counter-selection for introns that cannot be detected by the NMD machinery was previously shown, suggesting that introns rely on NMD to compensate for suboptimal splicing efficiency and accuracy (Jaillon et al. 2008; Saudemont et al. 2017). Whether nucleosome positioning or other factors, such as GC content, can regulate splicing efficiency and shape intron evolution in Paramecium has not been studied so far.
Here, we investigated a possible role of nucleosome positioning in the recognition of introns in P. tetraurelia. We mapped the nucleosome occupancy in the somatic MAC through paired-end (PE) MNase-seq. We compared the positioning of nucleosomes with that of introns, whose accurate splicing efficiency data were determined from NMD-depleted cells.
Results
Genome-wide nucleosome position profiling along the Paramecium somatic genome
Using MNase-seq, we derived a first nucleosome positioning profile of the MAC genome of P. tetraurelia during vegetative growth. Both chromatin samples and naked MAC DNA controls were digested to mononucleosome size (∼150 bp) (Fig. 1A,B; Supplemental Fig. S1A). The results obtained from two biological replicates were highly reproducible (Pearson's correlation R = 0.94) (Supplemental Fig. S1B). We therefore combined data from both biological replicates for downstream analyses. All the data presented in the main figures were obtained with the average of two chromatin samples and two naked DNA controls, respectively. The results of each individual sample are reported in the Supplemental Figures. Using the gene annotation, together with the TSSs identified by 5′ CAP-seq and TTSs identified by poly(A) detection (Arnaiz et al. 2017), we investigated the nucleosome occupancy along transcription units and around their extremities. As described in other eukaryotes, P. tetraurelia presents an enriched nucleosome density over the transcription units compared with the flanking regions, showing regular arrays of nucleosomes over transcription units (Fig. 1C,D; Supplemental Fig. S1C,D). As expected, we were able to identify NFRs upstream of the TSSs of Paramecium genes, followed by an array of well-positioned nucleosomes (Fig. 1C,D; Supplemental Fig. S1C). The analysis of TTSs shows regions with very low nucleosome occupancy downstream from the TTSs and a weakly organized array toward the gene body (Fig. 1D; Supplemental Fig. S1D). We further separated gene pairs into three groups based on their relative orientation: tandem (n = 20,233), convergent (n = 8876), and divergent (n = 8867) (Fig. 1E; Supplemental Fig. S1E–G). We found that nucleosome arrays are clearly visible upstream of the TTSs only when genes are positioned in tandem (Fig. 1E; Supplemental Fig. S1F), but not in convergent pairs (Fig. 1E; Supplemental Fig. S1G). This observation suggests that the nucleosome positioning at TTS observed for tandem genes might be owing to the downstream TSS, as suggested for Saccharomyces cerevisiae (Chereji et al. 2017). Alternatively, convergent genes might be influenced by the transcription readthrough of the gene in the opposite orientation.

Nucleosome occupancy along the Paramecium MAC genome. (A) Schematic representation of the MNase-seq experiment. (B) MNase digestion of MAC chromatin with increasing MNase enzyme concentration. (C) Heatmap showing nucleosome occupancy ±1 kb around the center of each gene ordered by gene size (small genes on top and large genes at the bottom) for 38,143 genes located on scaffolds that are at least 200 kb long. (Left) Average of two chromatin-treated samples (Chromatin); (right) average of two naked DNA control samples (Naked DNA). (D) Average nucleosome occupancy around transcription start sites (TSSs) identified by 5′ CAP-seq on the left and transcription termination sites (TTSs) identified by poly(A) detection on the right: in green is the average profile of the chromatin-treated sample (Chromatin); in blue, the average profile of the naked DNA sample (Naked DNA); and in magenta, the Chromatin/Naked DNA ratio, enrichment of which is shown on the second axis on the right (red axis). (E) Average nucleosome occupancy ±1 kb around the center of intergenic regions: same color-code as in panel D. Intragenic regions have been divided into three groups based on the relative positions of gene pairs: tandem (left), convergent (middle), or divergent (right). (F) Inter-center distance between well-positioned nucleosomes (Methods) on the same scaffold. In blue are distance distributions from actual data (from 1 bp to 2 kb, binning = 1); in orange, the Gaussian smoothed signal. Black dashed lines indicate the local maxima (peak centers) of the smoothed data (Methods). (G) In orange are the first eight local maxima from panel F ordered by increasing distance; in blue, the linear fitted model. At the bottom right, information about linear fitting and estimated NRL (Mean ± SD) is given. P-value is calculated using a two-sided Z-test.
Based on our nucleosome position calling and using only well-positioned nucleosomes identified in both replicates (see Methods), we calculated the NRL (Methods). We found that P. tetraurelia displays one of the smallest NRL reported in eukaryotes (150.89 ± 0.57 bp on average) (Fig. 1F,G; Supplemental Fig. S1H,I), close to the 156 ± 2 bp of S. pombe (Godde and Widom 1992), which is much smaller than the 167 bp of S. cerevisiae (see Discussion) (Vaillant et al. 2010). In humans, the NRL within gene bodies is smaller than outside (Valouev et al. 2011). We performed a similar analysis subsetting nucleosomes based on whether their centers overlap with gene bodies or not. We found a negligible difference between the NRL within gene bodies (151.00 ± 0.94 bp, >80% of the analyzed sequences) and those outside of genes (150.29 ± 1.29 bp) (Supplemental Fig. S1J).
The tiny introns of Paramecium genes are frequently associated with inter-nucleosomal DNA
We then analyzed nucleosome positioning over gene bodies. In P. tetraurelia, exons range from several nucleotides to a few kilobases (Fig. 2A; for transcription units identified by 5′ CAP-seq and poly(A) detection, see Supplemental Fig. S2A) and are interspersed with tiny introns, the majority spanning between 20 and 35 bp with a median size of 25 bp (Fig. 2B). The distribution of exon size shows a peak ∼150 bp, close to the size of nucleosomes in P. tetraurelia, which is smaller than the simulated exon size by assuming uniform exon sizes within each gene (Fig. 2A; Supplemental Fig. S2A). By visual inspection of the nucleosome occupancy profiles, we noticed a tendency of the MNase signal to be stronger over exons, leaving the introns preferentially between two nucleosome peaks (Fig. 2C; for each MNase-digested chromatin sample, see Supplemental Fig. S2B). This was especially visible when we examined the nucleosome density over introns sorted by the distance of each intron center to the closest nucleosome center (Fig. 2D; Supplemental Fig. S2C). This distance is significantly higher than what we would expect by calculating the distance of random positions inside gene bodies to the closest nucleosome center (P-value < 10−10 calculated with Mann–Whitney U test, one-sided, alternative H1: Intron distance from the closest nucleosome is higher than random chance) (Fig. 2E). Using this distance, we grouped introns into three categories: central, proximal, and distal (as illustrated in Fig. 2F; Supplemental Fig. S2C). We calculated their distribution and compared it with that of exons <300 bp (roughly the same sample size) categorized in the same way (Fig. 2G; Supplemental Fig. S2D). Introns were found enriched at distal positions, that is, located in the regions between two neighbor nucleosomes, compared with exons (45% vs. 22%, respectively). In contrast, exons were more enriched in central positions compared with introns (46% vs. 28%, respectively). These distributions are statistically significantly different: P-value < 10−10 calculated with a χ2 test (Fig. 2G). Moreover, P. tetraurelia exons seem to favor mononucleosome length sizes with 35% of exons having sizes composed of between 100 bp and 200 bp. Such a size distribution is significantly shorter than what would be expected if we simulated exon sizes as uniformly distributed within each transcript, in which case only 24% of the exons would fall in this range (P < 10−10, Mann–Whitney U test, one-sided, alternative H1: Simulated exons are bigger than real exons) (Fig. 2A). Similar results were obtained using only exons of transcription units whose extremities are identified by both 5′ CAP-seq and poly(A) detection (Supplemental Fig. S2A). This distribution of exon sizes might reflect some selective constraint keeping introns in phase in distal position, that is, at the edge of the nucleosome.

Inter-nucleosomal DNA is frequently associated with intron position. (A) Histogram showing exon size distribution (bin size = 25 bp): in blue are real exons; in orange, simulated exons created assuming uniform exon sizes within each gene. (B) Histogram showing intron size distribution (bin size = 1 bp). (C) Example track reporting nucleosome occupancy over genes with intron locations indicated by vertical dashed lines. We can observe nucleosome-free regions (NFRs) around the gene promoters and introns frequently associated with inter-nucleosomal DNA. (D) Heatmap showing nucleosome occupancy ±200 bp around intron centers. Introns are ordered based on increasing distances from their center to the closest nucleosome center, from top to bottom. The average of the chromatin samples is shown on the left and the average of the naked DNA samples on the right, with the same color-code as in Figure 1C. Vertical black dashed lines delineate the average size of an intron (25 bp). Individual samples are displayed in Supplemental Figure S2C. (E) Histogram reporting the distance of an intron center to the closest nucleosome center (red). For each intron, a random position inside the corresponding gene body was selected, and the distance to its closest nucleosome center is reported (green). Bin size = 5 bp. (F) Schematic representation of the criteria to assign features for each intron (or exon) into one of the three classes, based on the distance (d) between its center and the closest nucleosome center position: central, d ≤ 25 bp; proximal, 25 bp < d < 50 bp; and distal 50 bp ≤ d ≤ 75 bp. (G) Relative distribution of introns, exons, and both features over categories defined in panel F for the introns overlapping with a fixed nucleosome (∼70% of all introns; see Methods) and exons with a size <300 bp overlapping with fixed peaks. See Supplemental Figure S2D, including the features with d > 75 bp.
Higher splicing efficiency for introns at the edges of nucleosomes
Previous studies have described the effect of nucleosome positioning on mRNA maturation in multiple organisms (Andersson et al. 2009; Nahkuri et al. 2009; Schwartz et al. 2009; Spies et al. 2009; Tilgner et al. 2009; Iannone et al. 2015). To address whether nucleosome positioning affects intron splicing in P. tetraurelia, we examined the relationship between nucleosome positioning and intron splicing efficiency, using published data sets from both wild-type (WT) and NMD-depleted cells (by RNAi-mediated knock down [KD] of UPF genes), which provide a measurement of the splicing efficiency of P. tetraurelia introns (Saudemont et al. 2017). Because NMD has been shown to play an important role in removing misspliced transcripts and different evolutionary constraints have been observed for NMD-sensitive (presence of a premature termination codon [PTC] after retention) and NMD-insensitive (absence of a PTC after retention) introns (Saudemont et al. 2017), we further divided our three positional categories (central, proximal, distal) of introns into NMD-sensitive or NMD-insensitive groups (Fig. 3A).
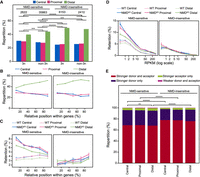
Nucleosome positioning is associated with intron splicing efficiency. (A) Relative distribution of different classes of introns. Introns are grouped based on their length (3n or non-3n) and whether their retention causes a premature termination codon making them sensitive to the nonsense-mediated decay (NMD) mechanism (NMD-sensitive) or not (NMD-insensitive). Within each group, introns are classified based on the distance to the closest nucleosome center as in Figure 2G. P-values are calculated using the χ2 test, and only the significant ones are indicated. (B) Intron repartition according to the categories defined in Figure 2F as a function of their relative position within a gene. Introns are grouped based on their NMD sensitivity. Bin size = 20%. A barplot representation with relative P-values is displayed in Supplemental Figure S3A. (C) The retention rate of introns in WT (dashed lines) and in NMD-depleted (NMDKD; solid lines) cells as a function of their relative position within a gene. Introns are grouped as in panel B. Error bars represent the SEM. P-values calculated using Mann–Whitney U test, and adjusted with a false-discovery rate (5%), are displayed in Supplemental Figure S3B. Bin size = 20%. (D) The retention rate of introns in WT (dashed lines) and in NMDKD (solid lines) cells as a function of gene expression levels. Error bars represent the SEM. Colors and groups are as in panel B. P-values calculated using Mann–Whitney U test, and adjusted with a false-discovery rate (5%), are displayed in Supplemental Figure S3C. (E) Relative characterization of introns, within the same categories as in panel B, based on the strength of splicing acceptor and donor sites. P-values are calculated using the χ2 test and adjusted with a false-discovery rate (5%). Tests were run between introns belonging to the same positional group or between introns belonging to the same NMD group. P-value in all the plots: (*) <0.05, (**) <10−2, (***) <10−3, (****) <10−4, (*****) <10−5, and (******) <10−6.
First, we observed that the proportion of distal introns is higher in NMD-insensitive introns compared with NMD-sensitive ones, independent of the introduction of a frameshift (3n vs. non-3n introns) (Fig. 3A). We could not observe statistically significant differences between 3n and non-3n NMD-insensitive intron distributions (P = 0.38, χ2 test), and only a minor significant increase of distal introns at the expense of central introns and proximal introns can be detected between 3n and non-3n NMD-sensitive introns (P < 10−3, χ2 test) (Fig. 3A). Because no major differences in the intron distribution between 3n and non-3n introns were observed, we decided to consider only the NMD state for subsequent analyses.
According to previous reports, a PTC is more likely to be recognized by the NMD system if it is located far away from the actual termination codon at the 3′ end of the gene (Brogna and Wen 2009; Vitali et al. 2019). We reasoned that an NMD-sensitive intron close to the TSS has a higher probability to induce a PTC far away from the actual termination codon. Therefore, we analyzed the distribution of intron positional categories with regard to nucleosomes (distal, central, proximal) as a function of their relative position within genes and of their NMD sensitivity. We found that, for NMD-insensitive introns, the proportion of distal introns is much higher than that of central introns for all distance classes, with only a slight increase of distal intron percentage toward the gene 3′ end (Fig. 3B; Supplemental Fig. S3A). However, for the NMD-sensitive introns, we observed a linear increase of the percentage of distal introns toward the gene 3′ end (Fig. 3B; Supplemental Fig. S3A). This indicates that introns close to the TTS are more frequently associated with distal positions, that is, at the edge of the nucleosome. To assess whether these introns close to the TTS are less sensitive to NMD, we monitored intron retention rates for the different intron groups. This confirmed that (1) the NMD pathway is more efficient for NMD-sensitive introns close to the TSS, that is, located at the beginning of a gene (Fig. 3C), and (2) much higher retention rates in NMD-depleted cells are observed for NMD-sensitive introns located near a TSS compared to those near a TTS (Fig. 3C, left panel). As expected, no difference can be observed for NMD-insensitive introns (Fig. 3C, right panel). For NMD-sensitive introns, we observed a higher splicing efficiency (i.e., lower retention rate) for introns located in distal positions compared with those in central and proximal positions independent of their relative position within a gene (Fig. 3C, left; Supplemental Fig. S3B). We conclude that NMD-sensitive introns located at distal positions, that is, at the edge of nucleosomes, are more efficiently spliced.
As shown by Saudemont et al. (2017), the intron retention rate is inversely correlated with the gene expression level and is higher for introns that can be detected by the NMD machinery than for those that cannot. In WT cells, both NMD-sensitive and NMD-insensitive introns showed similar retention rates, with higher retention rates for genes with lower expression levels (Fig. 3D). The retention rate of NMD-sensitive introns increased significantly upon NMD depletion, whereas it did not for NMD-insensitive introns (Fig. 3D). We extended this analysis to our intron positional categories. As expected, NMD-insensitive introns showed similar splicing efficiency for all intron classes in both WT and NMD-depleted cells (Fig. 3D, right panel; Supplemental Fig. S3C). We found that the retention rate of NMD-sensitive introns is lower for distal introns compared with the other two categories (Fig. 3D, left; Supplemental Fig. S3C), indicating again that NMD-sensitive introns located at the edges of nucleosomes are more efficiently spliced. This can already be observed in WT cells, whereas in NMD-depleted cells, in which nonsense mRNAs are no longer degraded, this difference is much stronger (Fig. 3D, left panel; Supplemental Fig. S3C). For the low-expressed genes (RPKM ≤ 1), the retention rate of central introns is 36.6% higher than that of distal introns, and it drops to 24.6% and 13.8% for the mid-expressed (1 < RPKM ≤ 10) and highly expressed (RPKM > 10) genes, respectively (Fig. 3D, left panel; Supplemental Fig. S3C). We further analyzed the proportion of intron positional categories within genes with different expression levels and found similar proportions for all expression classes (Supplemental Fig. S3D). Similar results were also observed for genes issued from the last whole-genome duplication (Aury et al. 2006) that have different expression levels (Supplemental Fig. S3E). Finally, after controlling both gene expression levels and the relative distance of the intron to the TSS, we still observed that the distal introns have a higher splicing efficiency than the central and proximal ones (Supplemental Fig. S3F).
It has been shown that the splicing efficiency of P. tetraurelia introns depends on the sequences at the donor and acceptor sites (Jaillon et al. 2008). We thus assessed whether this difference in splicing efficiency between our nucleosome-positional classes could be explained by a different distribution of stronger donor (5′-GTA) and/or stronger acceptor (3′-TAG) sites (Supplemental Fig. S3G) within different intron groups. As expected, NMD-insensitive introns were more frequently associated with both stronger donors and acceptors whatever the distance of the intron to the closest nucleosome center (Fig. 3E). In contrast, we found a minor increase, for the NMD-sensitive introns, in the association of distal introns with “weaker donor and acceptor” (0.82%) and “stronger acceptor only” (4.46%) intron groups compared with central introns (0.50% and 3.80%, respectively) with, respectively, 64% and 17% increases (Fig. 3E). This slight increase was not associated with a higher retention rate for distal introns compared with central introns. Instead, we did observe a reduced retention rate in distal introns (Fig. 3C,D). We conclude that the reduced retention rate in distal introns is not owing to a difference of donor/acceptor signals in this class.
GC content related to nucleosome positioning contributes to intron splicing efficiency at the edges of nucleosomes
It is well known that nucleosome positioning is highly associated with GC content: Nucleosome centers show higher GC than distal regions (Iyer and Struhl 1995; Peckham et al. 2007; Tillo and Hughes 2009; Vaillant et al. 2010; Lorch et al. 2014). In P. tetraurelia, we observed that NMD-sensitive introns have a higher GC content (18.9%, 18.4%, and 15.7% for central, proximal, and distal introns, respectively) than their NMD-insensitive counterparts (16.3%, 16.1%, and 13.2% for central, proximal, and distal introns, respectively) (Fig. 4A). Moreover, as we would expect, the central introns have the highest GC content followed by proximal and distal introns (Fig. 4A). We therefore analyzed the impact of GC content on intron retention rates. We found a direct correlation between GC percentage and retention rate in NMD-sensitive introns, yet no statistically significant difference could be observed between different intron groups (Fig. 4B; Supplemental Fig. S4A). This suggests that GC-content anticorrelates with intron splicing efficiency. To further evaluate how different parameters, such as GC content, gene expression level, intron relative position within genes, nucleosome positioning, and RNA secondary structure prediction (Supplemental Table S1), affect intron splicing efficiency, we first filtered the parameters by trying to lower the variance inflation factor (VIF) below five and then used the resulting parameters to train a multivariate regression model as previously described (Chen et al. 2010). Only the parameters with a statistically significant contribution were retained (Methods). The final fitted model has an R = 0.62, which explains 39% of the variation in intron splicing efficiency measured in NMD-depleted cells. The model allowed us to estimate the contribution of each parameter (for a full list of parameters, see Supplemental Table S1).
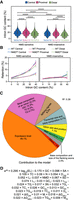
GC content related to nucleosome positioning contributes to intron splicing efficiency. (A) GC content (%) distribution of introns based on the distance to the closest nucleosome center and NMD sensitivity. Mean and standard deviation for each group is reported at the bottom. P-values were calculated using the Mann–Whitney U test and adjusted using the false discovery rate (5%). Tests were run between introns belonging to the same positional group or between introns belonging to the same NMD group. (*) P < 0.05, (**) P < 10−2, (***) P < 10−3, (****) P < 10−4, (*****) P < 10−5, (******) P < 10−6. (B) The retention rate of introns in WT and NMD-depleted (NMDKD) cells as a function of their GC content (excluded GT and AG dinucleotides at both extremities). Introns are classified based on their distance to the closest nucleosome center and on whether they are NMD sensitive or not. Binning = 10%. Error bars represent the SEM. P-values calculated using the Mann–Whitney U test, and adjusted using a false-discovery rate (5%), are displayed in Supplemental Figure S4A. (C) Modeling splicing efficiency (SE) in NMD-depleted cells: The pie chart reports the contribution of each parameter or group of parameters used in the final model. The full list of retained parameters, reporting their contribution and their statistical significance, is displayed in Supplemental Table S1 as well as in Supplemental Figure S4C. (D) The full fitted model in explaining intron splicing efficiency, indicating whether each parameter is positively or negatively correlated with splicing efficiency. The parameter abbreviations are explained in Supplemental Table S1.
The highest contribution came from the level of gene expression that accounts for ∼46% of the model (Fig. 4C). The GC content of the intron accounts for 15%, and together with other parameters associated with the base composition of the introns (e.g., TC% accounts for 6.2%), the total contribution of base composition reaches ∼22%. Although the difference between GC content of an intron and the flanking exons (ΔGC) has been previously reported to be linked to intron splicing efficiency (Amit et al. 2012), ΔGC was not retained in our final model (Supplemental Table S1). As expected, GC content and ΔGC (introns − flanking exons) are highly correlated (Supplemental Fig. S4B), and forcing the usage of the latter does not improve the model. Splicing signals account for 9.5% of the model, and the parameters associated with the size and base composition of the transcript account for ∼8.7%. Intron size and position in the transcript account for 7.1% of the model, followed by intron sensitivity to the NMD pathway (2.5%), the size and base composition of the flanking exons (2.4%), whether an intron is 3n or not (0.66%), and the parameters associated with the formation of secondary structures (0.35%). All the parameters relative to nucleosome positioning account for only 0.43% of the model (Fig. 4C–D; Supplemental Table S1). Moreover, if we divide the introns based on their NMD sensitivity, our model can explain 40% of the variation in intron splicing efficiency for the NMD-sensitive introns but only 27% for the NMD-insensitive ones (Supplemental Fig. S4C). We therefore conclude that the GC content, which is tightly linked to nucleosome positioning, contributes to intron splicing efficiency: a high GC content, which is correlated with high nucleosome occupancy, is associated with low splicing efficiency.
Discussion
We have performed the first nucleosome position profiling in the P. tetraurelia MAC genome during vegetative growth. Despite its high AT richness (72% AT), the P. tetraurelia MAC genome displays a very regular nucleosome positioning pattern as observed in other eukaryotes: NFRs at the TSSs and TTSs, as well as a regular nucleosome array along genes. An independent study reached the same conclusions (Drews et al. 2022). Unlike Tetrahymena thermophila, another AT-rich ciliate (78% AT; with an NRL of 199 bp) (Beh et al. 2015), the NRL in the P. tetraurelia MAC genome presents a smaller periodicity (151 ± 1 bp), very similar to that of S. pombe (156 bp) (Godde and Widom 1992) and that of Plasmodium falciparum (155 bp; >80%AT) (Kensche et al. 2016; Silberhorn et al. 2016). This short NRL means that the naked “linker” DNA between nucleosomes in Paramecium is extremely small (only a few base pairs) compared with that of most other eukaryotic genomes, at least tens of base pairs or even larger (Arceci and Gross 1980). A higher H1/core-histone ratio has been previously reported as being associated with a longer NRL (Fan et al. 2003, 2005; Woodcock et al. 2006). For the three eukaryotes with the smallest NRL, P. tetraurelia, P. falciparum, and S. pombe, no ortholog of histone H1 has been identified so far. This strongly suggests that the absence of H1 might contribute to the extremely short NRL observed in Paramecium chromatin organization in the somatic MAC genome.
In yeast and humans, actively transcribed genes tend to have a shorter NRL than transcriptionally inactive genes, partially owing to the binding of H1 generating inaccessible chromatin at inactive genes (Valouev et al. 2011; Correll et al. 2012; Barbier et al. 2021). With the separation of the germline MIC and the somatic MAC genomes in two distinct nuclei, the Paramecium MAC genome is characterized by very high coding density. Indeed, >80% of the MAC is covered by annotated genes, and 65% of the coding genes are expressed (RNA-seq coverage of at least 1 RPKM) during vegetative growth (Aury et al. 2006; Arnaiz et al. 2017), which might explain the extremely short length and narrow distribution of NRL. A significant difference in the nucleosome organization between MAC and MIC genomes has been reported for T. thermophila (Xiong et al. 2016). How nucleosomes are organized in the Paramecium MIC genome is unknown. At each sexual cycle of Paramecium, the parental MAC is destroyed, and the new MIC and MAC are generated from the parental germline MIC (Bétermier and Duharcourt 2014). During new MAC development, at least 30% of the germline DNA is eliminated during massive genome rearrangements (Guérin et al. 2017; Sellis et al. 2021). A large amount of extremely short (26- to ∼1000-bp) noncoding germline sequences, called internal eliminated sequences (IESs), need to be precisely excised to correctly assemble functional genes in the new MAC genome of Paramecium species (Sellis et al. 2021). How nucleosome positioning is organized in the germline MIC genome relative to IESs and whether nucleosome positioning and/or GC content might play a role in IES excision are open questions (Coyne et al. 2012; Lhuillier-Akakpo et al. 2014).
In multicellular eukaryotes, long introns are recognized through exon definition, and nucleosomes positioned along exons might contribute to the exon–intron architecture, possibly pointing to a function in exon definition (Andersson et al. 2009; Nahkuri et al. 2009; Schwartz et al. 2009; Spies et al. 2009; Tilgner et al. 2009; Iannone et al. 2015). In contrast, short introns are recognized through intron definition. With an average length of 25 nt, introns of P. tetraurelia are among the shortest reported in eukaryotes (Jaillon et al. 2008). The large number of introns (more than 90,000) are associated with weak splicing signals. In the current study, we examined the role of nucleosome positioning in intron splicing. We found a regular nucleosome array associated with intron positions within genes, with exons wrapped around nucleosomes and introns frequently located at the edge of nucleosomes. By using the accurate splicing efficiency data obtained from NMD-depleted cells (Saudemont et al. 2017), we performed a thorough investigation on the effect of nucleosome positioning on splicing efficiency. We showed that the NMD-sensitive introns located at the edge of nucleosomes display higher splicing efficiency than those at the nucleosome centers. However, we found that this higher splicing efficiency is owing to the fact that the introns located at the edges of nucleosomes display lower GC content. Our multiple regression analysis indicated that the nucleosome positioning has a minimal contribution (0.43%) to the intron splicing efficiency (Supplemental Fig. S4C; Supplemental Table S1). Our results strongly indicate that GC content and, more broadly, intron base composition, rather than nucleosome positioning, directly influence intron splicing efficiency in Paramecium. This conclusion may pave the way for future mechanistic studies to decipher how GC content impinges on intron splicing efficiency. Whether the effect of GC content and nucleosome positioning on intron splicing efficiency observed in Paramecium can be extended to other eukaryotes remains an open question.
We also observed that during evolution, nucleosome positioning has been displaced relative to introns, frequently locating the AT-rich intron sequences at the edge of nucleosomes (Fig. 4A). Although both NMD-sensitive and NMD-insensitive introns present a higher proportion of distal positions, NMD-insensitive introns show a significantly higher proportion (50% for 3n and 48% for non-3n introns) than do NMD-sensitive introns (40% for 3n and 44% for non-3n introns) (Fig. 3A). This strongly suggests that the NMD-insensitive introns not located at the AT-rich nucleosome edges, whose retention in transcripts cannot be cleaned up by the NMD pathway, are counter-selected during evolution. Whether introns in Paramecium might play a functional role is still unclear. These introns do not seem to contribute to alternative splicing to generate protein diversity or to encode ncRNAs as in large other genomes with long and abundant introns (Chen et al. 2003; Ruby et al. 2007; Lee and Rio 2015). Because of their extremely small size, it seems unlikely that these introns play a role in regulating transcription rate as suggested in recent publications (Alexander et al. 2010; Fong et al. 2014; Aslanzadeh et al. 2018). As the parameters analyzed in this study only explain ∼40% of the variation in intron splicing efficiency, other parameters remain to be identified, and perhaps other models would be necessary to fully understand what intron properties determine splicing efficiency. How such a large number of tiny introns in Paramecium is maintained during evolution and how these introns can be efficiently spliced need to be further investigated.
Methods
Paramecium strains, cultivation, and autogamy
All experiments were performed with the entirely homozygous WT strain 51 of P. tetraurelia. Cells were grown at 27°C in wheat grass powder (WGP) infusion medium bacterized the day before use with Klebsiella pneumoniae and supplemented with 0.8 mg/mL β-sitosterol (Beisson et al. 2010a, 2010b).
Macronuclei preparation
Cells were exponentially grown for 12 divisions, and then cultures at 1000 cells/mL were filtered through eight layers of sterile gauze. Cells were collected by low-speed centrifugation (550g for 1 min) and washed once with 10 mM Tris-HCl (pH 7.4). The pellet was diluted threefold by addition of lysis buffer (0.25 M sucrose, 10 mM MgCl2, 10 mM Tris at pH 6.8, 0.2% Nonidet P-40) and processed at 4°C as previously described (Arnaiz et al. 2012) with some modifications. Briefly, cells were lysed with 10 strokes of a Dounce homogenizer. Particular care was taken to make sure that macronuclei were still intact under the microscope. Washing buffer (0.25 M sucrose, 10 mM MgCl2,10 mM Tris-HCl at pH 7.4) was added to a final volume of 10 times the initial pellet. Macronuclei were collected by centrifugation at 2000g for 1 min and washed once in washing buffer. The pellet was diluted twofold in 2.1 M sucrose, 10 mM MgCl2, 10 mM Tris (pH 7.4); loaded on top of a 3-mL sucrose layer (2.1 M sucrose, 10 mM MgCl2, 10 mM Tris-HCl at pH 7.4); and centrifuged in a swinging rotor for 1 h at 210,000g. The macronuclear pellet was washed once, centrifuged at 2000g for 1 min, and resuspended in washing buffer at 107 nuclei/mL. The macronuclei recovery is quite low, of the order of 10%–20%.
MNase digestion of chromatin isolated from macronuclei
Samples containing 105 macronuclei were incubated in the digestion buffer (0.25 M sucrose, 10 mM MgCl2,10 mM Tris at pH 7.4, 1 mM CaCl2) with increasing amounts (0, 0.5, 1, 2, 5, 7.5, 10 U) of MNase (Sigma-Aldrich) for 10 min at 30°C. Reactions were stopped by the addition of three volumes of 0.5 M EDTA (pH 9.0), 1% N-lauroylsarcosine (Sigma-Aldrich), 1% SDS, 1 mg/mL Proteinase K (Merck) and incubated overnight at 55°C. DNA from each sample was gently extracted once with phenol and dialyzed twice against TE (10 mM Tris-HC1, 1 mM EDTA at pH 8.0) containing 25% ethanol and once against TE. Samples were then treated with RNase A, and DNA was quantified with a NanoDrop spectrophotometer (Thermo Fisher Scientific) and separated on a 1.2% agarose gel. The reactions containing mostly mononucleosomal DNA fragments (see Fig. 1) were selected, and mononucleosomal DNA fragments were purified from 3% low melting-temperature agarose gels and treated with β-agarase (Sigma-Aldrich) for sequencing.
MNase digestion of naked DNA
Following purification on a sucrose layer, the macronuclear pellet was washed once, centrifuged at 2000g for 1 min, and resuspended in three volumes of lysis solution (0.5 M EDTA at pH 9.0, 1% SDS, 1% N-lauroylsarcosine [Sigma-Aldrich], 1 mg/mL of Proteinase K [Merck]) and then incubated overnight at 55°C. DNA was gently extracted with phenol and dialyzed twice against TE (10 mM Tris-HC1, 1 mM EDTA at pH 8.0) containing 20% ethanol and once against Tris 10 mM (pH 8.0). Then 1.6 μg of DNA was digested with increasing amounts of MNase (0 to 1 × 10−3 U) in the digestion buffer for 10 min at 30°C. The reactions were stopped with 250 mM EDTA. The samples were analyzed on a 1.2% agarose gel, and reactions containing fragments of 100–200 bp were gel-purified for DNA sequencing (see Supplemental Fig. S1).
MNase library preparation and sequencing
Sequencing libraries were generated using the sequencing kit: TruSeq SBS kit v5–GA (36 cycle, Illumina FC-104-5001). Samples were then sequenced on an Illumina GA-IIx sequencer using a PE 74-bp setting. The MNase-seq data sets used in this study are from Hardy et al. (2021) and are available under accession number PRJEB39679 at the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena).
Alignment was performed using Bowtie 2 (v2.3.3 ‐‐local and other default parameters) (Langmead and Salzberg 2012) and mapping to the MAC genome of strain 51 v1.0 (ptetraurelia_ mac_51.fa), available at ParameciumDB (Arnaiz et al. 2019; https://paramecium.i2bc.paris-saclay.fr/).
Nucleosome positioning calling
After aligning reads to the reference MAC genome, PCR duplicates with the same start and end positions were removed. Only reads mapped in proper pairs with a mapping quality score equal or higher than 30 were kept. Filtering, sorting, and filling of mate-related flags were performed using SAMtools, version 1.9 (Danecek et al. 2021). BAM files were converted into BED using BEDTools, version 2.29.2 (Quinlan and Hall 2010), and a customized script. We aimed to use only reads deriving from mononucleosomes; therefore, read pairs >150 bp and <75 bp were excluded. We used only the data within the scaffolds >200 kb. A nucleosome score was calculated using the central 75 bp of each read pair. The signal was then smoothed with a Gaussian filter and a sigma of 30 over 90 bp for visual assessment of nucleosome position calling. Local maxima and local minima were identified by convoluting the nucleosome score with a first derivative of a Gaussian (sigma 30 over ±90 bp). The points of inflection were identified by convoluting the nucleosome score with a kernel containing the second derivative of a Gaussian (sigma 30 over ±90 bp). Peaks were called as a local maximum between two inflection points with opposite inclination. Peaks were called independently in the two chromatin samples, and then a list of well-positioned nucleosomes was compiled using those nucleosomes whose dyad (i.e., center) differs by <10 bp between the two biological replicates (∼75% of all nucleosomes). These well-positioned nucleosomes were used for downstream analyses.
Computation of NRL
To compute the NRL, we first calculated the distance of each nucleosome to all the other nucleosomes on the same scaffold and then used the distances obtained to generate the density distribution. This density distribution was then smoothed using a Gaussian filter (sigma = 10 over ±30bp) and local maxima identified convoluting the density distribution with the first derivative of a Gaussian (sigma = 10 over ±30bp). The first n local maxima were then ordered by increasing distances and fitted using a linear model. The slope of the fitted model corresponds to the NRL.
Nucleosome distribution calculation
Gene annotation v2.0 of MAC was from Arnaiz et al. (2019), and the TSSs and TTSs were from Arnaiz et al. (2017). The gene annotations and RNA-seq data are available at ParameciumDB (https://paramecium.i2bc.paris-saclay.fr/). To compare with the distribution of real exon sizes, a set of simulated exons was created assuming uniform exon sizes within each gene, that is, for a given gene with n exons, we divided its total exon length by n to get the length of n simulated exons of the corresponding gene. The NMD data were obtained from Saudemont et al. (2017); splicing efficiency of each intron was calculated as the splicing events/total number of observations (i.e., spliced + unspliced reads). The mean profiles and heatmaps were drawn using a customized script and plotting using Matplotlib (version 3.1.0) (Hunter 2007). All statistical analyses were performed with Python (version 3.7.4, https://docs.python.org/release/3.7.4/) using statsmodels (version 0.10.1) (Seabold and Perktold 2010) and SciPy (version 1.3.1) (Virtanen et al. 2020) modules.
Multilinear regression
The starting parameters used for the multiple linear regression can be found in Supplemental Table S1. Parameters were transformed using appropriate functions in order to maximize their linearity with intron splicing efficiency, for example, log transformation of expression levels. Values were then standardized. VIFs were calculated for the whole pool of parameters. If the parameter with highest VIF exceeded the threshold of five, it was excluded from the pool. Parameters VIF was then recalculated and the process repeated until VIF was greater than five. Parameters from this first selection were used to fit our linear regression model. A randomly selected set of introns (10% of all introns) was kept from the multilinear regression model fitting and used as a test data set to evaluate the model performance. We performed a two-sided Z-test for each coefficient with H0: C = 0 and H1: C ≠ 0. Statistically significant coefficients were then retained, and the linear model was trained again with the associated parameters. This step was repeated until the number of variables is stabilized. Estimation of the contribution of each parameter is calculated as previously described (Chen et al. 2010), which is based on the absolute value of the product of each coefficient and the Pearson's correlation value of its parameter with the splicing efficiency. Contributions were then converted to percentages. Using the intron test data set, we calculated the Pearson's correlation between real and predicted data. To calculate the Pearson's correlation between prediction and real data divided by NMD-sensitive and NMD-insensitive, all the introns belonging to either group were used. For this part, Python (version 3.7.4, https://docs.python.org/release/3.7.4/) was used with scikit-learn (version 0.21.3) (Pedregosa et al. 2011), statsmodels (version 0.10.1) (Seabold and Perktold 2010), and SciPy (version 1.3.1) (Virtanen et al. 2020) modules. The full list of parameters can be found in Supplemental Table S1.
Software availability
The customized script and Jupyter notebooks used for this study are available as Supplemental Code and at GitHub (https://github.com/CL-CHEN-Lab/Nucleosome).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Laurent Duret for useful suggestions and discussion and Laurent Duret and Eric Meyer for sharing with us the NMD data, and we acknowledge the high-throughput sequencing facility of I2BC for its sequencing and bioinformatics expertise. This work was supported by Centre National de la Recherche Scientifique, Agence Nationale pour la Recherche (ANR-10-BLAN-1603, ANR-18-CE12-0005, ANR-19-CE12-0015), LABEX Who Am I? (ANR-11-LABX-480 0071, ANR-11-IDEX-0005-02), ATIP-Avenir, and Plan Cancer.
Author contributions: L.S., M.B., C.T., C.-L.C., and S.D. conceived and planned the study. M.M., F.G., and S.D. conducted the experiments. S.G., M.W., O.A., and C.-L.C. performed the bioinformatics analyses. C.T. and C.-L.C. supervised the bioinformatics analyses. S.G., C.-L.C., and S.D. wrote the manuscript, and all the authors reviewed it.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276125.121.
- Received August 20, 2021.
- Accepted February 14, 2022.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








