
Natural variation in C. elegans short tandem repeats
Abstract
Short tandem repeats (STRs) represent an important class of genetic variation that can contribute to phenotypic differences. Although millions of single nucleotide variants (SNVs) and short indels have been identified among wild Caenorhabditis elegans strains, the natural diversity in STRs remains unknown. Here, we characterized the distribution of 31,991 STRs with motif lengths of 1–6 bp in the reference genome of C. elegans. Of these STRs, 27,667 harbored polymorphisms across 540 wild strains and only 9691 polymorphic STRs (pSTRs) had complete genotype data for more than 90% of the strains. Compared with the reference genome, the pSTRs showed more contraction than expansion. We found that STRs with different motif lengths were enriched in different genomic features, among which coding regions showed the lowest STR diversity and constrained STR mutations. STR diversity also showed similar genetic divergence and selection signatures among wild strains as in previous studies using SNVs. We further identified STR variation in two mutation accumulation line panels that were derived from two wild strains and found background-dependent and fitness-dependent STR mutations. We also performed the first genome-wide association analyses between natural variation in STRs and organismal phenotypic variation among wild C. elegans strains. Overall, our results delineate the first large-scale characterization of STR variation in wild C. elegans strains and highlight the effects of selection on STR mutations.
Short tandem repeats (STRs) are repetitive elements consisting of 1–6 bp DNA sequence motifs that provide a large source for genetic variation in both inherited and de novo mutations (Willems et al. 2016; Fotsing et al. 2019). The predominant mechanism of STR mutations is DNA replication slippage, which often causes expansion or contraction in the number of repeats (Mirkin 2007; Gemayel et al. 2010). Because of their intrinsically unstable nature, STRs have orders of magnitude higher mutation rates than other types of mutations, such as single nucleotide variants (SNVs) and short insertions or deletions (indels) (Lynch 2010; Sun et al. 2012; Willems et al. 2016; Gymrek et al. 2017). The precise mutation rates of STRs are highly variable across different loci and are affected by motif sequences and repeat lengths (Legendre et al. 2007). In humans, STRs are estimated to constitute about 3% of the genome and are associated with dozens of diseases (Mirkin 2007; Hannan 2018; Malik et al. 2021). Emerging studies have also revealed the role of STRs in regulation of gene expression and complex traits in humans and other organisms, which were suggested to facilitate adaptation and accelerate evolution (Fotsing et al. 2019; Jakubosky et al. 2020; Reinar et al. 2021).
The free-living nematode Caenorhabditis elegans is a keystone model organism that has been found across the world (Brenner 1974; Kiontke et al. 2011; Andersen et al. 2012; Félix and Duveau 2012; Cook et al. 2017; Crombie et al. 2019, 2022; Lee et al. 2021). The C. elegans Natural Diversity Resource (CeNDR) catalogs and distributes thousands of wild strains, genome sequence data, and genome-wide variation data, including SNVs and short indels (Andersen et al. 2012; Cook et al. 2017; Evans et al. 2021a). Numerous C. elegans population genomics studies and genome-wide association (GWA) studies have leveraged CeNDR resources, such as the genetic variant data across wild strains and the GWA mapping pipeline (Snoek et al. 2020; Lee et al. 2021; Evans et al. 2021a; Gilbert et al. 2022; Widmayer et al. 2022; Zhang et al. 2022). However, the natural diversity in C. elegans STRs and their impacts on organism-level and molecular traits among wild strains remain unknown because of the lack of STR variation characterization. STRs are challenging to genotype because of their repetitive nature causing errors such as “PCR stutters” (Gymrek 2017). Recent advances provided opportunities to identify genome-wide STR variation accurately in large scales using high-throughput sequencing data (Willems et al. 2017).
In this work, we focused on characterization of STRs with motif lengths of 1–6 bp in the reference genome of C. elegans and identified their natural variation across 540 genetically distinct wild strains. Using these data, we analyzed mutations, diversity, and how selection occurred in the STR loci. We also investigated the possible impacts of STR variation on phenotypic variation across wild C. elegans strains.
Results
Genome-wide profiling of STR variation in C. elegans
To investigate the natural variation of C. elegans STRs, we first identified 31,991 reference STRs in the C. elegans reference genome (Table 1; Supplemental Table S1). These STRs comprise motif lengths of 1–6 bp and a minimum repeat number of 11, 6, 5, 3.75, 3.4, and 3, respectively, for each ascending motif length. The reference STRs were unevenly distributed across the genome (Supplemental Fig. S1A) with higher density on chromosome arms and tips than centers, suggesting that higher recombination is associated with the increasing incidence of STRs (Rockman and Kruglyak 2009). Mono-STRs (1 bp STRs) that contributed more than half of the reference STRs were also denser on arms and tips than centers, whereas STRs with motif lengths of 2–6 bp distributed differently across the genome (Table 1; Supplemental Fig. S1B).
The distribution of STRs in C. elegans
We examined natural variation in reference STRs across 540 genetically distinct wild C. elegans strains (Cook et al. 2017; Evans et al. 2021a) and observed natural variation in 27,667 STRs (Supplemental Table S1). We further identified 9691 polymorphic STRs (pSTRs) with missing calls in <10% of all strains (Table 1; Supplemental Table S1). The composition of STR sequences was found to be more complicated than simple repeats of motifs (Urquhart et al. 1994). Here, we classified STRs into six groups based on the reference STR sequences and motif sequences: simple-perfect, simple-center-perfect, simple-interrupted, compound-perfect, compound-center-perfect, and compound-interrupted (see Methods; Supplemental Table S1; Supplemental Fig. S2). The simple-interrupted group comprised more than half of the 27,667 STRs with natural variation and the 9691 pSTRs (Supplemental Fig. S2). However, simple-interrupted and compound-interrupted STRs were significantly underrepresented (one-sided Fisher's exact test with Bonferroni-corrected P = 3.3 × 10−87 and 0.03, respectively) in the 9691 pSTRs than in the 27,667 STRs. Interrupted STRs might cause extra challenges to sequencing read alignment and STR variant calling, which could partially explain why about two-thirds of the 27,667 STRs had missing calls in equal or more than 10% of all strains. We further examined mutations closely for 887 simple-perfect pSTRs, which altogether were found with 2320 homozygous alternative alleles among the 540 wild strains. We found 1037 and 691 alternative alleles showed perfect “deletion” and “insertion”, respectively, by the number of repeats of the motifs; 144 and 149 alternative alleles showed imperfect “deletion” and “insertion”, respectively, with extra substitutions; 299 alternative alleles showed only substitutions. We examined nucleotide substitutions in alternative alleles of all pSTRs. A total of 5564 pSTRs were found with substitutions in their homozygous alternative alleles, among which 85% (7638 out of 8985) only had a single nucleotide substitution. The maximum number of nucleotide substitutions is 11, and the maximum proportion of substituted nucleotides compared with the reference allele is 73%.
We focused on the 9691 pSTRs. The density of pSTRs on arms and tips was not always higher than centers (Fig. 1A; Supplemental Fig. S3) likely because DNA slippage, not recombination, is the major source of STR mutations (Kunkel 1993). Poor alignment in hyper-divergent regions (Lee et al. 2021) might also hinder STR variant calling and reduce the density of pSTRs in certain regions, such as gaps at the left arm of Chromosome II and the right arm of Chromosome V (Fig. 1A). The bases A and T were the most abundant motif sequences in both reference and polymorphic STRs, which is consistent with previous findings in C. elegans and many other eukaryotic genomes (Fig. 1B; Supplemental Fig. S4A; Tóth et al. 2000; Denver et al. 2004; Saxena et al. 2019). We also found that different genomic features were enriched with STRs of different motif lengths (Fig. 1C,D; Supplemental Fig. S4B,C; Supplemental Table S2). For example, the most prevalent An and Tn mono-STRs were only enriched in introns, 3′UTR, and intergenic regions (Fig. 1D,E; Supplemental Fig. S4C,D; Supplemental Table S2). Tri-STRs and hexa-STRs were mostly enriched in coding sequence (CDS) regions (Fig. 1D; Supplemental Fig. S4C; Supplemental Table S2), suggesting purifying selection constrains these STRs to maintain the triplet code (Metzgar et al. 2000).
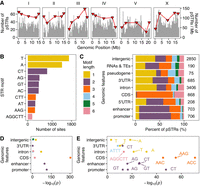
The distribution of polymorphic short tandem repeats (STRs) across the Caenorhabditis elegans genome. (A) The distribution of polymorphic STRs (y-axis on the left) in the C. elegans genome. Red triangles represent the number of STRs per Mb (y-axis on the right) in different genomic domains (tips, arms, and centers) (Rockman and Kruglyak 2009). (B) The top ten most frequent motif sequences in polymorphic STRs are shown on the y-axis, and the number of those sites on the x-axis. (C) Percent of polymorphic STRs with different motif lengths in each genomic feature are shown on the x-axis, and different genomic features on the y-axis. The total number of polymorphic STRs in each genomic feature is indicated. (D) Enriched STRs with different motif lengths (colored as in C) in different genomic features are shown. (E) The top three most enriched STR motif sequences (labeled) in each genomic feature (if enriched motifs were found) are shown. Statistical significance for enrichment tests (Supplemental Table S2) was calculated using one-sided Fisher's exact test and was corrected for multiple comparisons (Bonferroni method).
Polymorphic STRs are often contracted as compared to the reference genome
STR mutations by DNA slippage are more likely to cause length variation in multiples of the motif lengths (Metzgar et al. 2000; Mirkin 2007) than single nucleotide substitutions. Of alternative alleles among wild C. elegans pSTRs, we observed that 30.2%, 35.5%, and 34.3% were insertions, deletions, or substitutions, respectively (Fig. 2A). In the same 540 C. elegans strains, the proportions of SNVs and indels are 83.3% and 16.7%, respectively. To better understand STR mutations, we computed the expansion and contraction scores (Press et al. 2018) by comparing the longest and/or shortest alternative alleles to the median alleles for each of the 7506 pSTRs with length variation (Fig. 2B). We found significantly higher contraction scores than expansion scores when we compared their absolute values for mono-, tri-, and tetra-STRs (Fig. 2C; Supplemental Table S2). In di-STRs, however, the contraction scores were significantly lower than expansion scores (Fig. 2C). Di-STRs stood out as exceptions again in allele frequencies, in which contracted alleles were at significantly lower frequency than expanded alleles (Fig. 2D; Supplemental Table S2). We examined contraction and expansion in STRs with different motif sequences and focused on di-STRs (Fig. 2E; Supplemental Fig. S5). All di-STRs had 36.3% to 38.6% alternative alleles expanded (Fig. 2E), except (CG)n di-STRs, which only had 9.6% alternative alleles expanded. Because we used short-read sequencing data, which are more easily aligned to deletion loci than insertion loci, the actual differences between contraction and expansion might be underestimated. For example, we found significantly more aligned reads in loci of contracted alleles than expanded alleles (Wilcoxon test, P < 2 × 10−16) among all pSTR homozygous alleles (see Methods). Altogether, we found more STR contraction than expansion among wild C. elegans, with the exception of di-STRs.
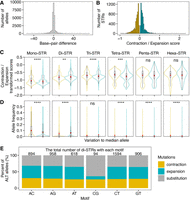
Contraction and expansion of polymorphic short tandem repeats (STRs). (A) The distribution of base-pair differences for polymorphic STR alleles compared with the reference alleles is shown. Positive and negative values on the x-axis indicate allele expansion and contraction, respectively, compared with the reference alleles. (B) The distribution of contraction (in yellow) and expansion (in blue) scores for each pSTR. Expansion score = [max(STR length) – median(STR length)]/median(STR length); contraction score = [min(STR length) – median(STR length)]/median(STR length). (C) Comparison of the log10 transformed absolute values between contraction (in yellow) and expansion (in blue) scores in polymorphic STRs with different motif lengths. (D) Comparison of allele frequencies between contracted (in yellow) and expanded alleles compared with the median allele length in polymorphic STRs with different motif lengths. The mean and median values in (C,D) are indicated as red points and horizontal lines in each box, respectively (Supplemental Table S2). Statistical significance was calculated using the two-sided Wilcoxon test and was corrected for multiple comparisons (Bonferroni method). Significance of each comparison (Supplemental Table S2) is shown above each comparison pair (ns: adjusted P > 0.05, [**] adjusted P ≤ 0.01, [***] adjusted P ≤ 0.001, [****] adjusted P ≤ 0.0001). (E) Percent of alternative alleles showing contraction, expansion, and substitution in di-STRs. The total number of di-STRs with different motif sequences is indicated above each stacked bar.
STR diversity is correlated with species-wide selective sweeps
The majority of pSTRs among the 540 wild C. elegans strains were multiallelic with a median of three alleles per STR (Fig. 3A). Only 4% of pSTRs had a major allele frequency less than 0.5 (Fig. 3B; Supplemental Table S3), likely because C. elegans reproduces primarily by hermaphroditic selfing and recent selective sweeps have reduced diversity across the species (Andersen et al. 2012). The selective sweeps were thought to have purged diversity from the centers of Chromosomes I, IV, and V, and the left arm of the X Chromosome from the C. elegans global population. However, recent sampling efforts of wild C. elegans revealed higher genetic diversity in strains from the Hawaiian Islands and other regions in the Pacific Rim, which were hypothesized as the geographic origin of the species (Crombie et al. 2019, 2022; Lee et al. 2021). We have previously classified wild C. elegans into swept and divergent strains based on the proportion of swept haplotypes that were identified using SNVs across the genome (see Methods; Crombie et al. 2019; Lee et al. 2021; Zhang et al. 2021). Here, we observed a much higher density of pSTRs with major allele frequencies close to 1 among the 357 swept strains than among all the 540 strains or among the 183 divergent strains (Fig. 3B). Within divergent strains, more than 9% of pSTRs had a major allele frequency less than 0.5 (Fig. 3B). We also found that divergent strains had a significantly higher percentage of homozygous alternative alleles and heterozygous alleles than swept strains (Wilcoxon test with Bonferroni-corrected P = 9.2 × 10−74 and 4.9 × 10−10, respectively) (Fig. 3C; Supplemental Table S4). Furthermore, principal component analysis (Price et al. 2006) using pSTRs and SNVs showed similar clusters using the 540 strains, which largely correspond to the geographic locations of these strains (Fig. 3D,E; Supplemental Table S5). The 163 Hawaiian strains, including 157 divergent strains, were mostly separated from the global strains that had experienced the selective sweeps (Fig. 3D,E). To further explore the STR diversity in C. elegans, we calculated the expected heterozygosity (HE) for each pSTR among all strains, only among swept strains, or only among divergent strains (Fig. 3F; Supplemental Table S3). Divergent strains showed higher diversity across the genome than swept strains and no signatures of selective sweeps (Fig. 3F). The swept strains showed the largest drop of HE in all the four swept regions (Fig. 3F), which is consistent with low levels of genome-wide genetic diversity using SNVs in previous studies (Andersen et al. 2012; Crombie et al. 2019; Lee et al. 2021). However, the left arm of Chromosome X, in which it was suggested to have experienced a selective sweep using 97 strains (Andersen et al. 2012), showed much stronger signs of selective sweeps using pSTRs than SNVs among the 357 swept strains (Fig. 3F; Supplemental Table S3), suggesting the potential use of STRs in population and evolutionary studies. Altogether, these results suggested that the diversity of STRs in C. elegans has been reduced in many strains by the selective sweeps, and divergent strains have retained high levels of STR diversity.
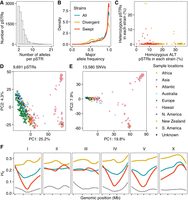
Genetic diversity of Caenorhabditis elegans short tandem repeats (STRs). (A) The distribution of allele counts per polymorphic STR. (B) Major allele frequencies of all polymorphic STRs (pSTRs) for all strains (in blue), divergent strains (in yellow), and swept strains (in red) are shown. (C) The percentage of pSTRs with heterozygous alleles is plotted against the percentage of pSTRs with homozygous alternative (ALT) alleles for each of the 540 strains. Divergent and swept strains are colored yellow and red, respectively. (D,E) Plots show the top two axes of variation, as determined by principal components analysis (PCA) of the genotype covariances using polymorphic STRs (D) and single nucleotide variants (SNVs) (E). Each dot represents a strain and is colored by the sampling location. (F) Chromosomal expected heterozygosity (HE) of pSTRs is shown as locally regressed lines for all strains (in blue), divergent strains (in yellow), and swept strains (in red). Chromosomal HE of SNVs among swept strains is shown as locally regressed lines (in gray). Tick marks on the x-axis denote every 5 Mb.
We further examined pSTR diversity in different genomic features and found that CDS had significantly lower HE than any other genomic features, indicating reduced pSTR diversity in these regions (Supplemental Fig. S6A; Supplemental Table S2). In addition to lower HE, pSTRs in CDS regions also had significantly lower variance in repeat number than most other genomic regions (Supplemental Fig. S6B; Supplemental Table S2), suggesting pSTR expansion and contraction might be limited in CDS regions. Increased slippage rates and STR instability were linked to high AT content rather than high GC content (Schlötterer and Tautz 1992; Brandström and Ellegren 2008). We observed the highest GC content among pSTRs in CDS regions (Supplemental Fig. S6C; Supplemental Table S2). Altogether, these results suggested that STR diversity was constrained in the conservative CDS regions to maintain proper gene function.
We also wondered whether the length of STR affected STR diversity in C. elegans. We calculated correlation between the length of the reference alleles and HE of pSTRs with different motif lengths. The correlation coefficient (adjusted P-value) for pSTRs with motif length of 1–6 bp is −0.18 (adjusted P = 3.18 × 10−44), 0.1 (adjusted P = 1.8 × 10−13), 0.12 (adjusted P = 1.56 × 10−13), 0.15 (adjusted P = 2.76 × 10−10), 0.14 (adjusted P = 1.08 × 10−3), and 0.23 (adjusted P = 2.1 × 10−25), respectively (Kendall rank correlation test, with Bonferroni correction). In conclusion, we observed weak correlation between STR length and STR diversity.
STR mutation rates in MA lines
In addition to the selective sweeps, other exogenous and endogenous factors might also influence STR diversity in C. elegans. For example, because of ample bacterial food and a stable environment (Crombie et al. 2019), Hawaiian strains might have gone through more generations and fewer bottlenecks than European strains, which might have had to enter the dauer diapause stage more frequently to survive starvation and overwinter (Frézal and Félix 2015). Therefore, Hawaiian strains might be able to accumulate more STR and other mutations than European strains. To better understand STR mutation and evolution in C. elegans, we examined STR variation in two mutation accumulation (MA) line panels that were derived from two strains, N2 and PB306 (Joyner-Matos et al. 2011; Matsuba et al. 2012; Saxena et al. 2019; Rajaei et al. 2021): (1) N2 MA lines include 67 O1MA lines that were propagated for ∼250 generations, and 38 O2MA lines that were derived from eight selected O1MA lines with high and low fitness and were propagated for an additional ∼150 generations; and (2) PB306 (a wild strain) MA lines include 67 O1MA lines that were propagated for ∼250 generations. We called STR variants using the same methods as for wild strains. We identified 2956 pSTRs with missing calls in <10% of all 172 MA lines and their two ancestors (Supplemental Fig. S7; Supplemental Table S6). The pSTRs of MA lines showed similar composition and enrichment features as pSTRs of our 540 wild strains (Supplemental Fig. S7).
O1MA lines in both MA line panels have undergone passage for about 250 generations with minimal selection (Joyner-Matos et al. 2011; Matsuba et al. 2012; Saxena et al. 2019; Rajaei et al. 2021). To investigate STR mutations in MA lines, we calculated mutation rates for total mutations and three different mutations (deletions, insertions, and substitutions) between the ancestor and each O1MA line (ANC-O1MA) (see Methods; Fig. 4A–C; Supplemental Table S7). We found a significantly lower total mutation rate in O1MA lines derived from the N2 strain than from the PB306 strain (Wilcoxon test with P = 0.017) (Fig. 4A). Among different types of mutations, N2 O1MA lines showed significantly higher deletion rates but significantly lower substitution rates than PB306 O1MA lines, which were likely driven by mono-STRs (Fig. 4B; Supplemental Fig. S8; Supplemental Table S2). Within each of the two O1MA line panels, we found the highest mutation rates in substitutions (Fig. 4B; Supplemental Table S2). N2 O1MA lines showed significantly higher deletion rates than insertion rates, indicating more contractions than expansions, whereas PB306 O1MA lines showed significantly higher insertion rates than deletion rates (Fig. 4B; Supplemental Table S2). Altogether, these results suggested that genetic variation between the N2 strain and the PB306 strain might affect STR mutation rates and types. Furthermore, we again found that the CDS had significantly lower mutation rates than most other genomic features (Fig. 4C; Supplemental Table S2).
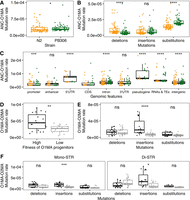
Mutation rates in mutation accumulation (MA) lines. (A,B) Comparison of total short tandem repeat (STR) mutation rates (A) and STR mutation rates of deletions, insertions, and substitutions (B) between O1MA lines derived from N2 (orange) and PB306 (green). (C) Comparison of STR mutation rates in coding sequence (CDS) regions and other regions using both N2 (orange) and PB306 (green) O1MA lines. (D–F) Comparisons of total STR mutation rates (D) and STR mutation rates of deletions, insertions, and substitutions using all pSTRs (E), or mono-STRs and di-STRs (F) between O2MA lines that were derived from N2 O1MA progenitors with high (black) and low (gray) fitness. Each dot represents the mutation rate between the ancestor strain (ANC) and one of O1MA lines (ANC-O1MA) or between one of the eight N2 O1MA lines and one of its derived O2MA lines (38 in total) (O1MA-O2MA). Statistical significance of difference comparisons (Supplemental Table S2) was calculated using the two-sided Wilcoxon test and P-values were adjusted for multiple comparisons (Bonferroni method). Significance of each comparison is shown above each comparison pair (ns: adjusted P > 0.05, [*] adjusted P ≤ 0.05, [**] adjusted P ≤ 0.01, [***] adjusted P ≤ 0.001, [****] adjusted P ≤ 0.0001).
Although minimal selection was maintained during propagation from the N2 ancestor to its O1MA derivatives, lines with consistently high and consistently low fitness at about 250 generations were selected as progenitors for O2MA lines (Matsuba et al. 2012; Saxena et al. 2019). These O2MA lines allowed us to explore how initial fitness (or the initial genomic load of spontaneous deleterious mutations) affects the mutation process of STRs. As for ancestors and O1MA lines, we calculated STR mutation rates between each O1MA line and its O2MA line (O1MA-O2MA) (Fig. 4D–F; Supplemental Table S7). We found a significantly higher total mutation rate in O2MA lines derived from high fitness O1MA lines than from low fitness O1MA lines (Wilcoxon test with P = 0.0056) (Fig. 4D). In contrast to ANC-O1MA, the difference in total mutation of O1MA-O2MA was primarily because of insertions rather than substitutions (Fig. 4B,E; Supplemental Table S2). The insertion rates in both mono-STRs and di-STRs were significantly higher in O2MA lines derived from high fitness O1MA lines than from low fitness O1MA lines (Fig. 4F; Supplemental Table S2), whereas deletion rates and substitution rates using all STRs, mono-STRs, and di-STRs showed no significant differences (Fig. 4E,F; Supplemental Table S2). Altogether, these results suggested that STR mutations might be fitness-dependent, in which high fitness O1MA lines accumulated more STR insertions than low fitness O1MA lines.
Impacts of pSTRs on phenotypic differences
We next investigated the impact of STR variation on phenotypic differences in wild C. elegans strains. We obtained phenotypic data of 11 traits, including lifetime fecundity, dauer formation, telomere length, and responses to eight different drugs or toxicants from ten previous C. elegans natural variation studies (Cook et al. 2016; Zdraljevic et al. 2017; Hahnel et al. 2018; Lee et al. 2019; Brady et al. 2019; Zdraljevic et al. 2019; Na et al. 2020; Evans et al. 2020, 2021b; Zhang et al. 2021). We performed genome-wide scanning between STR length variation and phenotypic variation using a likelihood-ratio test and the Bonferroni threshold (see Methods). Each phenotypic trait was tested for a median of 2495 pSTRs (ranging from 2271 to 3382) (Supplemental Table S8). We identified significant associations between STR variation and phenotypic differences in nine of the 11 traits (Fig. 5; Supplemental Table S8). A total of 202 pSTRs, with motif lengths of 1–6 bp, was linked to the nine traits, each of which was linked to one to 109 pSTRs (Fig. 5; Supplemental Table S8). Most of the significant pSTR peaks overlapped with quantitative trait loci (QTL) identified by SNV-based genome-wide association mappings in the original studies (Cook et al. 2016; Zdraljevic et al. 2017; Hahnel et al. 2018; Brady et al. 2019; Lee et al. 2019; Zdraljevic et al. 2019; Na et al. 2020; Zhang et al. 2021; Evans et al. 2021b), likely because of linkages between STRs and nearby SNVs. Furthermore, as expected, the effects of STR length variation on phenotypic variation were not always linear (Supplemental Fig. S9; Supplemental Table S9). Altogether, these results suggested that STR variation might contribute to phenotypic variation in C. elegans.
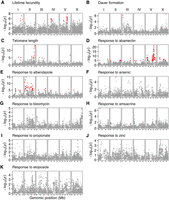
Genome-wide association of short tandem repeat (STR) length variation and phenotypic differences. Each point represents an STR that is plotted with its genomic position (x-axis) against its –log10(P) value (y-axis) in the likelihood-ratio tests for natural variation in C. elegans lifetime fecundity (A), dauer formation (B), telomere length (C), responses to abamectin (D), albendazole (E), arsenic (F), bleomycin (G), amsacrine (H), propionate (I), zinc (J), and etoposide (K). STRs with significant adjusted P-values using the Bonferroni threshold are colored red.
Discussion
Natural variation in C. elegans STR mutations
STRs have long been recognized as one of the most variable classes of genomic variation. The polymorphisms in few STRs have previously been studied in a limited number of C. elegans strains worldwide and in local populations (Sivasundar and Hey 2003; Barrière and Félix 2005, 2007; Haber et al. 2005). Here, we characterized the distribution of 31,991 STRs with motif lengths of 1–6 bp in the reference genome of C. elegans and identified 9691 polymorphic STRs across 540 genetically distinct wild strains. We found more STRs on chromosome arms than centers (Supplemental Fig. S1A), likely because recombination rates were higher on arms than centers (Rockman and Kruglyak 2009). Most pSTRs were multiallelic but had a predominant major allele (Fig. 3A,B), which might be caused by the self-fertilizing reproductive mode and deepened by the recent selective sweeps.
As previously shown in other species (Metzgar et al. 2000; Mirkin 2007), length variation caused by deletions or insertions was more common than substitutions among STR mutations in C. elegans (Fig. 2A). We found significantly more STR contraction than expansion (Fig. 2B,C) when we compared wild strain genomes to the reference genome. The reference strain N2 was isolated from Bristol, England and was identified as a swept strain (Andersen et al. 2012). To understand the evolution of STRs in C. elegans, a more informative comparison might come from choosing a reference from strains that avoided selective sweeps and was isolated from regions nearby the species origins. For example, the Hawaiian strain XZ1516, which likely carries the most ancestral genotypes (Crombie et al. 2019; Ma et al. 2021), has contraction in 66% of the 2400 pSTRs that showed length variation to the reference STRs. Therefore, STR expansion rather than contraction likely occurred from ancestors to descendants in C. elegans if we consider strains that might reflect the ancestral origin of the species.
Polymorphic STRs reflect species evolutionary history
The differences in SNV diversity across the genomes of wild strains revealed the species-wide selective sweeps and potential geographical origins of C. elegans (Andersen et al. 2012; Crombie et al. 2019, 2022; Lee et al. 2021). Our results in STR diversity across the C. elegans genome of the 540 wild strains agreed with previous discoveries using SNVs. STR diversity across the genome showed signatures of selective sweeps among the 357 swept strains (Fig. 3F) in similar genomic regions as previous results (Andersen et al. 2012). We found higher STR diversity in divergent strains than swept strains (Fig. 3B,C). The divergence in STRs across wild strains corresponded to their geographic locations as revealed by SNVs (Fig. 3D,E). Altogether, these results suggest natural variation in STRs reflects the evolutionary history of C. elegans. Because of the higher mutation rates of STRs than SNVs, exploring STR polymorphisms could further help to better resolve demography and short-scale genealogy in population genetic studies.
The impacts of selection on STR variation
The species-wide selective sweeps might have had significant influences on the STR diversity that we observed in the wild C. elegans strains (Fig. 3). Additionally, purifying selection might have constrained motif lengths and mutations of STRs in CDS regions to maintain proper functions in wild strains (Fig. 1D,E; Supplemental Figs. S4C,D, S6). We also observed constrained STRs in CDS regions of MA lines (Fig. 4C; Supplemental Fig. S7C,D), which in principle mostly experienced relaxed selection, indicating strong deleterious effects of STR variation on CDS functions. We found the highest mutation rates in substitutions of ANC-O1MA (Fig. 4B) and in insertions of O1MA-O2MA (Fig. 4E), which might be related to their different mutation loads in the progenitors, because the growth environment from the ancestor to O1MA and from O1MA to O2MA was essentially identical (Matsuba et al. 2012; Saxena et al. 2019). It would be interesting to investigate the mutation pattern in a narrow time range, for example, each 50 generations, to examine if mutation types and rates are associated with the load of mutations accumulated in the background during the spontaneous mutational process.
Among O2MA lines, we also found fitness-dependent STR mutations (Fig. 4D–F). O2MA lines derived from high fitness O1MA lines showed significantly higher insertion rates than those derived from low fitness O1MA lines (Fig. 4E,F). The original study found the short indel mutation rate was significantly greater in the high fitness lines than in the low fitness lines (Saxena et al. 2019). The authors proposed that high fitness lines might have higher tolerance than low fitness lines to harbor more indels because of synergistic epistasis (Saxena et al. 2019), which might also explain the fitness-dependent STR mutation that we observed here. Expansion could decrease the stability of STRs and has been widely associated with human disease and trait defects (Mirkin 2007; Sureshkumar et al. 2009). Assuming expanded STRs are more likely to have deleterious effects on fitness than contracted STRs, high fitness MA lines might be able to accumulate more expanded STRs than low fitness lines before being removed by selection. Future efforts should measure the fitness of O2MA lines and examine the correlation between STR mutation rates and fitness.
Methods
C. elegans genotype data
We obtained the reference genome of C. elegans from WormBase (WS276) (Harris et al. 2020) and alignment of whole-genome sequence data in the BAM format of 540 wild C. elegans strains from CeNDR (20210121 release) (Andersen et al. 2012; Cook et al. 2017; Evans et al. 2021a). These BAM files were generated using BWA (Li and Durbin 2009) incorporated in the pipeline alignment-nf (https://github.com/AndersenLab/alignment-nf) (Cook et al. 2017). We also acquired the hard-filtered isotype variant call format (VCF) file (CeNDR 20210121 release) for SNVs among the 540 wild C. elegans strains (Cook et al. 2017).
STR variant calling
We built an STR reference from the C. elegans reference genome using Tandem repeats finder (Benson 1999) and the STR reference construction framework described in HipSTR-references (https://github.com/HipSTR-Tool/HipSTR-references) (Willems et al. 2017). Then, we called STR variants using BAM files of the 540 strains, the STR reference, and HipSTR (v0.6.2) in the de novo stutter estimation mode (Willems et al. 2017). We filtered the VCF of HipSTR output using the script filter_vcf.py as recommended in HipSTR to have high-quality calls. In total, we found variation in 27,667 STRs among the 540 strains. We further filtered STR variants with equal or more than 10% missing data across all strains using BCFtools (v.1.9) (Li 2011) and came to 9691 polymorphic STRs, which we used in downstream analyses unless otherwise specified.
Composition of reference STRs
We used STR motif sequences estimated by Tandem repeats finder and STR reference genotypes in the VCF called by HipSTR to analyze composition of reference STRs for the 27,667 STRs with polymorphisms. We categorized STRs into six groups: simple-perfect [e.g., “(A)n” “(GGT)n”], which showed perfect repeats of single motif sequences; simple-center-perfect [e.g., “T(A)n” “C(GGT)nAA”], which was simple-perfect in the center but with flanking nucleotide(s); simple-interrupted [e.g., “TTG(A)nG(A)n” “CT(GGT)n T(GGT)nTT(GGT)n”], the repeats of which were interrupted at least once; compound-perfect [e.g., “(T)n(A)n”], which showed perfect repeats of several motif sequences; compound-center-perfect [e.g., “(AGGCG)n(AGGTT)nAG”]; and compound-interrupted [e.g., “(ACG)nAC(AAC)nAA”].
STR annotation and effect prediction
We determined genomic regions of reference STRs according to the general feature format (GFF3) file from WormBase (WS276) (Harris et al. 2020) and prediction of promoters and enhancers (Jänes et al. 2018). STRs with multiple annotated features were assigned to a single feature using the following priority: CDS > 5′UTR > 3′UTR > promoter > enhancer > intron > RNAs & TEs > intergenic regions. We also assigned each STR to the Watson strand or the Crick strand based on their assigned features. STRs that were assigned to the same features on both strands (e.g., introns of two genes on different strands) were assigned to the Watson strand. STRs in intergenic regions were assigned to the Watson strand. In analyses using motif sequences (Figs. 1B,E, 2E; Supplemental Figs. S4A,D, S5, S7A,D), we used the reverse complement sequences of the motif sequences for STRs that were assigned to the Crick strand.
Expansion and contraction
For each polymorphic STR with expanded and/or contracted alternative alleles, we calculated the expansion score = [max(STR length) – median(STR length)]/median(STR length) (Press et al. 2018) and/or the contraction score = [min(STR length) – median(STR length)]/median(STR length). We also compared the number of mapped sequencing reads between expanded alleles and contracted alleles. We focused on homozygous alleles and used the number of mapped reads for a sample's genotype for each variant (the “DP” column in the VCF called by HipSTR). We normalized “DP” for each variant of each sample by the reference allele length and the total number of reads that mapped to all pSTRs of each sample. Then, we compared the normalized number of reads between expanded alleles and contracted alleles.
Classification of swept and divergent strains
We acquired the sweep haplotype summary data of the 540 wild C. elegans strains from CeNDR (20210121 release) (Cook et al. 2017). We defined strains with greater than or equal to 30% of swept haplotype in any of the four chromosomes (I, IV, V, and X) as swept strains. Other strains were defined as divergent strains.
Principal components analysis (PCA)
For STRs, because only eight polymorphic STRs have no missing data for all 540 strains, we imputed the genotype of the 9691 polymorphic STRs for strains with missing data. For strains with homozygous alleles (e.g., “0|0”, “1|1”, “2|2”), a single character (e.g., “0”, “1”, “2”) was used to represent the genotype. For strains with heterozygous alleles (e.g., “0|1”, “1|2”, “3|2”), we treated the genotypes as numeric values and chose the smaller one as the genotype (e.g., “0”, “1”, “2”). Then we imputed missing genotypes using the R package missMDA (v1.18) (Josse and Husson 2016). For SNVs, we used the hard-filtered isotype VCF (CeNDR 20210121 release) and used BCFtools (Li 2011) to filter SNVs that had any missing genotype calls and those that were below the 5% minor allele frequency. We used PLINK v1.9 (Purcell et al. 2007; Chang et al. 2015) to prune the SNVs to 13,650 markers with a linkage disequilibrium threshold of r2 < 0.8. We further filtered the 13,650 markers to 13,580 markers with homozygous alleles among the 540 strains. Then, we used the generic function prcomp() in R (R Core Team 2013) to perform principal components analysis for both STRs and SNVs.
STR diversity
We calculated expected heterozygosity (HE) (Nei 1973) for STR diversity using the following equation:
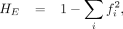 where the fi denotes the allele frequency of the ith allele for a specific STR. We calculated the HE for each of the 9691 pSTRs among all strains. We also selected 6976 and 9269 pSTRs that showed variation among swept strains
and divergent strains, respectively, and calculated HE for each pSTR within each group of strains. We further calculated HE using 195,993 SNVs among swept strains. These SNVs were obtained using the same VCF and method described in the PCA but without
filtering by the minor allele frequency of 0.05.
where the fi denotes the allele frequency of the ith allele for a specific STR. We calculated the HE for each of the 9691 pSTRs among all strains. We also selected 6976 and 9269 pSTRs that showed variation among swept strains
and divergent strains, respectively, and calculated HE for each pSTR within each group of strains. We further calculated HE using 195,993 SNVs among swept strains. These SNVs were obtained using the same VCF and method described in the PCA but without
filtering by the minor allele frequency of 0.05.
STR variants in mutation accumulation (MA) lines
We obtained whole-genome sequence data in the FASTQ format of 174 MA lines, including N2 MA lines: the N2 ancestor, 67 O1MA lines, and 38 O2MA lines; PB306 MA lines: the PB306 ancestor and 67 O1MA lines (NCBI BioProject database [https://www.ncbi.nlm.nih.gov/bioproject/] under accession numbers PRJNA395568, PRJNA429972, and PRJNA665851) (Saxena et al. 2019; Rajaei et al. 2021). We used the pipelines trim-fq-nf (https://github.com/AndersenLab/trim-fq-nf) and alignment-nf to trim raw FASTQ files and generate BAM files for each line, respectively. We called STR variants for the 174 lines as described above and identified 2956 pSTRs with missing calls in <10% of all strains.
Mutation rate of polymorphic STRs in MA lines
We calculated the STR mutation rate in MA lines using their 2956 pSTRs. For each O1MA line and the ancestor, we selected STR sites with data in both lines. Then, we compared the two alleles of each STR in the O1MA line to the two alleles in the ancestor, respectively, to identify insertion, deletion, substitution, or no mutation. If the genotypes of the ancestor and its derived line were different in length, we defined it as a “deletion” (derived line < ancestor) or an “insertion” (derived line > ancestor). If the genotypes of the ancestor and its derived line were same in length but different in sequences, we defined it as a “substitution.” Otherwise, we defined it as “no mutation.” Note that alleles defined as “deletion” or “insertion” might also have substitution(s). We did not limit the number of substitutions per STR locus. We also did not differentiate an STR locus with a single nucleotide substitution or with multiple substitutions. We further obtained the number of generations between the O1MA line and the ancestor from the original studies (Saxena et al. 2019; Rajaei et al. 2021). The mutation rate (per allele, per STR, per generation) µ for each type of mutation was calculated as m/2nt, where m is the number of the mutation, n is the total number of STR sites between the two lines, and t is the number of generations. We calculated the mutation rate of the three different mutations for each N2 O2MA line to its ancestral N2 O1MA line using the same method.
Identification of pSTRs underlying phenotypic differences
For phenotypic traits, we obtained 11 different phenotype data from ten previous C. elegans natural variation studies (Cook et al. 2016; Zdraljevic et al. 2017; Hahnel et al. 2018; Brady et al. 2019; Lee et al. 2019; Zdraljevic et al. 2019; Evans et al. 2020, 2021b; Na et al. 2020; Zhang et al. 2021). For STR variation, we calculated the mean allele length of the two copies of each pSTR for each strain. Then, for each of the 11 phenotype data, we selected pSTRs that had at least two common variants (frequency of a certain mean allele length > 0.05) among strains with both STR and the phenotypic data, and only retained strains with the common STR variants and the phenotypic data. We treated STR lengths as factorial variables and performed likelihood-ratio tests on two models, the full model lm(phenotype ∼ STR) and the reduced model lm(phenotype ∼ 1), using the lrtest() function in the R package lmtest (v0.9-39) (https://cran.r-project.org/web/packages/lmtest/index.html). Statistical significance was corrected using the Bonferroni threshold.
Statistical analysis
Statistical significance of difference comparisons was calculated using the Wilcoxon test and P-values were adjusted for multiple comparisons (Bonferroni method) using the compare_means() function in the R package ggpubr (v0.2.4) (https://github.com/kassambara/ggpubr/). Enrichment analyses were performed using the one-sided Fisher's exact test and were corrected for multiple comparisons (Bonferroni method).
Data access
The data set and code for STR variant calling and generating all figures can be found at GitHub (https://github.com/AndersenLab/WI-Ce-STRs) and as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
We thank Timothy A. Crombie and Ryan J. McKeown for helpful comments on the manuscript. We also thank WormBase because without it these analyses would not have been possible. G.Z. is supported by the National Science Foundation–Simons Center for Quantitative Biology at Northwestern University (awards Simons Foundation/Simons Foundation Autism Research Initiative 597491-RWC and the National Science Foundation 1764421). Y.W. was supported as a joint PhD student by China Scholarship Council (No. 201706910052). E.C.A. is supported by a Grant from the National Institutes of Health R01 DK115690. The C. elegans Natural Diversity Resource is supported by a National Science Foundation Living Collections Award to E.C.A. (1930382).
Author contributions: E.C.A. conceived of and designed the study. G.Z. and Y.W. analyzed the data. G.Z., Y.W., and E.C.A. wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.277067.122.
-
Freely available online through the Genome Research Open Access option.
- Received June 27, 2022.
- Accepted September 21, 2022.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








