
Characteristics, origin, and potential for cancer diagnostics of ultrashort plasma cell-free DNA
- Irena Hudecova1,2,9,
- Christopher G. Smith1,2,9,
- Robert Hänsel-Hertsch1,3,4,5,9,
- Chandra S. Chilamakuri1,
- James A. Morris1,2,
- Aadhitthya Vijayaraghavan1,2,
- Katrin Heider1,2,
- Dineika Chandrananda1,2,11,
- Wendy N. Cooper1,2,
- Davina Gale1,2,
- Javier Garcia-Corbacho6,
- Simon Pacey2,7,
- Richard D. Baird2,7,
- Nitzan Rosenfeld1,2,10 and
- Florent Mouliere1,2,8,10
- 1Cancer Research UK Cambridge Institute, University of Cambridge, Li Ka Shing Centre, Cambridge CB2 0RE, United Kingdom;
- 2Cancer Research UK Cambridge Centre, Cancer Research UK Cambridge Institute, Li Ka Shing Centre, Cambridge CB2 0RE, United Kingdom;
- 3Center for Molecular Medicine Cologne CMMC, University of Cologne, 50931 Cologne, Germany;
- 4Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases (CECAD), University of Cologne and University Hospital Cologne, 50931 Cologne, Germany;
- 5Institute of Human Genetics, University Hospital Cologne, 50931 Cologne, Germany;
- 6Clinical Trials Unit, Clinic Institute of Hematological and Oncological Diseases, Hospital Clinic, 170 08036 Barcelona, Spain;
- 7Department of Oncology, University of Cambridge, Cambridge CB2 0QQ, United Kingdom;
- 8Department of Pathology, Amsterdam UMC, Vrije Universiteit Amsterdam, Cancer Centre Amsterdam, 1081 HV Amsterdam, The Netherlands
-
↵9 These authors contributed equally to this work.
Abstract
Current evidence suggests that plasma cell-free DNA (cfDNA) is fragmented around a mode of 166 bp. Data supporting this view has been mainly acquired through the analysis of double-stranded cfDNA. The characteristics and diagnostic potential of single-stranded and damaged double-stranded cfDNA in healthy individuals and cancer patients remain unclear. Here, through a combination of high-affinity magnetic bead–based DNA extraction and single-stranded DNA sequencing library preparation (MB-ssDNA), we report the discovery of a large proportion of cfDNA fragments centered at ∼50 bp. We show that these “ultrashort” cfDNA fragments have a greater relative abundance in plasma of healthy individuals (median = 19.1% of all sequenced cfDNA fragments, n = 28) than in plasma of patients with cancer (median = 14.2%, n = 21, P < 0.0001). The ultrashort cfDNA fragments map to accessible chromatin regions of blood cells, particularly in promoter regions with the potential to adopt G-quadruplex (G4) DNA secondary structures. G4-positive promoter chromatin accessibility is significantly enriched in ultrashort plasma cfDNA fragments from healthy individuals relative to patients with cancers (P < 0.0001), in whom G4-cfDNA enrichment is inversely associated with copy number aberration-inferred tumor fractions. Our findings redraw the landscape of cfDNA fragmentation by identifying and characterizing a novel population of ultrashort plasma cfDNA fragments. Sequencing of MB-ssDNA libraries could facilitate the characterization of gene regulatory regions and DNA secondary structures via liquid biopsy. Our data underline the diagnostic potential of ultrashort cfDNA through classification for cancer patients.
Fragments of DNA are released into the blood circulation following cell death and by active secretion (Stroun et al. 2000) while maintaining genetic and epigenetic signatures of the cell of origin (Heitzer et al. 2020). The fragmentation of cell-free DNA (cfDNA) in blood plasma can reflect its tissue of origin and mechanisms of release (Ivanov et al. 2015; Snyder et al. 2016; Cristiano et al. 2019; van der Pol and Mouliere 2019). In plasma of healthy individuals, cfDNA is fragmented around a length of ∼166 bp, corresponding to DNA wrapped around a nucleosome plus linker (Lo et al. 2010). These fragmentation patterns can be altered in the plasma of cancer patients (Mouliere et al. 2011; Jiang and Lo 2016; Underhill et al. 2016), possibly because of the biological mechanisms at play in cancer cells. For example, modifications in chromatin structure (van der Pol and Mouliere 2019; Heitzer et al. 2020) may contribute to the observation that tumor-derived cfDNA (circulating tumor DNA [ctDNA]) is in general shorter than non-tumor DNA, with a mode of distribution ∼145 bp versus ∼166 bp, respectively (Mouliere et al. 2011, 2014; Jiang et al. 2015; Jiang and Lo 2016; Underhill et al. 2016). Methods that can better recover shorter cfDNA molecules may increase the recovery of ctDNA molecules and potentially enrich for tumor signal (Thierry et al. 2016; Wan et al. 2017; Mouliere et al. 2018; Jiang et al. 2020).
The apparent size of plasma cfDNA can be affected by the preanalytical methods used, such as the blood processing protocol (Wong et al. 2016; Rikkert et al. 2018), the DNA extraction chemistry (Jorgez et al. 2006; Pérez-Barrios et al. 2016; Kloten et al. 2017; Sorber et al. 2017; Markus et al. 2018; Lampignano et al. 2020), and type of library preparation (Barlow et al. 2016). cfDNA-based approaches have focused on analysis of double-stranded DNA (dsDNA) (Jiang et al. 2015; Underhill et al. 2016; Mouliere et al. 2018), but alternative methods have been developed, including protocols for the recovery of single-stranded DNA (ssDNA) (Meyer et al. 2012; Gansauge and Meyer 2013; Gansauge et al. 2017). Unlike dsDNA library protocols that only recover intact dsDNA molecules, ssDNA-based protocols enable the recovery of ssDNA and damaged dsDNA, alongside intact dsDNA molecules (Gansauge and Meyer 2013). In plasma, previous analyses using ssDNA library protocols revealed a slight enrichment in cfDNA molecules shorter than 100 bp relative to dsDNA-based methods (Burnham et al. 2016; Snyder et al. 2016; Vong et al. 2017). However, studies assessing ctDNA enrichment with ssDNA library approaches yielded inconsistent findings, with some showing evidence of a slight increase, and others showing none (Moser et al. 2017; Liu et al. 2019; Zhu et al. 2020). The importance of recovering short plasma cfDNA in understanding cancer biology has been highlighted in previous studies demonstrating that cfDNA fragments <100 bp in length may be associated with regulatory mechanisms (Snyder et al. 2016; Ulz et al. 2016, 2019). In addition, exploring alternative structural modifications contained in ssDNA or damaged dsDNA (Snyder et al. 2016; Hänsel-Hertsch et al. 2017) could improve our understanding of cfDNA biology offering further opportunities to develop novel diagnostic strategies.
Here, we shed light on the genomic profiles, structure, and diagnostic potential of a novel population of ultrashort (US) plasma cfDNA recovered through a combination of magnetic bead–based DNA extraction and ssDNA-based library preparation (MB-ssDNA). Through shallow whole-genome sequencing (sWGS, <0.5-fold coverage) and the analysis of somatic copy number aberrations (SCNA), we determined the landscape of genetic alterations in US cfDNA fragments ∼50 bp in length, abundantly present in the plasma of healthy individuals and cancer patients. In addition, we studied their potential link with regulatory regions by investigating genome-wide coverage patterns at transcription start sites (TSS). Furthermore, we explored whether US cfDNA fragments map to regions associated with the potential to form G-quadruplex (G4) DNA secondary structures, and we estimated their quantities in the plasma of healthy individuals and cancer patients.
Results
A high-affinity magnetic bead–based DNA extraction and ssDNA library preparation protocol (MB-ssDNA) identifies a novel population of ultrashort cfDNA fragments
To determine whether a bias in the recovery of US cfDNA fragments exists, we first characterized the plasma cfDNA size distributions inferred from paired-end sWGS in ssDNA and dsDNA libraries from healthy individuals (n = 82 samples) (Fig. 1A; Supplemental Table S1). Two DNA extraction methods were applied (Methods); the first extraction, herein referred to as the “SC” protocol, used silica gel membrane-based columns to capture DNA in the presence of chaotropic salts. The second approach, herein referred to as the “MB” protocol, used high-affinity magnetic beads. In total, four different protocols were used to analyze plasma samples from healthy individuals: SC-dsDNA (n = 14), SC-ssDNA (n = 10), MB-dsDNA (n = 30), and MB-ssDNA (n = 28) (Supplemental Table S1).
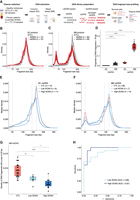
Differences in the recovery of ultrashort cfDNA fragments using different DNA extraction and DNA library preparation protocols. (A) Plasma samples from healthy individuals and cancer patients were processed by column (SC)-based and magnetic bead (MB)-based DNA extraction methods, followed by dsDNA and ssDNA library protocols. The ssDNA library protocol recovers ssDNA and damaged dsDNA molecules, along with intact dsDNA, whereas the dsDNA library protocol recovers only intact dsDNA molecules. Paired-end sWGS data were generated from those libraries and used to determine DNA fragment size distributions. (B,C) cfDNA fragment size distributions of plasma ssDNA and dsDNA libraries from healthy individuals processed by SC (B) and MB (C) protocols. The vertical dashed line indicates a DNA fragment size of 70 bp. (D) Proportion of fragments shorter than 70 bp in plasma ssDNA and dsDNA libraries from healthy individuals. (E,F) Median fragment size distributions of cfDNA from plasma samples of patients with advanced cancers, divided into two groups: low SCNA (t-MAD < 0.019) and high SCNA (t-MAD ≥ 0.019), and analyzed by SC-ssDNA (E) and MB-ssDNA (F). Vertical dashed lines correspond to DNA fragment sizes of 50, 145, and 166 bp. (G) Density of fragments found at 50 bp in plasma samples from healthy individuals and cancer patients with low and high SCNA, data generated using the MB-ssDNA protocol. (H) ROC curve analysis of fragment density at 50 bp, generated using the MB-ssDNA protocol, for discriminating cancer patients from the healthy individuals. Wilcoxon rank-sum test: (*) P < 0.05; (**) P < 0.01; (****) P < 0.0001.
Following dsDNA library preparation of cfDNA extracted by each method (i.e., SC-dsDNA and MB-dsDNA libraries) (Fig. 1B,C), we observed a consistent fragmentation pattern that mirrors the profile widely reported for cfDNA (Jiang et al. 2015; Underhill et al. 2016; Mouliere et al. 2018). Conversely, application of a ssDNA library protocol based on template-switching chemistry (Methods) revealed markedly different fragment size profiles depending on the DNA extraction method used (Fig. 1B,C; Supplemental Fig. S1). In agreement with previous studies (Burnham et al. 2016; Moser et al. 2017; Vong et al. 2017; Sanchez et al. 2018), plasma DNA extracted by SC followed by ssDNA library protocol (SC-ssDNA) showed a broadly similar fragmentation pattern, but with an enrichment in the recovery of US cfDNA fragments (<70 bp) relative to the dsDNA library protocol (SC-dsDNA) (P < 0.0001, Wilcoxon rank-sum test) (Fig. 1B,D). A more profound effect was observed in cfDNA extracted by MB-ssDNA protocol: this showed a bimodal distribution of DNA fragment sizes, with a population of DNA molecules centered at ∼50 bp, in addition to the mode previously observed at 166 bp (Fig. 1C). To confirm these observations, we tested an alternative ssDNA library approach based on the ligation of biotinylated adapters (Gansauge and Meyer 2013) in additional plasma cfDNA samples extracted by MB. This revealed a similar bimodal distribution (Supplemental Fig. S2A,B). Of note, data processing to exclude potential sequence artifacts associated with poly(A/T) genomic regions (Supplemental Fig. S3A–C; Supplemental Methods; Vardi et al. 2017) preserved the overall trend in DNA fragmentation patterns in MB-ssDNA libraries and further confirmed that US cfDNA fragments are not a technical artifact.
To quantify the relative enrichment of US cfDNA fragments in samples from healthy individuals, we estimated the proportion of molecules <70 bp (Fig. 1D). The US cfDNA fragments represented a median of 19.1% (interquartile range, IQR = 16.8%–22.0%) of all DNA fragments in plasma samples undergoing MB-ssDNA, whereas a median of only 3.4% (IQR = 2.6%–3.6%) of fragments recovered by SC-ssDNA were shorter than 70 bp (P < 0.0001, Wilcoxon rank-sum test) (Fig. 1D). In dsDNA libraries, the proportion of US cfDNA fragments was much lower (median = 0.7%, IQR = 0.3%–2.4%) than in ssDNA libraries (P < 0.0001, Wilcoxon rank-sum test), although there was still a significant difference between SC-dsDNA (median = 0.3%, IQR = 0.2%–0.3%) and MB-dsDNA protocols (median = 2.0%, IQR = 0.7%–2.6%; P < 0.0001, Wilcoxon rank-sum test) (Fig. 1D). Thus, by using MB-ssDNA we identified significantly more US cfDNA fragments in plasma than previously described.
The relative abundance of ultrashort cfDNA fragments recovered by MB-ssDNA is greater in plasma from healthy individuals than in plasma from cancer patients
We then analyzed cfDNA fragmentation patterns in plasma from patients with advanced cancers (n = 73 samples from 17 patients with seven different cancer types, all patients having at least one metastatic site) collected before initiation of treatment (Supplemental Table S1; Supplemental Fig. S4A,B), using the four protocols described above: SC-dsDNA (n = 16), SC-ssDNA (n = 17), MB-dsDNA (n = 19), and MB-ssDNA (n = 21) (Supplemental Table S1). Plasma samples from 12 patients (Pt01-Pt12) were processed by all four protocols for direct comparison (Supplemental Fig. S5). We stratified the patient samples into two groups based on their ctDNA fractions in MB-dsDNA libraries, quantified by a genome-wide score termed t-MAD (Methods). t-MAD estimates ctDNA fractions in plasma samples from cancers that show copy number alterations, by reflecting the extent of SCNAs across the genome from paired-end sWGS (Mouliere et al. 2018). Based on sequencing of plasma samples from healthy individuals, we defined a t-MAD threshold (0.019) and classified samples with t-MAD values below this threshold as “low SCNA”; samples with t-MAD values above this threshold were referred to as “high SCNA” (Methods).
The median distribution of fragment sizes in SC-ssDNA libraries for low SCNA samples was similar to that of samples from healthy individuals (P = 0.42, D = 0.076, Kolmogorov–Smirnov test), with no apparent population of US fragments centered at the 50-bp peak (Fig. 1E; Supplemental Fig. S4A,C). Conversely, in MB-ssDNA libraries, we observed significant differences between samples from cancer patients and healthy individuals in US fragments (<70 bp) (Fig. 1F; Supplemental Fig. S4B,D). We determined the proportion of fragments <70 bp and observed a significant decrease in low SCNA (median = 13.6%, IQR = 12.7%–15.8%) and high SCNA samples (median = 14.7%, IQR = 10.6%–17.5%) compared to samples from healthy individuals (median = 19.1%, IQR = 16.8%–22.0%; P < 0.01 and P < 0.001, respectively, Wilcoxon rank-sum test) (Supplemental Fig. S4D). The density of 50-bp fragments showed large and significant differences between samples from the two cancer patient groups versus those from healthy individuals (Fig. 1G). Analysis of this density, by receiver operating characteristics (ROC) curve, resulted in an area under the curve (AUC) of 0.97 and 0.89 for high SCNA and low SCNA samples, respectively (Fig. 1H). Taken together, these results suggest that the DNA fragmentation patterns recovered by MB-ssDNA are inversely correlated with ctDNA fractions, as inferred from SCNA analysis (Fig. 1G).
Ultrashort cfDNA fragments contain, but are not enriched for, tumor-derived signal when compared against other fragment size ranges
We next aimed to determine whether a particular combination of protocols could enrich for ctDNA, either in the overall representation of cfDNA molecules (Fig. 2A) or at selected fragment size ranges (Fig. 2B). First, to determine the ctDNA fractions across the entire distribution of fragment lengths (Supplemental Fig. S5), we assessed SCNA profiles and estimated t-MAD in samples from 12 patients, processed using different protocols (Supplemental Table S1). Overall, the estimated ctDNA fractions in high SCNA samples (n = 7) were not significantly different between ssDNA and dsDNA libraries processed by either DNA extraction method (SC-dsDNA vs. MB-dsDNA, P = 0.8; SC-dsDNA vs. SC-ssDNA, P = 0.8; SC-dsDNA vs. MB-ssDNA, P = 0.67; MB-dsDNA vs. SC-ssDNA, P = 0.8; MB-dsDNA vs. MB-ssDNA, P = 0.4; SC-ssDNA vs. MB-ssDNA, P = 0.67, Wilcoxon signed-rank test) (Fig. 2A).
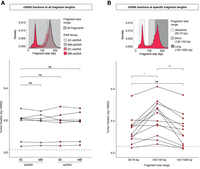
Plasma ctDNA fractions at all and specific DNA fragment lengths. (A) Schematic of the selection of all DNA fragment size ranges and comparison of t-MAD scores between different protocols. Tumor fractions as inferred by sWGS and t-MAD analysis, of samples from patients with high SCNA levels (seven patients and 28 plasma samples). The dashed line indicates a t-MAD threshold of 0.019 (inferred from a cohort of healthy individuals, sequence data down-sampled to 5 million reads). (B) Schematic of the selection of three different fragment size ranges. The t-MAD scores for plasma DNA from 12 cancer patients after in silico size selection of fragments between 30 – 70, 100 – 150, and 150 – 1000 bp in length. The t-MAD scores were plotted separately for different DNA size fragment bins. The dashed line indicates a t-MAD threshold of 0.0416 (inferred from a cohort of healthy individuals, sequence data down-sampled to 1.25 million reads). Wilcoxon signed-rank paired test: (*) P < 0.05; (**) P < 0.01; (ns) nonsignificant.
Then, we determined whether the population of US fragments discovered through MB-ssDNA contained and was relatively enriched in tumor-derived DNA molecules. We performed in silico size selection for cfDNA fragments in three size ranges: 30 – 70 bp, US cfDNA fragments; 100 – 150 bp, short fragments; and >150 bp, long fragments including mono- and dinucleosomal DNA (Fig. 2B). Using t-MAD, we observed an enrichment in ctDNA fractions within short fragments (median t-MAD = 0.227) in comparison to long (median t-MAD = 0.130) and US cfDNA fragments (median t-MAD = 0.139; P < 0.01 and P < 0.05, respectively, Wilcoxon signed-rank test) (Fig. 2B). The ctDNA fractions were not significantly different between the long and US cfDNA fragment groups (P = 0.46, Wilcoxon signed-rank test) (Fig. 2B). To confirm that US fragments contain ctDNA signal, we additionally performed in vitro size selection of select samples and showed that ctDNA fractions of US cfDNA were similar to those observed after in silico selection (Supplemental Fig. S6). These results established that the population of US cfDNA fragments, recovered by MB-ssDNA, carries tumor-specific signal (Supplemental Fig. S6) comparable in relative abundance to the mono- and dinucleosomal cfDNA fragments that make up the majority of cfDNA fragments, but lower compared to the ctDNA fractions in fragments of length 100 – 150 bp (Fig. 2B).
We wondered whether the size distribution of cfDNA would change if we exclusively recovered ssDNA molecules. We designed an experiment in which we omitted the initial denaturation step of the ssDNA-based protocol (MB-ssDNA). In these conditions, only ssDNA molecules would be expected to be ligated to adapters and therefore generate sequencing reads. We assessed the impact on DNA fragmentation profiles in two samples from healthy individuals (Supplemental Fig. S7A) and two samples from cancer patients (Supplemental Fig. S7B). In parallel, we conducted control experiments in which plasma DNA from the same individuals were processed using the unaltered protocol. In MB-ssDNA libraries produced without the denaturation step, the relative proportion of cfDNA fragments found at 166 bp was reduced, and a relative increase in the proportion of US fragments was observed (on average 2.2-times higher) (Supplemental Fig. S7A,B). The ctDNA fractions determined by t-MAD did not show substantial differences between the denatured and non-denatured conditions for selected size ranges after in silico size selection (Supplemental Fig. S7C). Although these findings are based on data from a limited number of samples, they suggest that in addition to intact or damaged dsDNA, the US fragments are enriched in ssDNA molecules, and that these cfDNA molecules also contain tumor-derived signal.
Ultrashort cfDNA fragments recovered by MB-ssDNA reveal distinct sequence coverage profiles near transcription start sites
Differences in the lengths of plasma cfDNA fragments can reflect changes in the chromatin structure of the genome (Snyder et al. 2016; Cristiano et al. 2019). Fragments shorter than 100 bp can indicate the presence of genomic regions that might be occupied by regulatory proteins, revealing the transcriptional activity of cells contributing to the cfDNA pool in plasma (Snyder et al. 2016). These studies were restricted by the small amounts of US cfDNA fragments recovered by standard column-based DNA extractions (Snyder et al. 2016) and thus we analyzed coverage patterns at TSS with MB-ssDNA (Fig. 3A).
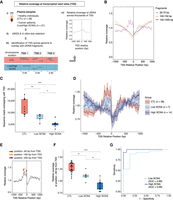
Assessment of plasma cfDNA sequence coverage near TSS in MB-ssDNA libraries. (A) Schematics of TSS coverage analysis in plasma cfDNA samples. (B) The relative coverage near TSS in MB-ssDNA libraries derived from pooled sWGS data of 10 healthy individuals assessed in different fragment size ranges. (C) The number of US cfDNA fragments overlapping with TSS regions in individual MB-ssDNA libraries derived from healthy individuals and cancer patients. (D) The relative coverage of US cfDNA fragments near TSS in samples from healthy individuals and cancer patients. The mean and standard deviation of the data are shown. (E) Schematic of the assessment of the density of US cfDNA fragments at specific positions relative to TSS. (F) The relative coverage of US cfDNA fragments at the position 40 bp upstream of TSS. (G) ROC curve analysis of the relative density at the “−40 bp” position for discriminating cancer patients from the healthy individuals. Wilcoxon rank-sum test: (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001.
Following in silico size selection of fragments corresponding to US, short, and long groups, we assessed sequence coverage near TSS regions (Methods) in pooled sWGS data from healthy individuals (Fig. 3B). In MB-ssDNA libraries, the relative coverage upstream of TSS showed a decreasing trend for fragments >100 bp in length (Fig. 3B). On the other hand, the relative coverage of US fragments showed the opposite pattern, with a sharp increase immediately upstream of the TSS (Fig. 3B). A similar trend was observed in pooled sWGS MB-dsDNA data from healthy individuals (Supplemental Fig. S8A). Following down-sampling of MB-ssDNA data to the same number of sequencing reads, the number of US fragments overlapping with unique TSS was significantly different between samples from healthy individuals (median = 11,981; IQR = 9605–15,661) and low SCNA samples (median = 8554; IQR = 7385 – 9080) and high SCNA samples (median = 5112; IQR = 4681–5498; P < 0.01 and P < 0.001, respectively, Wilcoxon rank-sum test) (Fig. 3C).
Given that cancer is associated with changes in gene expression programs (Ulz et al. 2016), we wondered whether coverage patterns of US fragments at TSS could reflect differences in the underlying pathological processes in cancer patients. We analyzed sWGS MB-ssDNA data derived from individual plasma samples of 17 cancer patients (n = 21 samples) and 22 healthy individuals (n = 28 samples) to assess TSS coverage patterns (Fig. 3D; Supplemental Fig. S8B). Although the trend in coverage profiles showed a sharp increase in the proportion of US cfDNA fragments upstream of TSS in healthy individuals, the intensity of TSS signal in cancer patients appeared to decrease with increasing extent of SCNA (Fig. 3D; Supplemental Fig. S8B). To control for potential batch effects, we selected a subset of plasma samples from healthy individuals (n = 18) and cancer patients (n = 6) processed in the same DNA extraction and DNA library preparation batch, and we observed the same patterns (Supplemental Fig. S8C).
Among the data derived from samples from healthy individuals, we observed three prominent peaks in coverage at positions 40, 160, and 250 bp upstream of TSS (Fig. 3E). We compared the relative coverage at these positions in samples from healthy individuals and low SCNA and high SCNA samples from cancer patients. The relative coverage at −40 bp from TSS was significantly higher in samples from healthy individuals compared to low SCNA and high SCNA samples (P < 0.01 and P < 0.0001, respectively, Wilcoxon rank-sum test) (Fig. 3F). The same trend was observed at positions −160 bp and −250 bp (Supplemental Fig. S9A,B). Using the −40 bp position as a parameter in ROC analysis, we observed a clear separation between the samples from healthy individuals and cancer patients, with an AUC in high SCNA and low SCNA group of 0.99 and 0.89, respectively (Fig. 3G). A clear separation was also observed when using the proportion of fragments at position −160 bp and −250 bp, with comparable AUCs in high and low SCNA groups (Supplemental Fig. S9C,D). We also confirmed the separability of the samples from healthy individuals and cancer patients by combining data from all three parameters together and using it as input for unsupervised clustering (Supplemental Fig. S9E).
To determine whether US fragments were associated with specific TSS regions and could thus inform as to differences in transcriptional programs between healthy individuals and cancer patients, we compared the relative coverage at different TSS, either associated with accessible chromatin regions of hematopoietic cells or accessible pan-cancer chromatin regions (each TSS group comprised approximately 3000 randomly selected genomic regions covering about 900 genes) (Methods). Because of low sequencing depth at a sample level, sequencing data from the three categories were merged separately for healthy individuals, low SCNA and high SCNA samples, and down-sampled to ∼5 million reads. The previously observed patterns at positions −40, −160, and −250 bp (as shown in Fig. 3D, including 90,273 TSS regions) were less evident in the subselected TSS regions, likely as a result of the lower sequencing depth within a smaller genomic region (∼30× fewer TSS regions) (Supplemental Fig. S10A). In line with our previous findings (Fig. 3D), we noted an overall decrease in relative coverage at position −40 bp in samples with high SCNA as compared to samples from healthy individuals and low SCNA samples (Supplemental Fig. S10A). Despite the limited resolution, the relative coverage at position −40 bp in TSS associated with accessible chromatin regions of hematopoietic cells (erythrocyte progenitors or lymphocytes) appeared to be higher in comparison with TSS regions associated with accessible pan-cancer chromatin regions in all three patient/control groups (Supplemental Fig. S10B).
To further explore differences in transcriptional programs between cancer patients and healthy individuals, we determined US cfDNA enrichment by comparing relative coverage of fragments in the selected TSS regions against relative coverage at random regions of the genome (Supplemental Fig. S11; Methods). The US cfDNA of healthy individuals was significantly more enriched in comparison to cancer patients with low SCNA and high SCNA content within each TSS category (P < 0.01 and P < 0.001, respectively, Wilcoxon rank-sum test) (Supplemental Fig. S11A). Of note, US cfDNA of healthy individuals was significantly more enriched in TSS associated with accessible chromatin regions of hematopoietic cells as compared to TSS associated with accessible pan-cancer chromatin regions (both P < 0.001, Wilcoxon signed-rank test) (Supplemental Fig. S11B). In contrast, we observed a less pronounced difference in US cfDNA enrichment between hematopoietic cells and cancer-related TSS in samples with low and high SCNA content (Supplemental Fig. S11B): in samples with high SCNA, we observed a lower fold difference in enrichment of US cfDNA in TSS associated with erythrocyte progenitors (Supplemental Fig. S11C) and lymphocytes (Supplemental Fig. S11D) as compared to cancer-related TSS regions. These findings indicate that the relative contribution of US cfDNA originating from hematopoietic cells in cancer patients appears to be reduced by the increased contribution of US cfDNA from cancer cells (Supplemental Fig. S11C,D).
Ultrashort cfDNA fragments recovered by MB-ssDNA map to accessible promoter regions with demonstrated potential to adopt G-quadruplex structures
Recent studies highlighted that the composition and topology of cfDNA could be more complex than initially thought (Kumar et al. 2017; Zhu et al. 2017; Sin et al. 2020). To assess the sequence context of cfDNA fragments recovered by MB-ssDNA, we analyzed the nucleotide composition of US, short, and long cfDNA (Supplemental Fig. S12A). We observed a higher GC content in US fragments in samples from healthy individuals (median = 44.3%; IQR = 43.1%–45.5%) compared to low SCNA (median = 41.8%; IQR = 40.6%–42.0%) and high SCNA samples (median = 39.6%; IQR = 38.8%–40.3%; P < 0.01 and P < 0.0001, respectively, Wilcoxon rank-sum test) (Supplemental Fig. S12A). Although fragments in the lengths of 100–150 bp and 150–1000 bp in MB-ssDNA libraries were similar in GC content (Supplemental Fig. S12B), we still observed significant differences between healthy individuals and low SCNA and high SCNA samples (both P < 0.05, Wilcoxon rank-sum test) (Supplemental Fig. S12A).
We hypothesized that the observed differences in GC content between healthy individuals and cancer patients in MB-ssDNA libraries could reflect the existence of GC-rich secondary structures in cfDNA. Single-stranded guanine-rich DNA sequences can fold into four-stranded DNA structures called G-quadruplexes (G4s) (Hänsel-Hertsch et al. 2017; Varshney et al. 2020), observed frequently in accessible chromatin of highly expressed promoter regions of amplified genes of cancer cells (Hänsel-Hertsch et al. 2016) or tumor tissue (Hänsel-Hertsch et al. 2020). We searched for evidence of enrichment for G4 sequences in cfDNA (G4-cfDNA enrichment) and compared their levels in healthy individuals and cancer patients (Fig. 4A). We determined G4-cfDNA enrichment by comparing the relative coverage of cfDNA fragments at predicted G4 loci against the relative coverage at random regions of the genome. In particular, we investigated the relative coverage of plasma cfDNA in accessible blood-related chromatin regions within gene promoters that, importantly, lack or contain predicted quadruplex sequences (PQS), which adopted G4 structures (observed quadruplex sequences [OQS]) in G4-promoting conditions (Chambers et al. 2015). Predicted quadruplex sequence/observed quadruplex sequence (PQS/OQS) represent experimentally validated G4-forming regions of higher thermodynamic stability than the remaining OQS (Hazel et al. 2004). To assess differences in the abundance of cfDNA fragments found in accessible blood-related chromatin regions within gene promoters that contain (Fig. 4B) or lack (Fig. 4C) PQS/OQS across different DNA fragment lengths, we performed in silico size selection (for US, short, and long cfDNA fragments) in samples from cancer patients and healthy individuals, processed by MB-ssDNA.
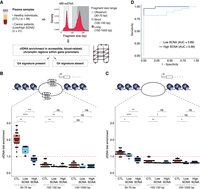
Assessment of cfDNA enrichment in accessible blood-related chromatin regions within gene promoters that contain or lack a G4 signature in MB-ssDNA libraries. (A) Schematics of the assessment of plasma cfDNA fold enrichment in genomic regions associated with predicted quadruplex sequences (PQS) that have been previously observed to adopt G4 structures (observed quadruplex sequences [OQS]), denoted as PQS/OQS regions. (B,C) Fold enrichment of cfDNA fragments in accessible blood-related chromatin regions within gene promoters that contain (B) or lack (C) a G4 signature at different fragment sizes (30–70, 100–150, 150–1000 bp) in samples from healthy individuals and cancer patients. The red dashed line indicates no enrichment relative to random permutation. (D) ROC curve analysis of US cfDNA fold enrichment in accessible blood-related chromatin regions within gene promoters that contain a G4 signature, for discriminating cancer patients from the healthy individuals using MB-ssDNA protocol. Wilcoxon rank-sum test: (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001; (ns) nonsignificant.
In the whole cohort (n = 49), the enrichment of US cfDNA fragments in accessible chromatin regions within gene promoters that contain PQS/OQS was substantially higher (median = 1.64-fold, IQR = 0.99–2.05-fold) than that for the short (median = 0.45-fold, IQR = 0.39–0.54-fold) and long fragments (median = 0.44-fold, IQR = 0.35–0.54-fold) (both P < 0.0001, Wilcoxon rank-sum test) (Supplemental Fig. S12C), but also in comparison to accessible chromatin regions within gene promoters lacking PQS/OQS (Supplemental Fig. S12D). Thus, MB-ssDNA revealed a higher enrichment of US cfDNA fragments in accessible chromatin regions associated with experimentally confirmed G4 sequences relative to longer cfDNA fragments.
We wondered whether this difference might be informative of the disease status. US cfDNA fragments derived from MB-ssDNA libraries from healthy individuals were substantially more enriched in accessible chromatin regions within gene promoters when PQS/OQS were present (median = 2.02-fold, IQR = 1.84–2.30-fold) (Fig. 4B) as compared to when they were absent (Fig. 4C; Supplemental Fig. S12E), and importantly relative to low SCNA (median = 1.56-fold, IQR = 1.20–1.64-fold) and high SCNA samples (median = 0.90-fold, IQR = 0.73–0.93-fold; P < 0.01 and P < 0.0001, respectively, Wilcoxon rank-sum test) (Fig. 4B). In addition, the G4-cfDNA enrichment was lower in the high SCNA samples compared to low SCNA samples (P < 0.001, Wilcoxon rank-sum test) (Fig. 4B). Analysis of the enrichment of US cfDNA fragments in accessible chromatin regions within gene promoters that contain PQS/OQS by ROC curve resulted in AUC of 0.99 and 0.89 for high SCNA and low SCNA samples, respectively (Fig. 4D). Taken together, this suggests the existence of fundamental biological differences between physiological blood plasma conditions and those in cancer.
To determine whether US cfDNA-enriched accessible PQS/OQS chromatin regions within gene promoters could originate from cellular G4 structures, we compared them with the same regions that adopt G4 DNA structural regions either in vivo or in cellulo, which we collectively define as in situ (Supplemental Fig. S12F; Supplemental Methods). This analysis revealed that 94% (6045/6429) of blood-related accessible PQS/OQS chromatin regions within promoters can form G4 in situ. In contrast, we found that the same regions that lack predicted G4 potential displayed negligible evidence (3%, 269/8810) for G4 formation in situ. These results support the possibility that US cfDNA may result from G4 secondary structures and thus could be a marker for single-stranded regulatory regions.
Discussion
Here, through the use of a novel workflow, MB-ssDNA, we identified and characterized a new population of plasma cfDNA molecules organized around ∼50 bp (Fig. 1C). In cancer patients, these fragments included tumor-derived molecules at relative levels similar to those detected in DNA fragments >150 bp (Fig. 2B). Using rapid and cost-effective sWGS, we showed that US cfDNA fragments could be used to identify samples from cancer patients based on their (1) relative abundance in plasma, (2) DNA sequence context, (3) relationship with TSS, and (4) abundance in regions associated with G-quadruplexes. The latter two observations suggest an association between US cfDNA fragments and regulatory mechanisms.
Our findings underline the impact of cfDNA processing workflows on downstream results, profoundly affecting the representation of cfDNA fragments obtained from plasma. In addition to the previously described bias of sequencing chemistry toward shorter (Tan et al. 2019) or longer fragments (Branton et al. 2008; Jain et al. 2018), this study showed that different DNA extraction protocols greatly affect the recovery of cfDNA molecules within specific size ranges (Fig. 1B,C). cfDNA extraction using magnetic beads carrying a greater positive charge captured significantly more US fragments (<70 bp) than extraction using silica gel membrane-based columns (Fig. 1D). The sequencing library preparation protocol additionally affected the representation of US cfDNA populations in downstream sequencing data (Fig. 1B–D). Although conventional dsDNA-based protocols recover intact dsDNA, damaged cfDNA are lost to downstream analysis. Here, we used ssDNA-based protocols that have potential to recover ssDNA and damaged dsDNA molecules and characterized their genomic profiles.
Using MB-ssDNA, we observed an average 35-fold higher recovery of US fragments compared to the other methods (86-, 13-, and fivefold higher as compared to SC-dsDNA, MB-dsDNA, SC-ssDNA, respectively). A report released during the revision of this manuscript using alternative chemistry for DNA extraction also suggested a better recovery of US fragments (Hisano et al. 2021). The US fragments observed in our study were relatively enriched in plasma from healthy individuals as compared to cancer patients (Fig. 1G). Using a modified MB-ssDNA protocol that omitted initial DNA denaturation, we showed that ssDNA fragments are also present in plasma and enriched in the size range 30–70 bp (Supplemental Fig. S7A,B). This experiment further indicated that ctDNA levels of these ssDNA molecules were consistent with those observed by the unaltered MB-ssDNA protocol that recovers ssDNA, dsDNA, and damaged dsDNA.
The observations reported in this study raise several questions. First, the biological origins of US cfDNA fragments are unknown. Second, it is unclear what causes a relative decrease in the abundance of US fragments in cancer patients as compared to healthy individuals. The US cfDNA could be more representative of open chromatin regions in hematopoietic cells, as characterized by ATAC-seq (Buenrostro et al. 2015; Sun et al. 2018) and as indicated by our findings (Supplemental Fig. S11). In healthy individuals, US fragments originating from such regions might be abundantly released into the circulation, because the majority of cfDNA is of hematopoietic origin (Lui et al. 2002; Moss et al. 2018). In cancer patients, the relative decrease in the 50-bp peak (Fig. 1F,G) might be related to a “dilution” by an excess of short fragments (100–150 bp) released from non-hematopoietic cells (Supplemental Fig. S11B–D), previously shown to be increased in patients with higher ctDNA levels (Jiang et al. 2015; Mouliere et al. 2018). The analysis of US cfDNA fragments, and the link to a particular cell lineage, could benefit from technological progress in determination of the cellular origin of plasma cfDNA (Cheng et al. 2019; Ulz et al. 2019).
To shed light on the biology of the US cfDNA, we explored and mapped their genomic and transcriptomic footprints. Here, we found that the number of US fragments overlapping with unique TSS in MB-ssDNA libraries was significantly higher in healthy individuals compared to cancer patients (Fig. 3C). Of note, we observed varying densities of US cfDNA coverage at specific positions upstream of TSS (−40, −160, and −250 bp) (Fig. 3F; Supplemental Fig. S9A,B). Despite uncertainty surrounding what physical associations these peaks might represent, we found that relative coverage at these positions varied significantly between healthy individuals and cancer patients, highlighting diagnostic potential for cancer detection (Fig. 3G; Supplemental Fig. S9C–E).
Prompted by a greater variability in GC content among US fragments as compared to longer fragments (Supplemental Fig. S12B), we explored the possible association of cfDNA with guanine-rich secondary structures, G4s (Fig. 4A). In the nucleus, G4 formation can occur in double-stranded G-rich regions when DNA becomes transiently single-stranded, during transcription and replication, and at single-stranded telomeric G-rich overhangs (Rhodes and Lipps 2015; Varshney et al. 2020). Endogenous G4 DNA mapping has recently been used to report on underlying cancer subtypes and vulnerabilities to G4-targeted treatments (Hänsel-Hertsch et al. 2020). However, the presence of G4s, or other DNA secondary structure, has not been studied in plasma. Here, we found that cfDNA fragments contain G4 sequences that may have adopted G4 structure in their cell of origin using a computational approach following these considerations: first, we focused on accessible chromatin regions in which G4 sequences can adopt G4 secondary structures (Hänsel-Hertsch et al. 2016). Second, we considered only accessible chromatin in promoter regions known to be highly enriched for endogenous G4 structure formation (Hänsel-Hertsch et al. 2016, 2020). Third, we focused on accessible chromatin regions from hematopoietic cells, considered the dominant source of cfDNA in healthy individuals and cancer patients (Lui et al. 2002; Moss et al. 2018). Our findings, based on accessible chromatin regions mapped in blood cells by DNase-seq (Oki et al. 2018), open up the possibility that G4s may not only form in cancer genomes but also in blood-related genomes. Furthermore, by considering various G4 regions that we and others have previously annotated by chromatin immunoprecipitation and sequencing (Hänsel-Hertsch et al. 2018, 2020; Spiegel et al. 2021; Zhang et al. 2021), our data highlights that some proportion of US cfDNA may indeed result from G4 secondary structures.
Reflecting our findings in DNA fragmentation and TSS features, we found that GC content and cfDNA enrichment in accessible chromatin containing G4 DNA sequences were significantly decreased in plasma from cancer patients as compared to healthy individuals. Previous studies indicated increased G4 formation in skin, stomach, liver, and breast cancer cells and tissue, as compared to healthy tissues from the same organs (Biffi et al. 2014). Contrary to these, our findings showed the opposite trend (i.e., decreased G4-cfDNA enrichment in cancer). The US cfDNA fragments could be digested to a greater extent in the plasma of cancer patients, as a result of the action of enzymes in different disease states (Keyel 2017), and differences in chromatin organization in different tissue types (Snyder et al. 2016; Moss et al. 2018; Sun et al. 2019) governing the accessibility of cfDNA to plasma enzymes. Owing to the nature of G4, these structures may be more resistant to nucleases known to play a role in cleaving cfDNA in healthy individuals (e.g., DNASE1, DFFB, and DNASE1L3) (Han et al. 2020). In cancer patients, it is possible that the presence or altered activity of these nucleases, and/or the action of additional nucleases, could explain the lower representation of G4 sequences in plasma cfDNA. Although progress has been made in the exploration of cfDNA biology (Serpas et al. 2019; Han et al. 2020), the spectrum of enzymes and mechanisms contributing to the fragmentation of cfDNA still needs to be established in its entirety. Overall, the observation that US cfDNA fragments in healthy individuals are significantly more enriched in G4 sequences relative to cancer patients suggests the potential to infer G4 content in plasma cfDNA for cancer diagnosis. Future strategies may selectively target US G4 cfDNA to enrich for cfDNA fragments informing on the regulatory processes at play in situ. Of note, exploration of secondary structures of cfDNA at larger fragment sizes is warranted.
Our results obtained from sequencing plasma cfDNA, extracted by high-affinity magnetic beads undergoing ssDNA library preparation protocol (MB-ssDNA), redraw the landscape of cfDNA fragmentation by unveiling and characterizing a population of US fragments with lengths ∼50 bp. Although these findings were based on a limited number of samples (n = 49), we showed their robustness (Supplemental Figs. S2A,B, S8C) and confirmed that the absence of these molecules carries cancer-specific information. We envision that, as shown in dsDNA-based approaches (Mouliere et al. 2018; Cristiano et al. 2019), this new cfDNA feature could be leveraged to further improve our ability to identify samples from individuals with cancer. By improving the recovery of US cfDNA fragments enriched in TSS and accessible chromatin, the MB-ssDNA approach could facilitate studies using transcription factor-bound cfDNA for applications such as forecasting cancer progression and tissue of origin prediction (Snyder et al. 2016; Ulz et al. 2016). Finally, by unveiling the presence of sequences with the potential to form G4s in plasma, our work highlights for the first time that more complex DNA secondary structures can be explored by cfDNA analysis, and their depletion in the plasma of cancer patients can potentially be used as a diagnostic marker for cancer research.
Methods
Patient recruitment
Cancer patients were recruited prospectively to the CALIBRATE study at Addenbrooke's Hospital, Cambridge, United Kingdom, approved by the local research ethics committee (REC reference numbers 14/SC/1170). A cohort of healthy individuals from the Cambridge Blood and Stem Cell Biobank, Department of Haematology, University of Cambridge and from BioIVT (https://bioivt.com) was included. Informed consent was obtained from all donors. The study was conducted in adherence to the principles outlined in the World Medical Association Declaration of Helsinki Research Involving Human Subjects.
Blood processing and DNA extraction
Blood samples were collected into EDTA-containing tubes and processed by a double-centrifugation protocol (1600g for 10 min; 14,000 rpm for 10 min). DNA was extracted using either the QIAamp Circulating Nucleic Acid Kit (QIAGEN; silica column-based, SC protocol) or QIAsymphony DSP Circulating Nucleic Acid Kit (QIAGEN; a version using high-affinity magnetic beads, MB protocol).
DNA library preparation and sWGS
Indexed sequencing libraries were prepared using the ThruPLEX Plasma-Seq Kit (Takara Bio; dsDNA protocol) and DNA SMART ChIP-seq Kit using template-switching chemistry (Takara Bio; ssDNA protocol). A subset of DNA libraries was prepared according to an adapted ssDNA-based protocol using ligation of biotinylated adapters (Gansauge and Meyer 2013; Burnham et al. 2016). Libraries were pooled and sequenced generating paired-end reads (2 × 50 bp, 2 × 75 bp, or 2 × 150 bp). Sequencing data was analyzed using an in-house pipeline, including a step for quantifying fragment size distributions (Supplemental Methods). t-MAD was determined as described previously (Mouliere et al. 2018), with a segmentation step summarized by median value. If applicable, BAM files were down-sampled to 5 million reads (or 1.25 million and 0.5 million using 500-kb genomic segments) for comparison between groups. A t-MAD threshold of 0.019 was set, defined using a cohort of plasma samples from healthy individuals using 5 million reads (Mouliere et al. 2018), and a threshold of 0.0416 was used for data down-sampled to 1.25 and 0.5 million sequencing reads. R code for t-MAD analysis is available at GitHub (https://github.com/sdchandra/tMAD) (R Core Team 2018). Significant differences in t-MAD and in the proportion of cfDNA fragments at specific size were calculated by Wilcoxon rank-sum test (Wilcoxon signed-rank test used for paired samples). Kolmogorov–Smirnov test was used to compare the empirical cumulative distribution function (ECDF) of fragment size distributions.
Sequence coverage analysis at TSS
TSS that characterize and map locations of functional elements were gathered from Ensembl (http://ftp.ensembl.org/pub/release-75/gtf/homo_sapiens/Homo_sapiens.GRCh37.75.gtf.gz). For coverage calculations we considered TSS from all isoforms of all protein coding genes (90,273 TSS), taking into account which strand the TSS was located on (Supplemental Table S2). ssDNA and dsDNA sWGS BAM files were down-sampled to the same number of sequencing reads. Coverage was calculated and normalized by mean depth in regions ±1 kbp of TSS. The Wilcoxon rank-sum test was used to determine whether there were significant differences in fragment numbers at specific size ranges overlapping with TSS, and in the relative proportion of fragments at positions 40/160/250 bp upstream of TSS. We also performed an analysis of relative coverage at TSS associated with accessible chromatin regions of different hematopoietic cells (erythrocyte progenitors and lymphocytes) or at TSS associated with accessible pan-cancer chromatin regions. The selection of these regions is described below. For TSS associated with accessible pan-cancer chromatin regions, we considered the top 5000 ATAC-seq peaks by its enrichment score (the normalized peak score of the given peak, i.e., score-per-million) (see Corces et al. 2018, Methods therein), derived from 410 tumor samples spanning 23 cancer types from The Cancer Genome Atlas (https://gdc.cancer.gov/about-data/publications/ATACseq-AWG). In total we identified 2982, 7453, and 16,235 TSS for erythrocyte progenitors, lymphocytes, and pan-cancer accessible chromatin regions, respectively. Each TSS group was normalized by the number of regions (2982 randomly selected genomic regions covering 879 genes). Furthermore, each control/patient category was normalized by the number of sequence reads (5,196,953). Code used for analysis of relative coverage at TSS is available in Supplemental Code 1.
Enrichment analysis of TSS containing cancer or hematopoietic cell-related accessible chromatin regions
Accessible chromatin regions were retrieved from ChIP-ATLAS (https://chip-atlas.org) using the following parameters: Antigen Class: DNase-seq; Cell type Class: Blood; Threshold for Significance: 500; Antigen: All; Cell type: Erythrocyte progenitors, Erythroid Cells, Lymphocytes, and Monocytes (Supplemental Table S3). We grouped erythrocyte progenitors and Erythroid Cells into “erythrocyte progenitors.” DNase-seq peaks of each experiment (SRX*) within either the erythrocyte progenitors or lymphocytes categories were sorted (sortBed), merged (mergeBed), and filtered for unique peaks that only appear in the respective categories (intersectBed). TSS for lymphocyte and erythrocyte progenitors were selected from overlaps with their specific unique accessible chromatin peaks (intersectBed). After filtering, monocyte-related TSS were excluded owing to small numbers. To reveal TSS containing pan-cancer accessible chromatin, a genomic coordinate pan-cancer ATAC-seq file was retrieved from https://gdc.cancer.gov/about-data/publications/ATACseq-AWG, sorted (sortBed), lifted to hg19 (https://genome.ucsc.edu/cgi-bin/hgLiftOver), sorted, and TSS (intersectBed) captured to reveal cancer-specific TSS. To compare pan-cancer-related TSS with blood-related TSS, we selected the top 5000 enriched ATAC-seq peaks by their enrichment score (the normalized peak score of the given peak, i.e., score-per-million) (see Corces et al. 2018, Methods therein). BEDTools shuffle was used to generate, respectively, 10 random distributions of the three TSS categories (pan-cancer, lymphocytes, erythrocyte progenitors). The plotEnrichment function of deepTools (Ramírez et al. 2016) was used to measure the coverage of individual BAM files in 11 BED files per condition (1 BED file + 10 randomized BED files). For each BAM file, enrichments in the three TSS categories were calculated using the average ratio of the real coverage divided by 10 different coverage values in the permuted TSS data sets. Code used for fold enrichment analysis of TSS is available in Supplemental Code 2.
Enrichment analysis of chromatin accessibility in transcription start sites and its relationship to G4s
Accessible chromatin regions were retrieved from ChIP-ATLAS (https://chip-atlas.org) using the same parameters as above but selecting all cells (Cell Type: All). A hg19 TSS file was generated as previously described (Hänsel-Hertsch et al. 2016). A maximum loop length of 7 nt, four G-repeats, and a minimum of three Gs per G-repeat were considered to predict PQS in the hg19 genome. The fastaRegexFinder tool (Python) was used with the following parameters to predict PQS: “‐‐quiet -r ‘([gG]{3,}\w{1,7}){3,}[gG]{3,}’”. The four available OQS files were retrieved (Chambers et al. 2015) and unified to a single OQS file using cat and the BEDTools functions mergeBed and sortBed (Quinlan and Hall 2010). BEDTools intersect was used to retrieve approximately 300,000 PQS that overlap with OQS, (PQS/OQS). BEDTools intersect was used to retrieve accessible chromatin regions in TSS that are either positive or negative for PQS/OQS. BEDTtools shuffle was used to generate 10 random distributions of accessible chromatin regions in TSS that are either positive or negative for PQS/OQS regions across the hg19 genome. The same enrichment software was used as described above. For each BAM file, enrichments in accessible chromatin regions in TSS that contain or lack PQS/OQS were calculated using the average ratio of the real coverage divided by 10 different coverages in the permuted accessible chromatin regions in TSS that contain or lack PQS/OQS. Code used for G4 cfDNA fold enrichment analysis is available in Supplemental Code 2.
Data access
The sequencing data generated in this study have been submitted to the European Genome-Phenome Archive repository (EGA; https://ega-archive.org), under accession number EGAS00001005093.
Competing interest statement
N.R. and D.G. are cofounders, shareholders, and present/former employees/officers of Inivata Ltd., a cancer genomics company that commercializes ctDNA analysis. C.G.S. has consulted for Inivata Ltd. Inivata Ltd. had no role in the conceptualization, study design, data collection, analysis, decision to publish, or preparation of the manuscript. N.R., F.M., and D.C. are inventors of the patent “Enhanced detection of target DNA by fragment size analysis” (WO/2020/094775), filed by CRUK Cambridge Institute. I.H., J.A.M., K.H., and D.G. are current employees of AstraZeneca, Inc. AstraZeneca was not involved with the study and had no role in the conceptualization or design of the clinical study or decision to publish the manuscript. The remaining authors declare no competing interests.
Acknowledgments
We thank the Genomics, Bioinformatics, and Compliance and Biobanking core facilities at CRUK Cambridge Institute, including James Hadfield, Paul Coupland, Hannah Haydon, Matthew Eldridge, and Jorgelina Trueba. We thank the Regional Computing Centre (RRZK) of the University of Cologne for support. We thank Alexander Wolf (QIAGEN) for his suggestions and advice regarding the DNA extraction procedure used. We thank Shankar Balasubramanian for feedback concerning the G4 analysis. We acknowledge support from the Cancer Research UK Cambridge Institute, the National Institute for Health Research (NIHR) Biomedical Research Centre, NIHR Cambridge Clinical Research Centre, and Experimental Cancer Medicine Centre, as well as the support from early phase and Cancer Clinical Trial Centre research teams including Duncan Jodrell, Bristi Basu, Sarah Loewenbein, Will Dott, Constanza Linossi, and Gary Doherty. For their assistance with collection of samples from healthy volunteers, we thank Joanna Baxter and Andreia Ribeiro Da Silva from the Cambridge Blood and Stem Cell Biobank, which is supported by the Cambridge NIHR Biomedical Research Centre, Wellcome Trust—Medical Research Council (MRC) Stem Cell Institute, and the Cambridge Experimental Cancer Medicine Centre, UK. For their assistance with DNA extraction from plasma samples, we thank Shubha Anand, Isart Roca, and Francesca Nice from the Cancer Molecular Diagnostics Laboratory/Blood Processing Laboratory, which is supported by Cambridge NIHR Biomedical Research Centre, Cambridge Cancer Centre, and the Mark Foundation of Cancer Research. Finally, we thank all patients and healthy volunteers for their contribution to this study. This study was supported by grants from Cancer Research UK (CRUK) Cambridge Institute (Core Grant, A29580) and by a funding from the European Research Council (ERC) under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC Grant Agreement n. 337905. R.H.-H. is supported by funding from the Center for Molecular Medicine Cologne and by the Deutsche Forschungsgemeinschaft (CRC1399). F.M. is supported by a Dutch Cancer Fund (KWF-12822).
Author contributions: I.H., C.G.S., R.H.-H., N.R., and F.M. conceptualized and designed the study; I.H., C.G.S., K.H., W.N.C., D.G., and F.M. performed experiments and collected data; I.H., C.G.S., R.H.-H., C.S.C., J.A.M., A.V., D.C., and F.M. performed bioinformatics analysis and analyzed sequencing data; S.P. and R.D.B. are co-chief investigators of the CALIBRATE trial; J.G.-C., S.P., and R.D.B. recruited patients and collected samples; I.H., C.G.S., R.H.-H., N.R., and F.M. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
-
↵10 These authors share senior authorship.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275691.121.
-
Freely available online through the Genome Research Open Access option.
- Received May 3, 2021.
- Accepted December 16, 2021.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








