
Reactivation of transposable elements following hybridization in fission yeast
- 1Division of Evolutionary Biology, Faculty of Biology, Ludwig Maximilian University of Munich, 82152 Planegg-Martinsried, Germany;
- 2National Institute of Biological Sciences, Beijing 102206, China;
- 3Tsinghua Institute of Multidisciplinary Biomedical Research, Tsinghua University, Beijing 102206, China
Abstract
Hybridization is thought to reactivate transposable elements (TEs) that were efficiently suppressed in the genomes of the parental hosts. Here, we provide evidence for this “genomic shock hypothesis” in the fission yeast Schizosaccharomyces pombe. In this species, two divergent lineages (Sp and Sk) have experienced recent, likely human-induced, hybridization. We used long-read sequencing data to assemble genomes of 37 samples derived from 31 S. pombe strains spanning a wide range of ancestral admixture proportions. A comprehensive TE inventory revealed exclusive presence of long terminal repeat (LTR) retrotransposons. Sequence analysis of active full-length elements, as well as solo LTRs, revealed a complex history of homologous recombination. Population genetic analyses of syntenic sequences placed insertion of many solo LTRs before the split of the Sp and Sk lineages. Most full-length elements were inserted more recently, after hybridization. With the exception of a single full-length element with signs of positive selection, both solo LTRs and, in particular, full-length elements carry signatures of purifying selection indicating effective removal by the host. Consistent with reactivation upon hybridization, the number of full-length LTR retrotransposons, varying extensively from zero to 87 among strains, significantly increases with the degree of genomic admixture. This study gives a detailed account of global TE diversity in S. pombe, documents complex recombination histories within TE elements, and provides evidence for the “genomic shock hypothesis.”
Hybridization is a pervasive evolutionary force with implications for adaptation and species diversification (Abbott et al. 2013). It entails disruption and novel arrangement of parental haplotypes with the potential to alter regulatory pathways (Turner et al. 2014). This includes regulation and epigenetic control of transposable elements (TEs) (Han et al. 2004) with proven consequences for speciation (Serrato-Capuchina and Matute 2018) and genome evolution (Kazazian 2004). Barbara McClintock hypothesized that hybridization could lead to a “genomic shock” reactivating the mobilization of TEs that were efficiently suppressed in the parental genomes (McClintock 1984). This hypothesis follows from the idea of a coevolutionary arms race (Van Valen 1973) between TEs striving to maximize proliferation and the host genome evolving suppression mechanisms to keep TE activity in check. By introducing elements of untested genetic variation into a naive genomic background, hybridization has the potential to disrupt genome stability with the possible effect of reactivating TEs (McClintock 1984).
Evidence for the “genomic shock hypothesis” is scarce, despite investigation in a diverse array of species. Results are often mixed, and outcomes differ even between closely related species. For example, intraspecific crosses between Drosophila melanogaster males containing the P-element transposon with naive females lacking expression of the suppressor gene result in hybrid dysgenesis (Kidwell et al. 1977; Bingham et al. 1982; Kidwell 1983; Bucheton et al. 1984). In other species of Drosophila this effect cannot be consistently replicated (Coyne 1985; Hey 1988; Lozovskaya et al. 1990; Vela et al. 2014). Hybridization between Arabidopsis thaliana and A. arenosa induces up-regulation of ATHILA retrotransposon expression and reduces hybrid viability (Josefsson et al. 2006). However, such an effect is not observed in crosses between A. thaliana and A. lyrata (Göbel et al. 2018). In sunflowers, contemporary crosses between Helianthus annuus and H. petiolaris show no evidence for increased large-scale TE mobilization (Kawakami et al. 2011; Ungerer and Kawakami 2013; Renaut et al. 2014). However, over evolutionary timescales, sunflower hybrid species combining ancestry from the same parental species show elevated number of LTR retrotransposons, indicating a role of hybridization for TE release in the past (Ungerer et al. 2006, 2009; Staton et al. 2009). Direct evidence for TE reactivation was observed from a 232-fold increase in TE expression in hybrids of incipient whitefish species (Dion-Côté et al. 2014). In other major groups like fungi, relatively little attention has been paid to study TE reactivation in the course of hybridization. In two recent studies in Saccharomyces species, no evidence supporting the genomic shock hypothesis was found (Hénault et al. 2020; Smukowski Heil et al. 2021).
Detailed investigation of the genomic shock hypothesis has long been hampered by technical difficulties of accurate TE characterization limiting studies for the most part to comparative genomics between high-quality assemblies of a few evolutionary divergent species (Hoban et al. 2016; Villanueva-Cañas et al. 2017; Bourgeois and Boissinot 2019). Long-read sequencing technology opens the opportunity to characterize TE variation at the resolution of multiple individual genomes from the same species. We here study the genomic shock hypothesis at microevolutionary resolution in the fission yeast Schizosaccharomyces pombe. S. pombe is a haploid unicellular ascomycete fungus of the Taphrinomycotina subphylum with facultative sexual reproduction (Jeffares 2018). Recent population genetic studies have shown that all globally known strains arose by recent admixture between two divergent ancestral lineages (described as Sk and Sp) (Tao et al. 2019; Tusso et al. 2019). These two lineages most likely diverged in Europe (Sp) and Asia (Sk) since the last glacial period. Human-induced migration at the onset of intensified transcontinental trade possibly induced hybridization of these ancestral lineages approximately 20–60 sexual outcrossing generations ago. Hybridization resulted in a broad range of ancestral admixture proportions predicting levels of phenotypic variation and reproductive compatibility between strains (Tusso et al. 2019).
To date, a detailed TE inventory has only been conducted for a single S. pombe strain (972 h−), which has been used for the assembly of the reference genome (Wood et al. 2002; Bowen et al. 2003) and is of pure Sp ancestry (Tusso et al. 2019). TEs found in the reference genome are all retrotransposons (class I TEs) with long terminal repeats (LTRs), which can be grouped into several LTR families (α−ι) on the basis of phylogenetic analyses (Bowen et al. 2003). The vast majority of TE elements in the reference genome only occurs in the form of solo LTRs (174 of 187 TE elements). Merely two types of full-length retrotransposons, called Tf1 and Tf2, containing the internal coding region (hereafter referred to as full-length elements) are known to exist in S. pombe (Levin et al. 1990; Levin 1995). Both Tf1 and Tf2 belong to the Ty3/Gypsy type of LTR retrotransposons, and their LTRs belong to the α and β family, respectively. In the reference genome, full-length elements (13 of 187 TE elements) are all Tf2 elements (Esnault and Levin 2015), but full-length Tf1 elements are known to exist in several wild strains (Levin et al. 1990). Short-read genome sequencing data have been used to investigate variation of TE insertions in S. pombe (Jeffares et al. 2015), but TE sequences cannot be reliably inferred from short reads.
In this study, using long-read sequencing data from strains spanning the worldwide diversity of fission yeast, we present a comprehensive description of the TE repertoire and place it in the context of recent hybridization between the Sp and Sk ancestors. Detailed phylogenetic and population genetic analyses were used to shed light on TE selection dynamics, assess the evidence of complex recombination, and test the genomic shock hypothesis.
Results
A previous large-scale analysis of global genetic diversity of S. pombe using 161 strains identified 57 clades differing by at least 1900 SNPs (Jeffares et al. 2015). We generated long-read sequencing data from a subset of 37 samples representing 29 of the 57 clades and two additional previously undescribed strains (Fig. 1; Supplemental Fig. 1; Supplemental Table S1). In six instances, two clones of the same strain accessed from different laboratories (with potentially different recent history) were independently sequenced. The data set was further complemented with the reference genome. Analyses of SNP variation place these 38 samples well within the global continuum of Sp to Sk ancestry (Fig. 1A,B; Supplemental Fig. 1; Tusso et al. 2019). Consistent ancestry profiles between clones of the same strain reflect high technical replicability (Supplemental Figs. 1, 2).
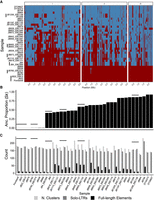
Genome composition by ancestry and LTR repertoire of a global collection of 38 samples corresponding to 31 nonclonal haploid S. pombe strains. (A) Heat map representing SNP-based haplotypes across all three chromosomes (I, II, and III) for the 38 samples used in this study. Six clonal samples derived from the same strain are indicated by vertical bars. Haplotypes are painted by Sp and Sk ancestry shown in red and blue color, respectively. For details on the inference of ancestry components, we refer to Tusso et al. (2019). (B) Distribution of Sk ancestry proportion per sample. (C) Number of solo-LTR sequences, full-length elements, and total number of clusters per strain. For a summary of the total number of TEs per strain, see Supplemental Figure 5. In all panels, clonal samples are grouped by horizontal black lines.
Global TE diversity of S. pombe
From long-read data averaging 85× sequence coverage per sample (range: 40×–140×), we generated 37 individual (near-)chromosome genome assemblies (Supplemental Table S3). Subsequent annotation allowed characterization of TE repertoires for each individual assembly. To establish synteny of TEs between the often highly rearranged genomes (Brown et al. 2011; Tusso et al. 2019), we translated the coordinates of TEs in each individual assembly to those in the reference genome. Locations of TE elements were highly collinear between samples (Supplemental Fig. 3) and often hosted multiple copies of TEs (as many as 16 copies). We refer to these local TE aggregations as TE clusters throughout. Within a cluster, synteny could not be unequivocally defined, restricting several subsequent analyses to TE clusters rather than individual TE sequences.
Across all samples we identified 8505 TE sequences that were contained in 719 TE clusters. Consistent with previous work, all TEs belonged to the Ty3/Gypsy LTR retrotransposon superfamily (Levin et al. 1990; Levin 1995). Additional identified helitron and TIR transposons overlapped with annotated genes in the reference genome and require further validation to guard against false positive inference. The vast majority of TE sequences occurred as solo LTRs, and only 1169 TE sequences were longer than 1.5 kb and contained internal sequences. The vast majority of the long sequences (924 sequences) had a length of around 4.9 kb, the expected length of full-length elements (Fig. 1C; Supplemental Fig. 4). The number of TE elements varied between strains both for solo-LTR sequences (range 115–198), as well as for full-length elements (range: 0–67) (Fig. 1C; Supplemental Fig. 5).
Methodological comparison
Variation between clones derived from the same strain was small but nonzero for the total number of clusters and TE sequences. A fraction of the differences was likely not owing to genome quality differences but may reflect biological variation acquired during a short period of time (Supplemental Fig. 6).
The overall high consistency of TE sequences in our clonal de novo genomes was in contrast to low congruence with TE inference from other sources (Supplemental Fig. 7). A comparison between our annotation of TE sequences in the reference genome and a BLAST-based annotation of TE sequences in an earlier version of the reference genome reported by Bowen et al. (2003) (Supplemental Table S6) showed consistency in 117 identified clusters. In either our Pomberef annotation or Bowen's annotation, 47 and 13 clusters showed unique evidence. In addition, comparing presence/absence of TE clusters as inferred from short-read data (Supplemental Fig. 7; Jeffares et al. 2015) to our annotations revealed a large proportion of inconsistent clusters ranging from 23% (JB22) to 49% (JB874). In summary, these results highlight the limitation of short-read data to infer TE insertions, confirm the robustness of long-read based inference, and tentatively suggest rapid mutation between clonal strains.
LTR diversity is superimposed on ancestral population divergence
Next, we extracted solo LTRs and flanking LTR sequences from full-length elements (both 5′ and 3′) amounting to 9503 LTR sequences altogether. Phylogenetic analysis of the resulting sequence alignment provided an overview of the global diversity of LTR sequences in S. pombe (Fig. 2A). Although bootstrap support was generally low, LTR sequences could be broadly grouped into previously reported families (Bowen et al. 2003). In most families, LTRs occurred exclusively as solo LTRs, had long terminal branches, and showed high intrafamily diversity. This phylogenetic signature reflects past transpositions of now extinct elements and is consistent with a long history of recombination-mediated conversion of full-length elements into solo LTRs followed by pseudogenization of the remaining LTR sequences. The α and β families constitute an exception. These two families hosted the large majority of full-length elements found in the data sets, Tf1 and Tf2, respectively (Fig. 2B). The other group of LTR sequences associated with full-length elements, here coined Sub-α/ζ, was closely related to the α family but showed evidence of recombination between LTR haplotypes from the α and β families, or between the β family and an ancestral sequence related to the ζ family (Supplemental Fig. 8). In total, we identified at least 24 recombinant solo-LTR haplotypes, several of which were found in multiple clusters (up to 64 and 40 clusters for the two most common recombinant haplotypes) (Supplemental Fig. 9).
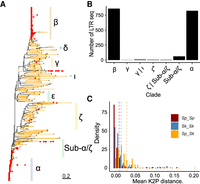
Phylogenetic relationship between LTR sequences. (A) Maximum-likelihood unrooted tree for solo LTRs and LTRs flanking full-length elements. Branches with bootstrap support higher than 95 are shown in yellow. LTR sequences associated with full-length elements are indicated with red points. Nomenclature of families follows Bowen et al. (2003) (Methods). (B) Number of flanking LTRs from full-length elements grouped by LTR family. Sequences without clear family membership are classified by the two most closely related families. (C) Pairwise divergence of LTR sequences belonging to the same family within syntenic clusters. Divergence between sequences is grouped by ancestral background: within ancestral background (Sp vs. Sp or Sk vs. Sk) or between ancestral backgrounds (Sp vs. Sk). Mean divergence per group is indicated by dashed vertical lines.
To relate the diversity of LTR sequences to global species diversity (Sp vs. Sk lineage), we inferred the ancestral genomic background of each syntenic cluster for each strain. In 581 of all 662 syntenic clusters, LTR sequences from the same family were exclusively present in clusters embedded by either of the two ancestral backgrounds. Consistent with higher overall genetic diversity in the Sk lineage (Tusso et al. 2019), the percentage of clusters with lineage-specific LTR insertions was higher for the Sk than for the Sp background (57% and 37%, respectively). In 152 clusters, sequences originating from the same LTR family occurred in at least one sample of each ancestral background and in 113 in at least two. Co-occurrence across ancestral backgrounds makes these sequences prime candidates of ancestral insertions before divergence of the two lineages. Across all shared 113 syntenic clusters, mean pairwise divergence was lower between sequences from the same LTR family inserted into the same ancestral background (Sp-Sp and Sk-Sk) than between different ancestral backgrounds (Sp-Sk) (Fig. 2C; Supplemental Fig. 10). Moreover, mean pairwise distance within the Sk group was higher than for the Sp group, which is consistent with higher effective population size inferred for the Sk group (Tusso et al. 2019). In summary, these results from solo-LTR sequences are consistent with the two-clade history inferred from genome-wide SNPs (Tao et al. 2019; Tusso et al. 2019) and show that a proportion of solo LTRs preceded Sp and Sk divergence and subsequent hybridization. This includes solo LTR sequences from the two most common α and β families that are characteristic of full-length elements (Supplemental Fig. 11).
Haplotype diversity of full-length elements documents a history of recombination
Next, we focused on full-length elements for which two haplotypes, Tf1 and Tf2 elements, have been previously described. Using window-based haplotype painting, all sequences were collapsed into 11 discrete haplotypes, each present in at least five sequences (Fig. 3A; Methods). These haplotypes can similarly be identified by means of phylogenetic analyses (Fig. 3B). Differentiation between haplotypes was primarily a result of divergence in the flanking LTR sequences and the first ∼2 kb of the internal sequence. Prevalence in the data set was highest for the Tf1 and Tf2 haplotypes occurring in 207 and 203 clusters, respectively. The remaining nine haplotypes populated 69 clusters. Despite relatively lower numbers, paralogous occurrence across different genomic regions (clusters) suggests that some of these recombinant haplotypes have recently been actively transposed. Prominent examples are haplotypes Tf2e, Tf2f, and Tf2g found in six, nine, and 28 independent clusters, respectively.
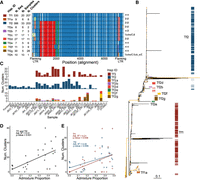
Diversity of full-length LTR elements. (A) Alignment of the 11 haplotypes identified by window-based haplotype painting in a global sample of S. pombe. For each haplotype we show haplotype ID, number of sequences found in all samples, number of samples, and number of independent TE clusters. Vertical black lines show boundaries of flanking LTRs. Colors per window (vertical comparison within the alignment) represent haplotype difference from TF1 used as reference. Owing to insertion–deletion polymorphism, the alignment exceeds the length of the individual full-length LTR elements. Flanking LTR families per haplotypes are shown at the right (5′ LTR:3′ LTR). (B) Maximum-likelihood unrooted tree for full-length LTRs. Branches with bootstrap support higher than 95 are shown in yellow. Colors correspond to the color assignment of the eight most common haplotypes in A. (C, bottom panel) Number of common haplotypes for full-length elements found in at least three independent clusters shown per sample. (Top and middle panels) Haplotypes Tf1 and Tf2 are shown in independent plots. Colors per haplotype ID as indicated. Samples are ordered by ancestral admixture from pure Sp to pure Sk as in Figure 1. (D) Relationship between ancestral Sp and Sk admixture proportions and the number of clusters with Tf1 and Tf2 full-length elements. Each point represents a nonclonal strain. The adjusted proportion of total variance explained (R2) and the type 1 error probability (P-value) are shown in the inset. (E) As in D but differentiating between haplotypes TF1 and TF2.
Haplotype diversity was larger for derivates from Tf2 (nine haplotypes) than for Tf1 with only one additional haplotype. Haplotype diversity was mostly governed by homologous recombination between the Tf1 and Tf2 haplotypes. For example, haplotype Tf1a contains an internal sequence of the Tf1 haplotype, but flanking LTRs are more similar to those of Tf2 (β LTR family). Conversely, haplotypes Tf2e and Tf2g are most similar to Tf2 in the internal sequence, but its flanking LTRs are more related to those found in Tf1 (α LTR family). Other haplotypes also suggest recombination of the internal sequence, as is illustrated in Tf2f.
Support for the genomic shock hypothesis
To estimate the age of insertion, we calculated pairwise divergence between the 5′ and 3′ flanking LTR sequence for each full-length element. Naturally, divergence in recombinant full-length elements was elevated with values exceeding 10% (Supplemental Fig. 12). For the vast majority of nonrecombinant full-length elements of both Tf1 and Tf2, however, divergence was consistently lower than 1%, suggesting that TEs mobilized over a short period of time in a “burst of activity” rather than accumulating gradually. Recent activity succeeding the Sp and Sk split was further supported by an uneven segregation of full-length haplotypes between ancestral backgrounds: The Tf2 haplotype was found in the majority of samples but was absent in strains with predominant Sk ancestry. The Tf1 haplotype showed the reverse pattern (Fig. 3C). Other haplotypes were either sample specific or were restricted to a few samples, with JB758 and JB953 being exceptionally prolific hosts of the nontypical full-length haplotypes. Haplotypes whose prevalence was restricted to only a few samples often showed high abundance within those samples. For instance, Tf2f and Tf2g were restricted to three and five samples but populated nine and 28 clusters, respectively.
Overall, this analysis suggests that full-length haplotypes can be grouped into two classes: (1) two common haplotypes (Tf1 and Tf2), which are characterized by flanking LTRs of the α and β family and are found in most samples, but segregate at different rates in the ancestral groups (with dominance of Tf2 in Sp and dominance of Tf1 in Sk); analysis of solo LTRs suggest likely presence of at least the β family before the split of Sp and Sk (Supplemental Fig. 11); and (2) a number of haplotypes restricted to few strains, often with evidence for recombination. Based on sequence similarity, at least some of these rarer haplotypes originated by homologous recombination between Tf1 and Tf2 and remained active thereafter.
We next examined whether the patterns of TE diversity in S. pombe conform to the “genomic shock hypothesis.” If recent hybridization (approximately 20–60 sexual generations ago) (Tusso et al. 2019) reactivated TE activity, strains with admixed genomic backgrounds should, on average, host a larger number of full-length elements relative to nonadmixed strains. This prediction was supported by the data. We observed a significant positive relationship between ancestral admixture proportion and the number of clusters containing full-length Tf1 or Tf2 sequences (including only nonclonal samples) (Fig. 3D). This correlation remained if each haplotype was considered independently (Fig. 3E) and when exclusively focusing on the most recent full-length element insertions (singleton clusters; P-value 0.004, adjusted R2: 0.24) (Supplemental Fig. 13). This correlation is consistent with increased TE activity in admixed samples. Alternatively, it may reflect a demographic signal of an increased population mutation rate in a pool of hybrids having recently experienced a population expansion. Under this scenario, we would expect to observe a general excess of low-frequency variants not only in TEs, but any type of genetic variation. Repeating the analysis using neutrally evolving SNPs, however, did not support a differential demographic explanation. There was no indication that the number of singleton SNPs was elevated in admixed samples (P-value 0.088, adjusted R2: 0.07) (Supplemental Fig. 13). Moreover, no correlation was found between the number of singleton SNPs and number of singleton TE clusters (P-value 0.374, adjusted R2: −0.01) (Supplemental Fig. 13). On the basis of these results, we propose that recent hybridization increased the rate of TE proliferation in S. pombe as is predicted by the “genomic shock hypothesis.”
Population genetic inference of selection
To shed further light on the evolutionary history of TE elements in S. pombe, we constructed unfolded site frequency spectra (SFS) scoring presence/absence of clusters as allelic states. First, we considered all clusters found in nonclonal strains (686 in total), of which 508 clusters (74.0%) were found in no more than five samples, 363 (52.9% of total) were restricted to single samples (singletons) (Fig. 4A), and 115 clusters (16.7%) occurred in 90% or more of the samples. These ubiquitously present clusters contained predominantly solo LTRs. Restricting the SFS to full-length elements, low-frequency variants were substantially more common and high-frequency clusters were drastically reduced. Of 415 clusters with at least one full-length element, 423 (97.4%) were found in no more than five samples and 258 (59.4% of total) were singletons. Only two full-length element-containing clusters exceeded a frequency of 30%.
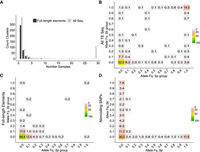
Allelic variation of LTR clusters between nonclonal strains. (A) One-dimensional site frequency spectrum summarizing the allelic frequency of TE cluster insertions across all 31 nonclonal strains. Bars differentiate between either all sequences or full-length elements. (B–D) Two-dimensional site frequency spectra showing the proportion of allele frequency bins shared between the ancestral Sp and Sk background for all TE sequences (B), full-length elements (C), and noncoding genome-wide SNPs (D). Site frequency spectra in B and C are unfolded, but folded in D. For folded spectra of B and C, see Supplemental Figure 14. Number and color range indicate the percentage out of all variants within each panel.
The excess of low-frequency and depletion of high-frequency clusters containing full-length elements can result from three non-mutually exclusive processes: a recent burst in transposition rate, a recent demographic population expansion resulting in an increase of rare variants, and purifying selection removing new insertions and maintaining variants in low frequency. To evaluate the effect of demographic variation, the history of admixture of the species has to be accounted for. We therefore scored variation of SNPs adjacent to each cluster and inferred the ancestral origin for each cluster (Sp or Sk). Subsequently, we calculated the two-dimensional site frequency spectrum (2dSFS) for both Sp and Sk ancestry using only nonclonal samples (Fig. 4B). Considering all TE elements (solo LTR and full length), ∼15% of the clusters were fixed in both ancestral lineages likely representing ancestral insertions present before the split of the two lineages. The majority of clusters, however, were lineage specific and in low frequency, with 43% of the clusters having frequencies below 0.1 in both lineages (∼68% in folded SF) (Supplemental Fig. 14). For full-length elements, low-frequency variants were substantially more common (Fig. 4C; Supplemental Fig. 14, folded 2dSFS). Here, the percentage of low-frequency variants increased to ∼64%, and clusters at intermediate frequency were reduced. To relate these patterns to genetic variation presumably evolving neutrally, we constructed the folded 2dSFS of genome-wide noncoding SNPs (Fig. 4D). Here, the percentage of low-frequency variants for noncoding SNPs was ∼30%, which was below values of the folded 2dSFS from all-LTR (67.9%) and full-length variants (64.6%) (Supplemental Fig. 14). The relative increase of rare alleles in LTRs over neutral SNPs cannot be explained with demographic expansion alone. Instead, these results are best explained by a recent increase in TE proliferation (in admixed genomes, see above) and a likely additional component of purifying selection against LTR elements in general. Consistent with the latter prediction, the percentage of fixed clusters was higher in the Sp ancestral population where a lower Ne has been predicted (Tusso et al. 2019), reducing the efficacy of selection (Charlesworth and Charlesworth 1983; Charlesworth and Langley 1989).
Discussion
Support for the genomic shock hypothesis on a microevolutionary scale
This study provides empirical evidence for a role of hybridization in the evolution of transposable elements. It adds to the evidence of TE reactivation as a consequence of hybridization in fungi (Hénault et al. 2020; Smukowski Heil et al. 2021). Our results are consistent with the idea that hybridization of two closely related S. pombe lineages, Sp and Sk (Dxy ∼0.005), activated TE proliferation in the admixed genomes (“genomic shock”) (McClintock 1984). The recent timing of hybridization allowed us to witness the sudden burst of transposition in natural populations before signal loss by re-establishment of novel repression mechanisms. Sudden bursts of transposition generated a large cohort of insertions with roughly the same age, as has similarly been documented in Drosophila (Vieira et al. 1999), rice (Piegu et al. 2006), piciformes (Manthey et al. 2018), salmonids (de Boer et al. 2007), and primates (Pace and Feschotte 2007).
Hybridization may have contributed to increased numbers of full-length TEs in admixed genomes in three ways. The first is by import of active, full-length elements from one pure parental background into the other. This was illustrated by transfer of Tf2 full-length elements from the Sp genomic background into the originally Tf2-free Sk background in admixed samples. Second, hybridization may have disrupted the allelic inventory of coadapted control mechanisms impeding TE mobilization in the parental backgrounds, for example, CENPB protein homologues (Cam et al. 2008), Set1 histone methyltransferase (Lorenz et al. 2012), histone deacetylases and chaperones (Murton et al. 2016), and to a low degree the RNAi machinery (Woolcock et al. 2011; Chapman 2018). Although the precise molecular mechanisms underlying activity, repression, and copy number of TEs have been extensively studied in the reference strain of S. pombe (Hansen et al. 2005; Cam et al. 2008; Lorenz et al. 2012; Murton et al. 2016), the mechanism underlying the observed reactivation of TEs in natural populations remains elusive and will require further study.
A third way hybridization may have contributed to TE proliferation is by reduction of Ne in the founder population of hybrids, reducing efficiency of purifying selection against TE (Charlesworth and Charlesworth 1983; Charlesworth 2009). Although a bottleneck as recent as 20 outcrossing generations ago is difficult to reconstruct, patterns of TE frequency overall confirmed the relationship between effective population size and purifying selection. Contrary to findings of other hybridizing yeast lineages, we found that putatively active full-length elements are only ubiquitously present in one of the pure parental backgrounds (Sp). Strains predominated by Sk ancestry had an overall reduced number of solo LTRs, less-fixed LTR insertions, and hosted fewer, if any, copies of full-length LTR elements (with only few, slightly admixed strains hosting Tf1). More efficient TE control and removal in the Sk lineage is consistent with its higher effective population size predicting higher efficiency of purifying selection of deleterious TE elements compared with the Sp ancestral lineages (Lynch and Walsh 2007). Similar variation in TE content as a result of changes in Ne has been observed in Brachypodium (Stritt et al. 2018), Arabidopsis (Lockton et al. 2008), Caenorhabditis (Dolgin et al. 2008), Drosophila (García Guerreiro et al. 2008), sticklebacks (Blass et al. 2012), Anoles (Tollis and Boissinot 2013), humans, and mice (Xue et al. 2018).
Transposition–selection balance was further reflected by site frequency spectra skewed toward low frequency of TE insertions (Barrón et al. 2014; Bourgeois and Boissinot 2019). Analysis in Drosophila found that 48%–76% of TEs had frequencies lower than expected under neutrality as a consequence of purifying selection (Cridland et al. 2013; Barrón et al. 2014; Blumenstiel et al. 2014). Deviation from neutral expectations has also been observed in other systems like Anoles (Ruggiero et al. 2017), mice (Xue et al. 2018), or Arabidopsis (Hazzouri et al. 2008; Lockton et al. 2008). In this study, we similarly found 68.4% (85.6% in folded 2dSFS) of TE insertion in frequencies below 0.2, in contrast to 44.1% found for presumably neutral, noncoding SNPs.
Overall, the data are best explained by a burst in transposition rate upon hybridization on a background of continuous purging of both solo LTRs, and to a larger degree full-length elements. To quantify the contribution of purifying selection on TE expansion during and after hybridization, explicit demographic reconstruction and fitness data would be necessary.
TE dynamics through time: the role of homologous recombination
The combination of phylogenetic and population genetic analyses of this study further contributes to our understanding of TE dynamics in natural populations. Under expectations of the neutral evolutionary theory, younger TE insertions are on an average expected to segregate at lower allele frequencies than older insertions (Kimura and Ohta 1973). Moreover, under the assumption of a molecular clock, progressively older insertions will have accumulated increasingly more mutations (Petrov et al. 1996). Thus, both allele frequency and sequence divergence provide information on the age of TE insertions (Blumenstiel et al. 2014). Across our global sample of S. pombe strains, the vast majority of full-length LTR elements segregated at very low frequencies, and sequence divergence of flanking LTRs within and between copies was shallow. In contrast, solo LTRs segregated at higher frequencies and were diversified into multiple divergent families. These results are consistent with full-length elements being mostly of young age and solo LTRs being of much older origin.
In the S. pombe reference strain, ∼70% of Tf2 mobilization events involve homologous recombination between newly synthesized cDNA and a pre-existing copy of Tf2; the remaining ∼30% of cases integrate in novel chromosomal locations (Hoff et al. 1998). Homologous recombination and recycling of target sites has two consequences for TE dynamics. First, older TE insertions may be replaced by more recent events, hampering the reconstruction of TE dynamics over long evolutionary timescales. Second, homologous recombination can be an important mechanism for TE diversification, resulting in new recombinant haplotypes for which this study provides copious examples. Importantly, recombination has occurred between elements of divergent families (Tf1 and Tf2) generating chimeric elements. Several of these recombinant haplotypes were found in multiple genomic clusters suggesting they have been recently active. Although most chimeric elements contained identical flanking LTRs, this was not always the case. In several instances, LTRs differed between the 3′ and 5′ end (Supplemental Fig. 12). However, with the only exception of haplotype Tf2b found in three clusters, all other haplotypes were found in single clusters. Because flanking LTR are known to be homogenized during reverse transcription, chimeric elements with divergent flanking LTR are more likely the result of homologous recombination between cDNA and a previous TE insertion.
Adaptive evolution of TEs
Transposable elements do not exclusively cause damage to the host. As sources of molecular variation, they can reroute regulation of gene expression (Trizzino et al. 2017; Sundaram and Wysocka 2020) and contribute to adaptive evolution (Schrader and Schmitz 2019). Environmental change can influence transposition rate as observed in S. cerevisiae (Paquin and Williamson 1984) or provide opportunities for positive selection of novel TE insertions (Aminetzach et al. 2005; Gresham et al. 2008; van't Hof et al. 2016; Esnault et al. 2019). Over long evolutionary timescales, beneficial TE insertions can become domesticated by the host genome (Miller et al. 1999). In S. pombe, for instance, CENPB protein homologues involved in TE silencing (Cam et al. 2008) are believed to have evolved from a domesticated pogo-like DNA transposase (Irelan et al. 2001; Casola et al. 2008).
TE insertions in S. pombe have also been discussed in the context of adaptation to environmental disturbance, or stress response. TE expression has been shown to be induced under stress conditions (Chen et al. 2003; Sehgal et al. 2007), and an enhancer sequence contained in Tf1 induces expression in adjacent genes (Leem et al. 2008). Artificially induced Tf1 insertions in the reference strain preferentially occurred upstream of stress response genes (Guo and Levin 2010). In natural strains, TE insertions are also enriched in the proximity to promoters of genes for stress response (Jeffares et al. 2015), which has been interpreted as evidence for TE-induced adaptive response (Esnault et al. 2019). Additionally, analysis of experimental populations under different environments shows variation in the genomic distribution of Tf1 integrations, as well as increased transposition rates under stress conditions (Esnault et al. 2019). Competition assays of the same evolved populations showed a selective advantage for several TE insertions under stress conditions with heavy metals.
In the context of this study, it is conceivable that the human-associated, hybrid strains that have been rapidly dispersed across the globe experienced a range of novel suboptimal environments inducing stress response (Jeffares 2018). Indeed, most strains used in this study were isolated from diverse substrates (Jeffares et al. 2015). Despite the apparent opportunities for adaptive evolution, the vast majority of full-length elements segregated at lower than expected frequency. This suggests that most of them were inserted recently (transposition burst) and rapidly removed by purifying selection and/or reduced to solo LTRs by homologous recombination, limiting the time frame of active proliferation. Our results do not exclude the adaptive potential of TEs, but instead suggest limitation of adaptive evolution to short periods of stress after which the selective advantage is lost. In this study, we identified only one full-length element candidate for pervasive long-term positive selection present in both ancestral backgrounds. However, the functional significance of this insertion is not clear and warrants future experimental exploration.
Overall, this study offers a comprehensive characterization of the global diversity of transposable elements in S. pombe. Phylogenetic and population genetic approaches provide evidence for the genomic shock hypothesis in fungi and copious examples of homologous recombination among full-length and solo-LTR sequences. Consistent with established principles of molecular evolution, TE insertions were generally subject to purging with the exception of a single locus. These results contribute to the debate on the role of TEs in evolution, notably in speciation and adaptation (Serrato-Capuchina and Matute 2018).
Methods
Previously, a worldwide collection of 161 naturally occurring S. pombe strains (JB strains) has been grouped into 57 clades differing by at least 1900 SNPs (Jeffares et al. 2015). Within each clade, strains are near clonal. In this study, we compiled single-molecule long-read sequencing data for strains representing 29 clades that cover the spectrum of genetic variation in the species by (1) selecting samples along the phylogeny (Supplemental Fig. 2), (2) including previously reported genetic groups (Jeffares et al. 2015; Tusso et al. 2019), and (3) considering genomic variation of strain ancestry (Fig. 1; Supplemental Fig. 1). For 17 strains that correspond to 16 clades, data are publicly available (Tusso et al. 2019). Two of the 17 strains, previously referred to as EBC131_JB1171 and EBC132_JB1174, were found to belong to the same clade represented by the strain JB900, and were thus referred to as JB900_EBC131 and JB900_EBC132 in this study. Another set of single-molecule long-read sequencing data from 20 strains were generated in this study. These 20 strains include strains from 13 additional clades, two strains not belonging to the 57 clades, and five strains sharing cluster affiliation with five of the 17 previously published strains. With the inclusion of the already assembled reference genome, the final data set comprised 38 samples (Supplemental Table S1).
Genome assemblies
Additional to previously published genome assemblies, Pacific Biosciences (PacBio) long-read sequencing data were generated for the other 20 strains. We performed de novo assembly using two assemblers: Canu 1.8 and wtdbg 2.4 (Koren et al. 2017; Ruan and Li 2020). The parameters settings were “genomeSize = 12.5m” for Canu and “-x sq -L 3000 -g 12.5m” for wtdbg. For JB4 and JB1180, which were sequenced using the PacBio RS II platform, we used the option “-x rs” when running wtdbg. QUAST 5.0.2 was used to evaluate the assembly quality (Gurevich et al. 2013). For each strain, only the assembly with the superior quality was kept for further improvement. GCpp 0.0.1 was run to polish the assemblies using the Arrow algorithm and long reads. Subsequently, finisherSC 2.1 was applied to further improve the assembly (Lam et al. 2015), followed by another round of GCpp polishing. For JB1180, the assemblies generated were of poor quality, and we instead used SMRT analysis software 2.3.0. This pipeline resulted in general near-chromosome-level genomes with a median number of contigs of 8, average N50, total assembly length, and recovery of annotated genes of 2.48 Mb, 12.56 Mb (reference genome 12.55 Mb), and 97.0%, respectively (Supplemental Table S2).
Phylogenetic analyses and the inference of ancestry blocks
We performed two analyses using SNP variants: first, phylogenetic analyses using genome-wide SNP data (Supplemental Fig. 2), and second, a reported pipeline to identify the composition of Sp and Sk ancestral haplotype blocks along the genome (Supplemental Fig. 1; Tusso et al. 2019). For SNPs derived from short-read sequencing data, we used a publicly available data set in variant call format (VCF) (Jeffares et al. 2015). SNP variation of the de novo genomes (this study) was inferred via alignment to the reference genome (ASM294v264) (Wood et al. 2002) and subsequent characterization of variants with the package MUMmer 3.23 (function “show-snps”) (Kurtz et al. 2004). Repetitive sequences in the reference were identified with RepeatMasker 4.0.8 (Smit et al. 2013) and excluded for SNP variant calling. For the phylogenetic analyses, genome sequences were reconstructed by editing the reference genome with the SNP information for each sample using a customized Python script (Van Rossum and Drake 2009). The analysis was performed independently for each chromosome. Alignments of all samples, including short- and long-read data, were used to build a maximum-likelihood tree using RAxML 8.2.10-gcc-mpi (Stamatakis 2014) with default parameters, GTRGAMMAI approximation, final optimization with GTR + GAMMA + I, and 1000 bootstraps.
For inference of ancestral Sp and Sk haplotype blocks along the genome we followed Tusso et al. (2019). Strain identities and ancestral block distribution were consistent between short-read data (Jeffares et al. 2015) and long-read data (this study).
Genome annotation of transposable elements
For each de novo genome, we followed the CARP wrapper (Zeng et al. 2018) to identify and annotate repetitive sequences. Each family was annotated, and TEs were identified from other repeat sequences using a reference of known TE sequences for S. pombe and other fungi, obtained from the database Repbase (Bowen et al. 2003; Bao et al. 2015). Unidentified sequences were compared to protein sequences, transposable elements in other species, and retrovirus sequences using BLAST 2.7.1. Sequence references were obtained from the database hosted by the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov) using the following search terms: reverse transcriptase, transposon, repetitive element, RNA-directed DNA polymerase, pol protein, non-LTR retrotransposon, mobile element, retroelement, polyprotein, retrovirus, and polymerase (access date January 2020).
To reduce ascertainment bias introduced by comparison with published elements, we complemented this final set with novel repeat sequences obtained from LTR_Finder 1.0.7 (Xu and Wang 2007), RepeatMasker 4.0.8 (Smit et al. 2013), and EDTA 1.9.9 (Ou et al. 2019). Identified sequences were pooled and used as reference in a second round of the whole pipeline, aiming to extend the finding of repeats that may differ from the already known reference sequences. Annotations of combined sequences retrieved from all packages were merged based on overlapping coordinates using BEDTools (Quinlan and Hall 2010). Additional to LTR elements, in the reference genome EDTA inferred 16 novel sequences belonging to TIR and non-TIR elements, not reported for S. pombe previously. However, 15 of these overlapped with annotated functional genes suggesting false positive inference.
Different strains are known to have large structural variants, including inversions exceeding 1 Mb in length and interchromosomal translocations (Brown et al. 2011; Teresa Avelar et al. 2013; Zanders et al. 2014; Tusso et al. 2019). To establish synteny between samples, all annotated coordinates were translated to the reference genome. For this, we produced a liftover of all genomic positions between each de novo genome and the repeat-masked reference genome using flo (Pracana et al. 2017) and liftOver 2017-03-14 (Kent et al. 2002) requiring a minimum match of 0.7. Then we used customized Python and R scripts (R Core Team 2021) to identify translated coordinates of flanking sequences to breaking points for each TE element. Because TEs could occur in tandem, several sequences will share the same adjacent nonrepetitive sequences within the sequence space of the masked reference genome and were grouped as clusters. In the case of several TE sequences per cluster, the position of the breaking points in the original de novo genome were then shifted along the corresponding 3′ and/or 5′ axis until finding the first nonrepetitive base in the liftOver (Supplemental Fig. 15). As a result, TE sequences within a cluster will all share the same flanking insertion breakpoint coordinates of the cluster. The final list of transposable sequences, the position of the cluster they belong to, and their individual location and direction can be found in Supplemental Table S3.
We compared the list of TE elements extracted in previous work to validate our pipeline in two ways. First, we compared LTRs detected in the reference genome by Bowen et al. (2003) and our annotation of TE sequences in the reference genome. Second, we compared presence/absence scores from our data with scores based on paired-end short-read Illumina data from Jeffares et al. 2015 (Supplemental Table S4). In the first case, we converted the cosmid-based coordinates of the LTRs annotated by Bowen et al. (2003) to coordinates in the current version of the reference genome by BLAST and manual adjustment (Supplemental Table S5) and grouped sequences in clusters as we did for long-read assemblies. We counted the number of sequences per cluster. For the second comparison in other samples and Jeffares et al. (2015) data set, we restricted this comparison to samples showing consistent strain ID between short- and long-read data. Differences observed between short- and long-read data (Supplemental Fig. 7) were compared with differences observed between clonal strains using only de novo genomes from long reads (Supplemental Fig. 6).
Phylogenetic analyses
We used a customized Python script to extract TE sequences of minimum length 100 bp from de novo genomes. Consensus sequences from all samples produced were then used as a reference for each query sequence. Query sequences were differentiated between solo LTRs, fragmented TEs with one or no flanking LTR sequence, and full-length elements longer than 4.5 kb, containing the full polyprotein sequence, and both flanking LTRs. Two alignments were produced using MAFFT 7.407 (Katoh and Standley 2013): one for full-length TE, and another one for LTR sequences including solo LTRs and all flanking LTRs associated to full-length TE elements. These alignments were used to produce a maximum-likelihood tree with IQ-TREE 1.6.10-omp-mpi (Nguyen et al. 2015) using the incorporated model prediction with ModelFinder (Kalyaanamoorthy et al. 2017) and 1000 ultrafast bootstrap (UFBoot) (Minh et al. 2013). Because different LTR families have been previously identified in the reference genome (Bowen et al. 2003), we used genomic coordinates of known LTR sequences to place solo-LTR families for other strains in the phylogeny.
Recombinant TE haplotypes
To identify potential recombinant haplotypes, the alignment of full-length elements was divided into windows of 30 bp. Other window sizes like 20 and 10 bp were also tested with similar results. For each window, pairwise comparisons between sequences were performed. If sequences within windows differed by more than 2 bp, they were classified as different. When a sequence was equidistant to two already identified haplotypes, it was grouped to the first comparison. However, these cases were rather rare and do not have major impact on the general results. Then, to identify whole sequence haplotypes (all windows), pairwise comparisons between whole sequences was performed (Supplemental Fig. 16). Haplotypes were scored as identical if they contained the same succession of identical 30-bp windows. We allowed one window to be different between sequences. Haplotypes were filtered, considering only haplotypes with at least 50% of the entire sequence and with at least five sequences (either paralogs or orthologs). This reduced the data set to 11 common haplotypes.
A similar analysis was performed for solo LTRs and flanking LTRs. LTR fragments were short (∼350 bp or ∼1060 bp in the alignment including insertions and deletions), precluding the windows-based approach. Instead, we used the full alignments focusing on the most common LTR families (α and β). We identified potential recombinant haplotypes by looking first for diagnostic variants of each family. For this, diagnostic variants constituted those near-fixed between families (>0.8 frequency in one family; <0.2 in the other) (Supplemental Fig. 9). These diagnostic variants were compared with sequences in the Sub-α/ζ group and used to identify recombinant haplotypes. Sequences were grouped into haplotypes on the basis of pairwise comparisons of diagnostic variants. Two sequences were considered from a different haplotype if they differed in up to two diagnostic variants.
Testing the genomic shock hypothesis
To test the hypothesis of TE reactivation in admixed genomes, we performed linear models to assess the relationship between admixture proportions (from pure Sp or Sk to 0.5 admixture) as explanatory variable and number of clusters containing at least one full-length TE as response. The normality assumption of the residuals held as assessed by the R package olsrr 0.5.3 setting a maximum P-value threshold of 0.05 (https://olsrr.rsquaredacademy.com). Additionally, we restricted the analyses to the presumably more recent insertions including only those clusters consisting of a single full-length element in a single sample (Supplemental Fig. 13). Analyses were performed including both TF1 and TF2 haplotypes, as well as by haplotype independently.
Population genetic analyses
The frequency distribution of syntenic TE sequences among nonclonal strains was summarized in one- and two-dimensional site frequency spectra (SFS). Clusters sharing the same start and end coordinates, allowing for a 100-bp error margin on each side of the repetitive cluster, were defined as syntenic loci. Error margins of 150 and 200 bp yielded similar results. The error margin was necessary to account for variation introduced by the lift over. Spacing between clusters exceeded a minimum of 500 bp in all cases to guard against false positive inference of synteny of adjacent clusters (Supplemental Fig. 17). Translating coordinates from de novo genomes onto the reference genome could lead to a potential underestimation in the number of clusters. For cases in which flanking sequences are absent in the reference genome or overlap with longer stretches of repetitive sequences, the range of a cluster will be expanded until the next nonrepetitive anchor in the reference genome is found. As a consequence, nonorthologous neighboring sequences of multiple query strains may be collapsed into a single genomic cluster spanning the “difficult to align region” of the reference genome. However, even if this bias exists, it would be rare and will not affect the main conclusions because (1) TE insertions were distributed along the whole genome without gravitating toward repetitive regions (Supplemental Fig. 18), (2) Tf1 insertions have been shown to be preferentially integrated into nucleosome-free promoters of genes (Bowen et al. 2003; Guo and Levin 2010), and (3) the vast majority of clusters contained a single sequence per sample (Supplemental Fig. 3C,D). Presence/absence of orthologs clusters was then scored as allelic state of the locus. Allele frequencies were summarized for all clusters by the derived state and summarized in an unfolded one-dimensional site frequency spectrum (Fig. 4A). Only nonclonal strains were included to produce the SFS.
In addition, we obtained a two-dimensional site frequency spectrum (2dSFS) considering allele frequency sharing between syntenic clusters surrounded by either Sp and Sk ancestry. Ancestry was inferred by classifying SNP information in the flanking sequences of insertion breakpoints for each cluster and sample (Tusso et al. 2019). For each locus, allele frequencies were separately estimated for each ancestral background resulting in an unfolded 2dSFS. In addition to simple presence/absence scoring, alleles were scored according to the number of sequences within cluster, their family, and direction of insertion, yielding comparable results (Fig. 4B; Supplemental Fig. 19; Supplemental Table S6). In both cases, only variants with at least four samples per genetic background were considered.
One- and two-dimensional site frequency spectra were calculated for all TE sequences (Fig. 4B) and separately for full-length elements (Fig. 4C). A two-dimensional site frequency spectrum was likewise produced from noncoding genome-wide SNPs using the same set of nonclonal strains and removing repetitive regions. This resulted in a final data set of 209,690 variant sites. Allele frequencies to produce a folded two-dimensional site frequency spectrum were calculated using the R package SNPRelate 1.24.0 (Zheng et al. 2012). In the absence of an appropriate outgroup, SNP variants cannot be polarized into an ancestral and derived state. To allow direct comparisons of SFS between TEs and SNPs, the two-dimensional site frequency spectrum of TEs was also folded (Supplemental Fig. 14).
Sequence divergence of LTRs
To assess the levels of sequence divergence of TEs, we calculated divergence between all solo LTR and flanking LTR sequences as a function of the genomic background (Sp or Sk) they are embedded in. We divided sequences by cluster and family according to phylogenetic reconstruction (Fig. 2A) and used the R package ape 5.4-1 (Paradis and Schliep 2019) to calculate pairwise Kimura's two-parameter distance (Kimura 1980) within and between ancestral backgrounds. Additionally, we measured divergence between the 5′ and 3′ flanking LTR sequences of each full-length TE element as a proxy of the age of its individual insertion (Supplemental Fig. 12).
Data access
The PacBio sequencing data and genome assemblies generated in this study have been submitted to the CNGB Sequence Archive (CNSA) (https://db.cngb.org/cnsa) under Project ID CNP0001878. All code used for the analyses are available in Supplemental Code and at Zenodo (https://zenodo.org/record/5747176).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank S. Lorena Ament-Velásquez, Fidel Botero-Castro, Bart P.S. Nieuwenhuis, Claire Peart, Ricardo Pereira, Alexander Suh, Vera Warmuth, Matthias Weissensteiner, and members of the Wolf and Du laboratories for providing intellectual input on the various analyses and comments on the manuscript. Funding was provided by LMU Munich (J.B.W.W.). The computational infrastructure was provided by the Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) Sequencing Cluster and Storage project funded by the Knut and Alice Wallenberg Foundation and the Swedish National Infrastructure for Computing.
Author contributions: S.T., L.-L.D., and J.B.W.W. conceived the study. All analyses were performed by S.T. and F.S. with contributions from Y.L. in genome annotation of transposable elements analyses. S.T. and J.B.W.W. wrote the manuscript with input from F.S., Y.L., and L.-L.D.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.276056.121.
- Received July 29, 2021.
- Accepted December 9, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








