
Antibody-free profiling of transcription factor occupancy during early embryogenesis by FitCUT&RUN
- Institute for Regenerative Medicine, Shanghai East Hospital, Shanghai Key Laboratory of Signaling and Disease Research, Frontier Science Center for Stem Cell Research, School of Life Science and Technology, Tongji University, Shanghai, 200092, China
-
↵1 These authors contributed equally to this work.
Abstract
Key transcription factors (TFs) play critical roles in zygotic genome activation (ZGA) during early embryogenesis, whereas genome-wide occupancies of only a few factors have been profiled during ZGA due to the limitation of cell numbers or the lack of high-quality antibodies. Here, we present FitCUT&RUN, a modified CUT&RUN method, in which an Fc fragment of immunoglobulin G is used for tagging, to profile TF occupancy in an antibody-free manner and demonstrate its reliability and robustness using as few as 5000 K562 cells. We applied FitCUT&RUN to zebrafish undergoing embryogenesis to generate reliable occupancy profiles of three known activators of zebrafish ZGA: Nanog, Pou5f3, and Sox19b. By profiling the time-series occupancy of Nanog during zebrafish ZGA, we observed a clear trend toward a gradual increase in Nanog occupancy and found that Nanog occupancy prior to the major phase of ZGA is important for the activation of some early transcribed genes.
Metazoans undergo a rapid and well-orchestrated process called zygotic genome activation (ZGA) in which developmental control is transferred from maternal to zygotic genetic material during early embryo development. Dozens of transcription factors (TFs) have been shown to be key activators in this process as indicated by lethality or developmental delay upon their depletion in several species (Lunde et al. 2004; Liang et al. 2008; Okuda et al. 2010; Bogdanović et al. 2012; Xu et al. 2012; De Iaco et al. 2017; Fogarty et al. 2017). Profiling genome-wide binding sites of key TFs is a key step to understanding the mechanisms of essential TF functions during ZGA. Chromatin immunoprecipitation followed sequencing (ChIP-seq) (Johnson et al. 2007) is widely used to profile genome-wide TF occupancy. However, to the best of our knowledge, ChIP-seq data on only a few factors obtained during ZGA have been published (Xu et al. 2012; Leichsenring et al. 2013; Nelson et al. 2014; Dubrulle et al. 2015; Koenecke et al. 2016, 2017; Stevens et al. 2017; Meier et al. 2018), mainly due to the following two limitations. First, TF ChIP-seq requires 106 or more cells, and harvesting this number of cells for studying human or mouse ZGA is unrealistic, particularly because the major phases of ZGA occur at the eight-cell and two-cell stages. Even when studying zebrafish ZGA, the major phase of ZGA occurs at the 1k-cell stage, and collecting 106 or more cells for a single TF ChIP-seq experiment is tedious and challenging for most laboratories. Second, profiling TF ChIP-seq requires the availability of high-quality antibodies, but verified antibodies are available for only a few well-studied TFs in nonmammalian species (Park 2009; Partridge et al. 2016). To avoid these limitations, TF ChIP-seq data can be produced by adding a tag to the studied TF and using an antibody targeting tag (Xu et al. 2012; Leichsenring et al. 2013; Dubrulle et al. 2015); nevertheless, the number of cells required remains an obstacle for studying TF functions during ZGA, especially for the stages prior to the major ZGA phase. Therefore, to determine the functional mechanisms of key TFs during early embryogenesis, a new technical approach is needed to profile TF occupancy antibody-free with a limited number of cells.
Cleavage Under Targets & Release Using Nuclease (CUT&RUN) was recently developed as an alternative technology for profiling TF occupancy (Skene and Henikoff 2017). Compared with ChIP-seq, CUT&RUN requires much less input material (Skene et al. 2018; Hainer et al. 2019), which makes it applicable to profiling TF occupancy during ZGA. However, performing CUT&RUN requires high-quality antibodies, which limits its application with nonmammalian species. In this study, we proposed using an Fc fragment of immunoglobulin G tagging approach followed by CUT&RUN (FitCUT&RUN) as an antibody-free version of CUT&RUN to map TF occupancy and verified its feasibility and reliability in K562 cells, even with low levels of input material. We further applied FitCUT&RUN to zebrafish embryogenesis to profile the time-series occupancy of Nanog during ZGA.
Results
Performance of FitCUT&RUN
To generate an antibody-free profile of TF occupancy, we developed FitCUT&RUN, a modified CUT&RUN method. In contrast to CUT&RUN, FitCUT&RUN requires the target TF to be tagged by rabbit Fc (rFc), on the presumption that the tagged rFc can be efficiently recognized by protein A-MNase (pA-MN) or protein A-protein G-MNase (pAG-MN) (Fig. 1A). To confirm the accuracy of this assumption and to optimize the tagging with rFc, which contains 227 amino acids (aa), we constructed fusion proteins with GST and different truncated rFc fragments (GST-rFc) and pAG-MN in Escherichia coli and performed a pull-down experiment (Fig. 1B; Supplemental Fig. S1A; Supplemental Table S1; see Methods for details). The results showed that the pAG-MN protein can interact with GST-rFc with the full-length rFc, proving the feasibility of the FitCUT&RUN method. Moreover, we did not observe apparent interactions between pAG-MN and GST-rFc with the truncated rFc fragments (160 aa, 130 aa, and 110 aa), indicating that the full-length rFc is required for interaction with pAG-MN. The results implied that FitCUT&RUN using the fusion protein consisting of full-length rFc and a target TF can be a practical antibody-free approach to profiling TF occupancy.
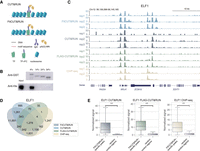
Feasibility and robustness of FitCUT&RUN with K562 cells. (A) Adaptation of FitCUT&RUN based on the common CUT&RUN method. (B) Western blot analysis of the in vitro pull-down assay to determine the interaction of pAG-MN with different truncated rFc fragments. (C) The genome browser view shows ELF1 FitCUT&RUN, CUT&RUN, FLAG-CUT&RUN (three replicates for each), and ChIP-seq (two replicates) signals around RPL41 loci as a representative example of the consistency among these methods. (D) Venn diagram showing the overlap status between ELF1 FitCUT&RUN, CUT&RUN, FLAG-CUT&RUN, and ChIP-seq peaks. The majority of ChIP-seq, CUT&RUN, and FLAG-CUT&RUN peaks were also detected by FitCUT&RUN. (E) Boxsplots showing that the normalized ELF1 ChIP-seq, CUT&RUN, and FLAG-CUT&RUN signal on FitCUT&RUN-specific peaks is significantly greater than that on random control regions, confirming the reliability of the FitCUT&RUN data.
To evaluate the performance of FitCUT&RUN, we performed ELF1 and ATF1 FitCUT&RUN, standard CUT&RUN, and FLAG-CUT&RUN (FLAG tagging followed by CUT&RUN) with K562 cells, for which there are publicly available high-quality ENCODE eGFP-ELF1 ChIP-seq and FLAG-ATF1 CETCh-seq data (Landt et al. 2012; Savic et al. 2015; Sloan et al. 2016; Davis et al. 2018). For both FitCUT&RUN and FLAG-CUT&RUN experiments, we introduced fusion protein by transfecting plasmid into K562 cells with limited effects on cell viability (Supplemental Fig. S1B). For each replicate of FitCUT&RUN, CUT&RUN, and FLAG-CUT&RUN, 1 × 105 cells were used, and the signals between replicates were found to be highly reproducible and consistent with their ENCODE ChIP-seq/CETCh-seq counterparts, which were based on 2 × 107 cells (Fig. 1C; Supplemental Fig. S1C–E; Supplemental Tables S2, S3). For both TFs, FitCUT&RUN identified the highest number of peaks (17,842 for ELF1 and 15,459 for ATF1). Motif analysis for FitCUT&RUN peaks showed that the top hits were indeed the expected motifs (GGAAG for ELF1, TGAC for ATF1). The majority of the ChIP-seq/CETCh-seq peaks (73.6% for ELF1 and 82.7% for ATF1) were also detected by FitCUT&RUN (Fig. 1D; Supplemental Fig. S1F). At the FitCUT&RUN specific peaks, the ChIP-seq/CETCh-seq data showed moderate enrichment, confirming the feasibility of FitCUT&RUN for use in profiling the occupancy of target TFs (Fig. 1E; Supplemental Fig. S1G). Similarly, the majority of the CUT&RUN and FLAG-CUT&RUN peaks were also detected by FitCUT&RUN, and at the FitCUT&RUN specific peaks, the FLAG-CUT&RUN and CUT&RUN data also showed moderate enrichment (Fig. 1D,E; Supplemental Fig. S1F,G). As shown in the heat maps, for each TF, the occupancy profiles from four methods were generally consistent, whereas FitCUT&RUN displayed the highest sensitivity (Supplemental Fig. S2A,B). We further calculated fraction of reads in peaks (FRiP) values, normalized strand coefficient (NSC) values, and average signals at all putative binding sites for each data set, to evaluate the signal-to-noise ratio for each method (Landt et al. 2012; Nizamuddin et al. 2021). Among six metrics, FitCUT&RUN ranked first four times, including ELF1's FRiP value, both factors’ NSC values, and ELF1's average signal, whereas CUT&RUN and FLAG-CUT&RUN ranked first once, respectively (Supplemental Fig. S2C–F), demonstrating the high signal-to-noise ratio of FitCUT&RUN. To further confirm the high signal-to-noise ratio of FitCUT&RUN at low sequencing depth, we sampled the fragments of ELF1 FitCUT&RUN, CUT&RUN, FLAG-CUT&RUN, and ChIP-seq data down to 5-M fragments, respectively, and FitCUT&RUN discovered more peaks than others and displayed the highest sensitivity (Supplemental Fig. S2G,H).
It has been reported that the CUT&RUN method requires much less input material than ChIP-seq (Skene et al. 2018; Hainer et al. 2019). To investigate whether FitCUT&RUN can be applied to low-input samples, we generated ELF1 and ATF1 FitCUT&RUN data using 1 × 104 and 5 × 103 cells, respectively (Fig. 2A; Supplemental Figs. S1E, S3A; Supplemental Table S2). Although the number of peaks detected based on the low-input material were much less than that using 1 × 105 cells, the FitCUT&RUN signals based on the low-input material showed high reproducibility with those obtained from 1 × 105 cells; peaks identified based on 104 and 5 × 103 cells corresponded to those with a higher signal based on 105 cells (Fig. 2B–D; Supplemental Figs. S1D, S3B–D). Furthermore, we also generated ELF1 and ATF1 FLAG-CUT&RUN data using 5 × 103 cells, and the correlations between FLAG-CUT&RUN signals based on 5 × 103 and 1 × 105 cells were much lower than that for FitCUT&RUN (Supplemental Figs. S1D, S3E,F). The results demonstrated the robustness of FitCUT&RUN with reduced input material.
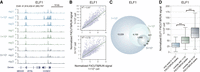
Robustness of FitCUT&RUN with reduced input material in K562 cells. (A) The genome browser view shows the ELF1 FitCUT&RUN signal for different input materials (three replicates for each). A similar signal represents the reproducibility of FitCUT&RUN with low-input material. (B) Scatterplots presenting the high correlation of the ELF1 FitCUT&RUN signal between different input materials. (C) Venn diagram showing the overlap of ELF1 FitCUT&RUN peaks with different input cells. The majority of peaks based on the low-input material showed high overlap with those obtained from 1 × 105 cells. (D) Box plots showing the normalized ELF1 FitCUT&RUN signal using 1 × 105 cells in three groups of peaks, that is, only detected in the FitCUT&RUN using 1 × 105 cells, overlap with FitCUT&RUN using 1 × 104 cells, and overlap with FitCUT&RUN using 5 × 103 cells. Peaks detected with reduced input material (5 × 103 cells) show a higher signal than those in the other two groups, proving the reliability of FitCUT&RUN with low-input material.
Application of FitCUT&RUN to early zebrafish embryos
As FitCUT&RUN performed robustly with low-input material, we applied it to profile TF occupancy during zebrafish ZGA. First, we constructed mRNAs encoding the fusion proteins of rFc with Nanog, Pou5f3, or Sox19b, which are known activators of zebrafish ZGA (Lee et al. 2013; Pálfy et al. 2020). To minimize the disruption of embryonic development upon mRNA injection, for each mRNA, we microinjected gradient volumes into one-cell embryos and counted the proportion of embryos with a normal phenotype at 24 h postfertilization (hpf). The results showed that there were few defects when injecting as much as 25 pg of constructed mRNA for these three TFs (Supplemental Fig. S4A), and in this study, we microinjected 25 pg of TF-rFc mRNA to perform FitCUT&RUN in zebrafish. Next, we produced three replicates of the FitCUT&RUN experiment for each studied TF using 50 dome stage embryos for each replicate (Fig. 3A; Supplemental Fig. S4B–D; Supplemental Table S2). The high correlation between replicates confirmed the reproducibility of FitCUT&RUN performance; for each TF, we combined replicates and then called peaks as its binding sites (Supplemental Table S4). It has been reported that these activators are critical for the establishment of accessible chromatin at their binding sites (Liu et al. 2018; Veil et al. 2019; Pálfy et al. 2020), and we indeed observed significantly decreased ATAC-seq signals at the binding sites after corresponding knockout (KO) of nanog, pou5f3, or sox19b (Fig. 3B; Supplemental Fig. S4E,F). Taken together, the data show that the FitCUT&RUN method can produce reliable TF occupancy profiles during zebrafish ZGA.
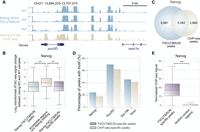
Reliable quality data obtained using the FitCUT&RUN method with early zebrafish embryos. (A) The genome browser view shows Nanog FitCUT&RUN (three replicates) and ChIP-seq (one replicate) signals around pou5f3 loci as a representative example of the consistency between these two methods. (B) Box plots showing the ATAC-seq signal (oblong stage) change between nanog-KO and wild-type samples (Supplemental Table S3; Pálfy et al. 2020) in three different regions. A significant decrease in Nanog FitCUT&RUN peaks compared to those of accessible regions (oblong stage) (Supplemental Table S3; Liu et al. 2018) without overlap with Nanog FitCUT&RUN peaks indicates that Nanog is critical for the establishment of accessible chromatin in these Nanog FitCUT&RUN peaks. A significant decrease in Nanog FitCUT&RUN-specific peaks compared to those of accessible regions (oblong stage) (Supplemental Table S3; Liu et al. 2018) without overlap with Nanog FitCUT&RUN peaks shows the reliability of the FitCUT&RUN-specific peak data, indicating that FitCUT&RUN can produce reliable TF occupancy profiles during zebrafish ZGA. (C) Venn diagram showing the overlap status between Nanog FitCUT&RUN and ChIP-seq data. The majority of ChIP-seq peaks were also detected by FitCUT&RUN; and FitCUT&RUN could detect many more peaks than ChIP-seq (Supplemental Table S4). (D) Bar plots showing the higher motif frequency of Nanog, Pou5f3, and Sox19b in the FitCUT&RUN-specific peaks compared to that in the ChIP-seq-specific peaks, indicating the reliability of the FitCUT&RUN data (Supplemental Table S4). (E) Box plots showing that the normalized Nanog ChIP-seq signal on FitCUT&RUN-specific peaks is significantly greater than that on random control regions, confirming the reliability of the Nanog FitCUT&RUN data.
Using publicly available ChIP-seq data for Nanog, Pou5f3, and Sox2 obtained from samples near the dome stage (Supplemental Table S3), we compared the binding profiles generated by FitCUT&RUN to those obtained by ChIP-seq. We used Sox2 ChIP-seq data in the comparison, because there is no Sox19b ChIP-seq data publicly available, and Sox19b and Sox2 belong to the same TF family with similar motifs. To fairly compare the data from these different technologies, for each studied TF, we randomly sampled the same number of unique mapped reads or fragments (see Methods for details). Using FitCUT&RUN, we detected many more peaks than had been obtained by ChIP-seq under the same peak-calling criteria (Fig. 3C; Supplemental Fig. S4G,H; Supplemental Table S4; see Methods for details). For each TF, we examined the frequency of binding motifs at the detected peaks (see Methods for details), and we found that the FitCUT&RUN-specific peaks exhibited moderately higher motif frequency than the ChIP-seq-specific peaks (Fig. 3D). Furthermore, at the FitCUT&RUN-specific peaks, we observed significantly decreased ATAC-seq signals after corresponding KO of nanog, pou5f3, or sox19b (Fig. 3B; Supplemental Fig. S4E,F; Pálfy et al. 2020), confirming the reliability of the FitCUT&RUN-specific peaks. The ChIP-seq data showed moderate enrichment at the FitCUT&RUN-specific peaks, and more FitCUT&RUN peaks were detected by ChIP-seq when greater sequencing depth of ChIP-seq was applied (Fig. 3E; Supplemental Fig. S4I–M). Considering that we used much less input materials for the FitCUT&RUN method (50 embryos per experiment) than had been used to obtain the publicly available ChIP-seq data (2000 embryos per experiment) (Xu et al. 2012; Leichsenring et al. 2013), the extensiveness and reliability of the FitCUT&RUN-specific peaks demonstrate the advantages of FitCUT&RUN with superior sensitivity for detecting TF occupancy.
Emergence of Nanog occupancy during zebrafish ZGA
Profiling the occupancy of key activators is essential to understand the transcriptional regulatory mechanisms during ZGA. However, to the best of our knowledge, no time-series occupancy profiles of known activators are publicly available for the whole process of zebrafish ZGA, whose major phase occurs at the 1k-cell stage and coincides with the midblastula transition (MBT). Nanog was reported to contribute to the establishment of accessible chromatin and is involved in regulatory processes during ZGA (Lee et al. 2013; Liu et al. 2018; Veil et al. 2019; Pálfy et al. 2020). With the reliability of the FitCUT&RUN method demonstrated, in this study, we profiled the occupancy of Nanog at the 256-cell and 1k-cell stages with three biological replicates for each stage, and 200 embryos and 100 embryos were used for each replicate for 256-cell and 1k-cell stages, respectively (Supplemental Table S2). We observed a clear trend of a gradually increasing number of Nanog binding sites during ZGA (Fig. 4A), from 2288 sites at the 256-cell stage and 5684 sites at the 1k-cell stage to 15,474 sites at the dome stage (Fig. 4B; Supplemental Table S4). To exclude the possibility that more Nanog binding sites derived from the dome stage are caused by using more cells in the experiment, we performed Nanog FitCUT&RUN using 20 embryos at the dome stage, that is, with similar cell number used for 256-cell stage FitCUT&RUN, and we indeed observed that the signals were highly consistent between FitCUT&RUN experiments from 20 embryos and from 50 embryos (Supplemental Fig. S5A,B; Supplemental Table S2). To evaluate the reliability of Nanog-binding sites at the 256-cell and 1k-cell stages, we examined the ATAC-seq signals of wild-type (WT) embryos at corresponding stages (Supplemental Table S3; Liu et al. 2018) and observed that promoters with bound Nanog displayed significantly higher ATAC-seq signals than other promoters (Supplemental Fig. S5C). We further compared the ATAC-seq signal changes upon the microinjection of nanog-rFc mRNA between ATAC-seq peaks with and without Nanog binding, and we found no significant differences between the two groups at the 256-cell and 1k-cell stages (Supplemental Fig. S5D; Supplemental Table S2). We also performed RNA-seq at the 256-cell, 1k-cell, and dome stages for WT, H2O injection, and nanog-rFc injection embryos, with two replicates for each condition, and we found that injecting nanog-rFc mRNA did not clearly alter gene expression of zebrafish embryos, at least until the dome stage (Supplemental Fig. S5E–G; Supplemental Table S2). These results demonstrated that the Nanog-binding sites detected at 256-cell and 1k-cell stages were unlikely to be false-positive results induced by the microinjection of nanog-rFc mRNA.
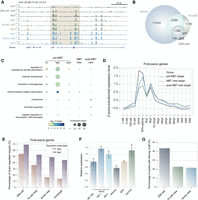
Nanog occupancy as detected by FitCUT&RUN during zebrafish ZGA. (A) The genome browser view shows a gradual increase in the Nanog FitCUT&RUN signal during the zebrafish ZGA process (256-cell, 1k-cell, and dome stage; three replicates of each). Multiple adjacent Nanog binding sites gradually accumulated as Nanog-binding cluster (NBC) regions; a representative example is indicated by the dashed box. (B) Venn diagram showing the overlap status of Nanog binding sites at different stages (256-cell, 1k-cell, and dome stage). (C) Dot plots showing the Gene Ontology (GO) enrichment analysis for Nanog target genes. Genes targeted by Nanog at pre-MBT had higher enrichment scores in the embryonic morphogenesis and regulation of cell fate commitment function categories, especially for NBC target genes. (D) Line plots showing the z-score-transformed expression level (Supplemental Table S3; White et al. 2017) of the first-wave genes (Lee et al. 2013) targeted by Nanog (pre-MBT target, MBT new target, and post-MBT new target) at different developmental stages. Pre-MBT target genes reached their expression peak (dome) earlier than the MBT new target genes (8 hpf) and post-MBT new target genes (50%-epiboly). (E) Bar plots showing the percentage of first-wave genes down-regulated at 4 hpf and 6 hpf upon nanog MO compared to that of the wild-type samples (Lee et al. 2013). The earlier the genes were targeted by Nanog, the larger the proportion with a down-regulation status, indicating that Nanog pre-MBT binding is critical for zebrafish ZGA. (F) Relative expression level of genes corresponding to the mutated Nanog-binding sites detected by RT-qPCR at the 1k-cell stage (dharma, fgfr4, and hist1h4l) or 4 hpf (fox3). Out of the four genes, foxa3 has two Nanog binding sites near its TSS; whereas injecting one gRNA did not change its expression, co-injecting two gRNA targeting two binding sites together displayed significant expression decrease. For the remaining three genes, two showed a significant expression decrease upon corresponding gRNA injection. (G) Bar plots showing the Nanog motif frequency in three groups of peaks within NBCs, that is, emerged from the 256-cell stage, 1k-cell stage, and dome stage. Peaks that emerged earlier show higher Nanog motif frequency.
Because a subset of Nanog-binding sites were detected prior to the major phase of ZGA, we next investigated the regulatory features of Nanog occupancy initiated at different developmental stages. The inferred putative target genes of Nanog during ZGA were categorized into three groups based on the timing of their position near Nanog-binding sites: pre-MBT targets (1317 genes), MBT new targets (1891 genes), and post-MBT new targets (4047 genes) (Supplemental Table S5; see Methods for details). Compared to the MBT new and post-MBT new targets, the pre-MBT target genes showed much greater enrichment in functions of embryonic morphogenesis and regulation of cell fate commitment (Fig. 4C), suggesting that Nanog may initiate to regulate the activation of developmental genes as early as the 256-cell stage. To study the regulatory effects of the timing of Nanog occupancy, we focused on two reported lists of early transcribed genes: first-wave transcribed genes (Lee et al. 2013) and early ZGA genes (Heyn et al. 2014). In both lists of genes, the proportion of pre-MBT Nanog targets was higher than that of the MBT new or post-MBT new targets (the percentage of first-wave transcribed genes was 37.8% in the pre-MBT group vs. 18.4% in MBT new group and 21.9% in the post-MBT new group, and the percentage of early ZGA genes in the corresponding groups was 28.7% vs. 13.2% and 18.7%, respectively), and the pre-MBT targets tended to reach their maximum expression level during ZGA earlier than the MBT new or post-MBT new targets (Fig. 4D; Supplemental Fig. S6A; Supplemental Table S5) (White et al. 2017). Furthermore, among the reported early transcribed genes, the largest proportion of genes with considerably reduced expression was in the pre-MBT target group, as indicated 4 hpf and 6 hpf upon nanog morpholino (MO) knockdown (KD) (Fig. 4E; Supplemental Fig. S6B; Supplemental Table S5; Lee et al. 2013). To validate the regulatory effects of Nanog binding prior to the major phase of ZGA, we designed five gRNAs, whose target sites were associated with four genes (Supplemental Table S6; see Methods for details). We injected gRNA and Cas9 protein at 1-cell stage embryos and used RT-qPCR to examine the expression level of associated gene at the 1k-cell stage (dharma, fgfr4, and hist1h4l) or 4 hpf (fox3). Out of the four genes, foxa3 has two Nanog binding sites near its TSS; whereas injecting one gRNA did not change its expression, co-injecting two gRNAs targeting two binding sites together displayed significant expression decrease. For the remaining three genes, two showed significant expression decrease upon corresponding gRNA injection (Fig. 4F). Taken together, our results demonstrated that the occupancy of Nanog prior to the major phase of ZGA is important for the activation of some early transcribed genes.
As multiple adjacent Nanog-binding sites gradually accumulated at some genomic loci (Fig. 4A), we identified 188 Nanog-binding clusters (NBCs) containing Nanog-binding sites since the 256-cell stage. Compared to that of other pre-MBT targets, the inferred putative pre-MBT target genes of NBCs showed much higher functional enrichment in embryonic morphogenesis and regulation of cell fate commitment (Fig. 4C; Supplemental Table S5), and a higher percentage of these genes were highly expressed (Supplemental Fig. S6C), indicating that the NBCs may be strongly associated with transcription activation and development control. Within the NBCs, Nanog-binding sites since the 256-cell stage displayed a much higher Nanog motif frequency and greater binding intensity at the dome stage than Nanog-binding sites since later stages (Fig. 4G; Supplemental Fig. S6D), indicating that motif may have influence on the sequential binding of Nanog.
Discussion
Fusing the Fc fragment of immunoglobulin G with the target protein has been applied to the purification of target proteins and the promotion of protein stability (Huang 2009; Czajkowsky et al. 2012). In this study, we presented FitCUT&RUN as an antibody-free method to map the genome-wide occupancy of TFs by introducing the fusions of target TFs with rFc. FitCUT&RUN was adapted from CUT&RUN, maintaining its advantages over ChIP-seq, such as a better signal-to-noise ratio and use under in situ conditions. Furthermore, the antibody-free feature of FitCUT&RUN expands its application to profiling TFs without the need for verified antibodies, which are scarce for nonmammalian species. Our results demonstrated the reliability and robustness of FitCUT&RUN using as few as 5000 K562 cells and its feasibility for use in producing reliable TF occupancy profiles during zebrafish ZGA.
Other approaches have been developed to avoid the requirement for verified antibodies in TF occupancy profiling, including DamID (Jacinto et al. 2015; Thambyrajah et al. 2016; Tosti et al. 2018), ChEC-seq (Zentner et al. 2015; Grünberg and Zentner 2017), and fusion of a tag with a target TF (Xu et al. 2012; Leichsenring et al. 2013; Dubrulle et al. 2015). For DamID, the resolution of TF-binding sites was limited to their distribution at GATC sites, and the excessive methylation activity of the fused DNA adenine methyltransferase (Dam) may lead to false-positive enrichment (Aughey and Southall 2016). To date, only ChEC-seq applications to yeast have been reported, and the feasibility of ChEC-seq for use in complex organisms needs to be demonstrated. The approach of fusing a tag to the target TF requires an additional step of antibody incubation for targeting the fusion tag, which may introduce additional noise. In this study, we compared the performance of FitCUT&RUN, CUT&RUN, FLAG-CUT&RUN, and ChIP-seq/CETCh-seq, and FitCUT&RUN displayed a higher signal-to-noise ratio than other approaches, together with high robustness with reduced input material. Taken together, FitCUT&RUN provides an easy and reliable solution for profiling TF occupancy, especially under circumstances of limited cell numbers.
Nevertheless, the current version of FitCUT&RUN has the following limitations. In this study, we performed FitCUT&RUN by transiently overexpressing the fusion protein of TF and rFc at a low level. Although we attempted to minimize the disturbance caused by injecting mRNA during zebrafish embryogenesis and demonstrated the reliability of the detected binding sites by using independent data sets, we cannot fully exclude the possibility that some of the detected binding sites are the result of false-positives induced by the microinjection of the fused mRNA. In addition, we cannot fully exclude the possibility that the better signal-to-noise ratio of FitCUT&RUN could be due to a high abundance of the overexpressed TF. To overcome that limitation, examining the levels of fusion protein and its endogenous counterpart can provide an assessment of overexpression, and it would be valuable to titrate the microinjection to endogenous levels. However, due to the lack of qualified TF antibodies, we failed to perform Western blot analysis in zebrafish early embryos. Alternatively, constructing knock-in strains of model organisms by fusing rFc with endogenous genes is an ideal approach. When knock-in strains are available, in addition to easily profiling TF occupancy in diverse tissues or during biological processes, it will also be possible to take advantage of the strains to investigate the functions of the targeted TFs by developing rFc-mediated protein degradation or chemiluminescence imaging technologies.
Profiling genome-wide binding sites of key TFs is a central step for understanding the important features of the transcriptional regulatory networks that promote ZGA. However, to the best of our knowledge, no time-series occupancy profiles of known activators during the whole process of zebrafish ZGA are publicly available. In this study, taking advantage of the superior sensitivity of FitCUT&RUN, we profiled the time-series occupancy of Nanog, one of the known key activators, before, during, and after the major phase of zebrafish ZGA. Our results demonstrated that the occupancy of Nanog prior to the major phase of ZGA is important for the activation of some early transcribed genes. Other TFs may have important roles in the occupancy of Nanog, probably due to co-occupancy, and such an interaction is worth investigating when more TFs binding sites are profiled in the future. Considering our results, we speculated that the proper occupancy of key TFs before the major phase of ZGA must be essential for the burst of transcription activity, and FitCUT&RUN is suitable for use in the comprehensive profiling of the occupancy of key TFs before the major phase of ZGA.
Methods
Cell culture and zebrafish husbandry
K562 cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum, penicillin, and streptomycin. Wild-type Tübingen-strain zebrafish were maintained, raised, and crossed under standard conditions (Westerfield 2000). Zebrafish embryos were collected at the one-cell stage and allowed to develop to the desired stage at 28.5°C. Zebrafish care and experiments were approved by the Institutional Animal Care and Use Committee of Tongji University.
Vector construction
The full-length Fc fragment of rabbit immunoglobulin G was customized by a gene synthesis service (GENEWIZ). Different truncated rFc fragments were amplified by PCR with different primer pairs. For the in vitro pull-down assay, linker fragment encoding peptide (G4S)3 (GGGGSGGGGSGGGGS) and rFc fragments of different lengths were sequentially cloned into a pGEX-6p-1 containing GST with BamHI and EcoRI, called pGEX-G4S-rFc. The plasmid expressing the pAG-MNase protein and 6xHis-tag was purchased from Addgene (plasmid #123461). Similarly, the (G4S)3 linker and full-length rFc fragment or 3 × FLAG (DYKDDDK) were cloned into pCS2+ with ClaI to obtain the pCS2-G4S-rFc or pCS2-3 × FLAG plasmid. To introduce ATF1/ELF1-rFc or ATF1/ELF1-FLAG into K562 cells, the full-length cDNAs of human ATF1 and ELF1 were cloned into pCS2-G4S-rFc or pCS2-3 × FLAG with BamHI. To express Nanog, Pou5f3, and Sox19b-rFc in zebrafish, the full-length cDNAs of zebrafish nanog, pou5f3, and sox19b were similarly cloned into pCS2-G4S-rFc with BamHI. All fragment clones were obtained using an In-Fusion HD cloning kit (Clontech 639636) according to the instructions. All primer sequences are listed in Supplemental Table S1.
In vitro pull-down assays
The constructs of pGEX-G4S-rFc and pAG-MNase were transformed into E. coli BL21, and the GST-rFc and protein A-protein G-MNase-His fusion proteins were induced at 37°C for 3 h using 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG). In vitro pull-down assays were performed using a Pierce Pull-Down PolyHis Protein:Protein Interaction kit (Thermo Fisher Scientific 21277) and analyzed by Western blotting with 6xHis-tag antibody (Invitrogen MA1-21315) and GST tag antibody (Invitrogen MA4-004).
Transfection and cell viability
To introduce ATF1/ELF1-rFc or ATF1/ELF1-FLAG into K562 cells, we used 4D-Nucleofector system and SF Cell Line 4D-Nucleofector X kit (Lonza V4XC-2024) for plasmid transfection. Briefly, 5 µg of each vector were mixed with 1 × 106 K562 cells and the cells were harvested at 20 h after transfection. To determine cell viability, cells were stained with Calcein-AM and PI (Beyotime C2015), then the percentages of viable cells were measured by flow cytometric analysis. To perform FitCUT&RUN, FLAG-CUT&RUN, or standard CUT&RUN, transfected cells or WT cells were diluted to the desired cell number.
In vitro transcription, mRNA injection, and embryo collection
The constructs of pCS2-nanog-rFc and pCS2-sox19b-rFc were linearized with SacII, and linearization of pCS2-pou5f3-rFc was digested with NsiI. Then, TF-rFc mRNA was transcribed in vitro using an mMESSAGE mMACHINE SP6 kit (Invitrogen AM1340). TF-rFc mRNA (100 pg, 50 pg, and 25 pg) was injected into zebrafish embryos at the 1-cell stage, and the proportions of the embryos with a normal phenotype 24 hpf were counted. To prepare samples for use with the FitCUT&RUN approach, 25 pg of the TF-rFc mRNA were injected, and embryos at the appropriate developmental stages were collected according to time after fertilization and morphology (200 embryos in the 256-cell stage, 100 embryos in the 1k-cell stage, and 50 embryos in the dome stage were collected).
FitCUT&RUN and CUT&RUN procedures and library preparation
The desired number of cells or embryos was collected, and CUT&RUN was performed with some modifications based on the original protocol (Skene and Henikoff 2017); for FitCUT&RUN, the antibody incubation step was skipped. Before binding cells with concanavalin A beads (Bangs Laboratories BP-531), zebrafish embryos were removed from their chorions, and the cells were suspended in wash buffer (20 mM HEPES [pH 7.5], 150 mM NaCl, 0.5 mM spermidine, and protease inhibitor). Beads-bound embryonic cells and K562 cells were permeabilized, and calcium ions were chelated with wash buffer containing 0.02% digitonin (Invitrogen BN2006) and 2 mM EDTA. The samples were washed twice in digitonin buffer (wash buffer containing 0.02% digitonin), and then, 2.5 µL 20× pAG-MNase (EpiCypher 15-1016) were added with gentle vortexing. Next, the samples were incubated overnight with rotation at 4°C. The samples were washed twice in digitonin buffer and equilibrated to 0°C. CaCl2 (2 mM) was added and incubated at 0°C for 30 min. Reactions were stopped with the addition of STOP buffer (200 mM NaCl, 20 mM EDTA, 4 mM EGTA, 50 µg/mL RNase A, 40 µg/mL glycogen, and 10 pg/mL yeast spike-in DNA). The samples were incubated at 37°C for 20 min to release DNA fragments and digest RNA. SDS (0.1%) and 250 µg/mL Proteinase K were added to the supernatants and incubated at 55°C for 30 min. The DNA was purified using phenol/chloroform/isoamyl alcohol (PCI) extraction and precipitated with glycogen and ethanol. For FLAG-CUT&RUN or standard CUT&RUN, anti-FLAG (Sigma-Aldrich F1804), anti-ATF1 (Santa Cruz Biotechnology sc-270), or anti-ELF1 antibodies (Santa Cruz Biotechnology sc-133096) were incubated overnight with rotation at 4°C. For negative control, two types of rabbit IgG (I: abcam ab6709, II: Cell Signaling Technology 2729S) were added and incubated overnight. Libraries were amplified from purified DNA using a HyperPrep kit (KAPA Biosystems KK8504), and the PCR program was run as follows: 98°C for 45 sec, 98°C for 15 sec, and 60°C for 10 sec, step two and three were repeated 12–18 times, and the final step was run at 72°C for 1 min. The libraries were sequenced by the Illumina HiSeq X Ten sequencing platform.
ATAC-seq library preparation and sequencing
The ATAC-seq libraries of early zebrafish embryos were prepared as previously described with minor modifications (Wu et al. 2016, 2018). In brief, embryos developing to desired stages were collected (20 embryos at the 256-cell stage, five embryos at the 1k-cell stage, and 12 embryos at the oblong stage) and then lysed in 200 µL of lysis buffer (10 mM Tris-HCl [pH 7.4], 10 mM NaCl, 3 mM MgCl2, and 0.5% NP-40) for 10 min on ice to prepare the nuclei. After lysis, nuclei were collected by centrifugation at 500g for 5 min and then incubated with Tn5 transposase and tagmentation buffer (4 μL ddH2O, 4 μL 5 × TTBL, 5 μL TTE mix V5) at 37°C for 30 min (Vazyme TD502). After tagmentation, 5 μL of 5 × TS stop buffer were added and incubated at room temperature for 5 min to end the reaction. Fragmented DNA was then purified using a Qiagen MinElute kit (Qiagen 28004). PCR was performed to amplify the library by mixing 5 μL of N5XX primer, 5 μL of N7XX primer (Vazyme TD202) with 10 μL of 5 × TAB and 1 μL of TAE (Vazyme TD502) at the following PCR conditions: 72°C for 3 min; 98°C for 30 sec; 14 cycles at 98°C for 15 sec, 60°C for 30 sec and 72°C for 3 min; and 72°C for 5 min. After PCR, the libraries were purified with 1.4× Agencourt AMPure XP beads (Beckman Coulter Genomics, p/n A63881) and sequenced by the Illumina HiSeq X Ten sequencing platform.
RNA-seq library preparation and sequencing
Embryos developed to desired stages were collected and removed from their chorions (20 embryos for each sample). The RNA-seq libraries of zebrafish embryos were prepared according to the protocols for the KAPA mRNA HyperPrep kit (KAPA Biosystems KK8580). The amplified libraries were sequenced by the Illumina NovaSeq platform.
Mutation of Nanog binding sites and RT-qPCR
The designs of gRNAs were based on the following criteria: targeting Nanog motif hits within Nanog binding sites at the 256-cell stage, and the binding sites are close to the TSS of the first-wave transcribed genes. To exclude the impact of maternal loading mRNAs, we only considered first-wave transcribed genes without maternal loading transcripts. In total, we designed five gRNAs, whose target sites are associated with four genes. For the selected peaks of Nanog binding sites, CRISPR targets were designed at the Nanog motif, and the guide RNAs (gRNAs) were synthesized by PCR amplification and in vitro transcription using the MEGAshortscript T7 kit (Invitrogen AM1354). One nanoliter of Cas9 protein (GenScript Z03389) (4 µM) and gRNAs (150 ng/µL) or Nuclease-free water were injected into zebrafish embryos at the 1-cell stage; 20 embryos developed to desired stages were then collected and removed from their chorions to extract total RNA. The cDNA library was synthesized using EvoScript Universal cDNA Master (Roche 07912374001). Two microliters of cDNA mix were used for RT-qPCR (Roche 4913850001), then relative expression levels of the corresponding Nanog target gene between mutants and water-injected samples were measured by 2(−ΔΔCt) using actb1 as an internal control gene. All primer sequences for mutation and RT-qPCR are listed in Supplemental Table S6.
Ensembl gene annotation on new Chromosome 4 assembly
A newly assembled Chromosome 4 was obtained from Yang et al (2020). The sequences of the Ensembl genes in Chromosome 4 were obtained using fastaFromBed (version 2.27.1) (Quinlan and Hall 2010) based on the genome assembly of danRer11. We mapped the sequence to the new assembly with Bowtie 2 (version 2.4.2; for mir-430 genes) (Langmead and Salzberg 2012) using the parameters “-f -a” or minimap2 (version 2.17; for other genes) (Li 2018) using the parameters “-ax asm5”. The transcription start site, transcription end site (TES), coding region start (CDS), coding region end (CDE), exon start position, and exon end position for each gene in the new Chromosome 4 assembly were generated based on the mapping details (Supplemental Table S7).
Processing of FitCUT&RUN, CUT&RUN, and FLAG-CUT&RUN data
Raw sequenced read pairs were filtered by Trim Galore! (version 0.6.5, http://singlecellqc.com/projects/trim_galore/) with cutadapt (version 3.3) (Martin 2011) using the following parameters: “‐‐trim-n”. The filtered reads were mapped back to the genome (hg38 for the K562 cell data and danRer11 with new Chromosome 4 assembly for the zebrafish data) using Bowtie 2 (version 2.4.2) (Langmead and Salzberg 2012) with the following parameters: “‐‐no-mixed ‐‐no-discordant ‐‐no-unal”. Mapped read pairs (fragments) with MAPQ < 30 were discarded and converted to BAM format using SAMtools (version 1.6) (Li et al. 2009; Danecek et al. 2021). BAM files were converted to BEDPE files using BEDTools (version 2.27.1) (Quinlan and Hall 2010). Fragment lengths ≤ 120 bp were kept. Replicates were merged, sampling down (10 M fragments for ATF1 and ELF1 when sequencing depth larger than 10 M, 5 M fragments for Nanog, Pou5f3, and Sox19b), and peak calling was performed by MACS (version 2.1.1.20160309) (Zhang et al. 2008) with the following parameters: “-f BEDPE -g 3.09e9 (for the K562 cell data) or 1.34e9 (for the zebrafish data) -q 0.05”. Peaks with fold ≥5 (ATF1 and ELF1, IgG) or 10 (Nanog, Pou5f3, and Sox19b) and Q-value ≤ 1 × 10−10 were maintained for the downstream analysis. The middle halves of fragments were piled up and transformed into bigWig format for visualization and subsequent analysis using custom scripts and BEDTools (version 2.27.1) (Quinlan and Hall 2010). Peaks based on public IgG CUT&RUN data in K562 (Supplemental Table S3) and IgG CUT&RUN data in zebrafish embryos at dome stages (Supplemental Table S2) were regarded as “negative” peaks. For any FitCUT&RUN, CUT&RUN, or FLAG-CUT&RUN data generated in this study, peaks overlapped with “negative” peaks were removed in the following analysis.
Processing of ATAC-seq data
ATAC-seq data were processed as previously described (Liu et al. 2018) with little modification. Raw sequenced read pairs were filtered by Trim Galore! (version 0.6.5, http://singlecellqc.com/projects/trim_galore/) with cutadapt (version 3.3) (Martin 2011) using the following parameters: “‐‐trim-n”. The filtered reads were mapped back to the genome (danRer11 with new Chromosome 4 assembly) using Bowtie 2 (version 2.4.2) (Langmead and Salzberg 2012) with the following parameters: “‐‐trim-to 3:40 ‐‐no-mixed ‐‐no-unal”. Mapped read pairs (fragments) with MAPQ < 30 were discarded and converted to BAM format using SAMtools (version 1.6) (Li et al. 2009; Danecek et al. 2021). BAM files were converted to BEDPE files using BEDTools (version 2.27.1) (Quinlan and Hall 2010). Fragment lengths ≤ 100 bp were kept and transformed to pseudo-single-end reads by the left 25-bp 5′ or 3′ part randomly. Replicates were merged, and peak calling was performed using MACS (version 1.4.2 20120305) with the following parameters: “-f BED -g 1.34e9 ‐‐keep-dup all ‐‐nomodel ‐‐shiftsize 25”. Peaks with P-value ≤ 1 × 10−10 and fold ≥10 were retained. Pseudo-single-end reads were extended to 50 bp, piled up, and transformed into bigWig format for visualization and subsequent analysis by custom scripts and BEDTools (version 2.27.1) (Quinlan and Hall 2010).
Processing of ChIP-seq/CETCh-seq data
Both ELF1 ChIP-seq data and ATF1 CETCh-seq data in K562 cells were obtained from the ENCODE portal (Sloan et al. 2016) (https://www.encodeproject.org/) by identifiers ENCSR975SSR for ELF1 and ENCSR159OCC for ATF1. For the eGFP-ELF1 data set, the stably transfected eGFP-ELF1 K562 cell lines was constructed and then a standard ChIP-seq experiment was performed. The FLAG-ATF1 data set was generated by CETCh-seq (CRISPR Epitope Tagging ChIP-seq) which introduced the FLAG tag to endogenous locus of ATF1 by CRISPR and then performed a traditional ChIP-seq experiment. ChIP-seq/CETCh-seq data were filtered by Trim Galore! (version 0.6.5, http://singlecellqc.com/projects/trim_galore/) with cutadapt (version 3.3) (Martin 2011) with the following parameters: “‐‐trim-n”. The filtered reads were mapped back to the genome (hg38 for K562 and danRer11 with new assembly Chromosome 4 for zebrafish data) using Bowtie 2 (version 2.4.2) (Langmead and Salzberg 2012) with the following parameters: “‐‐no-mixed ‐‐no-discordant ‐‐no-unal”. Mapped reads with MAPQ < 30 were discarded and converted to BAM format using SAMtools (version 1.6) (Li et al. 2009; Danecek et al. 2021). BAM files were converted to BED files using BEDTools (version 2.27.1) (Quinlan and Hall 2010). Replicates were merged, sampling down (10 M fragments for ATF1 and ELF1, 5 M fragments for Nanog, Pou5f3, and Sox2), and peak calling was performed by MACS2 (version 2.1.1.20160309) (Zhang et al. 2008) with the parameter “-f BED -g 3.09e9 (for the K562 cell data) or 1.34e9 (for the zebrafish data) -q 0.05”. Peaks with fold ≥5 (ATF1 and ELF1) or ≥10 (Nanog, Pou5f3, and Sox2) and Q-value ≤ 1 × 10−10 were retained for downstream analysis. Estimated fragment lengths were obtained from the log file of MACS2. Reads were extended to estimated fragment length, and the middle halves were piled up and transformed to bigWig format for visualization and subsequent analysis by custom scripts and BEDTools (version 2.27.1) (Quinlan and Hall 2010).
Processing of RNA-seq data
RNA-seq data (Supplemental Table S3) were filtered by Trim Galore! (version 0.6.5, http://singlecellqc.com/projects/trim_galore/) with cutadapt (version 3.3) (Martin 2011) using the following parameters: “‐‐trim-n”. The filtered reads were mapped back to the zebrafish genome (danRer11 with newly assembled Chromosome 4) using HISAT2 (version 2.1.0) (Kim et al. 2015) with the following parameters “‐‐no-mixed ‐‐no-discordant”. FPKM and read counts for each gene were calculated using StringTie (version 1.3.3b) (Pertea et al. 2015, 2016). DEseq2 was applied to call differentially expressed genes, using adjusted P-value = 0.05 and log2(fold_change) 0.58 (i.e., fold change 1.5) as a cutoff.
List of early transcribed genes
First-wave transcribed genes (Lee et al. 2013) and early ZGA genes (Heyn et al. 2014) were downloaded from the previous study. The genes were retained when their ID could be found in our annotation table (Supplemental Table S7). Finally, 196 first-wave transcribed genes and 342 genes early ZGA genes were used for subsequent analyses.
Motif scan
The motif position weight matrix in MEME format was collected from the JASPAR (Fornes et al. 2020) and footprintDB (Sebastian and Contreras-Moreira 2014) databases. Pou5f3 motifs were identified using MA0142.1 (JASPAR) and MA1115.1 (JASPAR). Sox motifs were identified using MA0142.1 (JASPAR) and MA0143.3 (JASPAR). Nanog motifs were identified using M0926_1.02 (footprintDB), NANOG_3 (footprintDB), NANOG_methyl_1 (footprintDB), and NANOG_methyl_2 (footprintDB). Motif scans were performed using the FIMO tool (version 5.0.5) (Grant et al. 2011) against the genome sequence with the following parameter “‐‐max-stored-scores 1000000”. If the peak (summits ± 100 bp) overlapped with either the MA0142.1 or MA1115.1 motif, it was regarded as a peak representing the Pou5f3 motif. Similarly, if the peak (summits ± 100 bp) overlapped with either the MA0142.1 or MA0143.3 motif, it was regarded as a peak representing the Sox19b motif. If the peak (summits ± 100 bp) overlapped with any M0926_1.02, NANOG_3, NANOG_methyl_1, or NANOG_methyl_2 motif, it was regarded as a peak representing the Nanog motif (Supplemental Table S4).
Nanog target gene definition and Gene Ontology analysis
If there is a Nanog-binding site lying within 5 kb around a gene TSS, this gene was regarded as the target gene of that Nanog-binding site. We further categorized genes based on the timing of target by Nanog: pre-MBT targets (1317 genes, targeted since the 256-cell stage), MBT new targets (1891 genes, targeted since the 1k-cell stage), and post-MBT new targets (4047 genes, targeted since the dome stage) (Supplemental Table S5). Functional annotation clustering analysis was performed using gprofile-official (version 1.0.0) (Raudvere et al. 2019) to find enriched terms with the following parameters “organism = ‘drerio’, sources = [‘GO:CC’,‘GO:MF’,‘GO:BP’]”. Terms with fewer than 15 targets, with more than 500 targets, or with adjusted P-values >0.001 were removed from the analysis. Terms with Jaccard similarity of intersection genes >0.6 were regarded as term clusters. The top three term clusters with the highest enrichment scores were presented.
UCSC Genome Browser
The genome browser view was obtained using the UCSC Genome Browser (Kent et al. 2002) with Track data hubs (Raney et al. 2014) and visualized with smoothing with a mean of pixels.
Statistical analysis
P-values were calculated by the two-sided Mann–Whitney U test using SciPy (version 1.31) (Virtanen et al. 2020) if not clarified in figure legends. Asterisks represent statistical significance; (***) P < 0.001, (n.s.) not significant.
Data access
The raw sequence data of K562 cell line and zebrafish generated in this study have been submitted to the Genome Sequence Archive (GSA) (Wang et al. 2017) in the Beijing Institute of Genomics (BIG) Data Center (https://bigd.big.ac.cn/gsa) under accession numbers CRA004074 and HRA00767 (https://ngdc.cncb.ac.cn/gsa-human). Codes used for analyses are available as Supplemental Code and at GitHub (https://github.com/TongjiZhanglab/FitCR).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Steven Henikoff for the kind gifts of protein A-MNase (pA-MN) and protein A/G-MNase (pAG-MN). We thank Richard Myers for generating data set ENCLB076XWH and the Kevin White laboratory for ENCSR975SSR, and we thank the ENCODE Consortium for providing the accessibility of these two data sets. This work was supported by the National Natural Science Foundation of China (32030022, 31970642, 31721003, 31601161), National Key Research and Development Program of China (2017YFA0102600), National Program for Support of Top-notch Young Professionals, Major Program of Development Fund for Shanghai Zhangjiang National Innovation Demonstration Zone (ZJ2018-ZD-004).
Author contributions: Y.Z. designed the study. X.W. and G.L. performed the experiments, with the help of J.C. The data were analyzed by W.W. and Y.W. The manuscript was written by X.W., W.W., G.L., and Y.Z.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275837.121.
- Received June 1, 2021.
- Accepted December 22, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








