
Long-read assembly and comparative evidence-based reanalysis of Cryptosporidium genome sequences reveal expanded transporter repertoire and duplication of entire chromosome ends including subtelomeric regions
- Rodrigo P. Baptista1,2,
- Yiran Li2,
- Adam Sateriale3,
- Mandy J. Sanders4,
- Karen L. Brooks4,
- Alan Tracey4,
- Brendan R.E. Ansell5,
- Aaron R. Jex5,
- Garrett W. Cooper6,
- Ethan D. Smith6,
- Rui Xiao2,
- Jennifer E. Dumaine3,
- Peter Georgeson6,7,8,
- Bernard J. Pope6,7,9,10,
- Matthew Berriman4,
- Boris Striepen3,
- James A. Cotton4 and
- Jessica C. Kissinger1,2,11
- 1Center for Tropical and Emerging Global Diseases, University of Georgia, Athens, Georgia 30602, USA;
- 2Institute of Bioinformatics, University of Georgia, Athens, Georgia 30602, USA;
- 3Department of Pathology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA;
- 4The Wellcome Sanger Institute, Hinxton, CB10 1SA, United Kingdom;
- 5Faculty of Veterinary and Agricultural Sciences, The University of Melbourne and Population Health and Immunity Division, The Walter and Eliza Hall Institute of Medical Research, Parkville 3052, Australia;
- 6Department of Clinical Pathology, The University of Melbourne, Victorian Comprehensive Cancer Centre, Melbourne VIC 3000, Australia;
- 7Melbourne Bioinformatics, The University of Melbourne, Parkville VIC 3010, Australia;
- 8University of Melbourne Centre for Cancer Research, Victorian Comprehensive Cancer Centre, Melbourne VIC 3000, Australia;
- 9Department of Surgery (Royal Melbourne Hospital), Melbourne Medical School, Faculty of Medicine, Dentistry and Health Sciences, The University of Melbourne, Melbourne 3010, Australia;
- 10Department of Medicine, Central Clinical School, Faculty of Medicine Nursing and Health Sciences, Monash University, Melbourne 3004, Australia;
- 11Department of Genetics, University of Georgia, Athens, Georgia 30602, USA
Abstract
Cryptosporidiosis is a leading cause of waterborne diarrheal disease globally and an important contributor to mortality in infants and the immunosuppressed. Despite its importance, the Cryptosporidium community has only had access to a good, but incomplete, Cryptosporidium parvum IOWA reference genome sequence. Incomplete reference sequences hamper annotation, experimental design, and interpretation. We have generated a new C. parvum IOWA genome assembly supported by Pacific Biosciences (PacBio) and Oxford Nanopore long-read technologies and a new comparative and consistent genome annotation for three closely related species: C. parvum, Cryptosporidium hominis, and Cryptosporidium tyzzeri. We made 1926 C. parvum annotation updates based on experimental evidence. They include new transporters, ncRNAs, introns, and altered gene structures. The new assembly and annotation revealed a complete Dnmt2 methylase ortholog. Comparative annotation between C. parvum, C. hominis, and C. tyzzeri revealed that most “missing” orthologs are found, suggesting that the biological differences between the species must result from gene copy number variation, differences in gene regulation, and single-nucleotide variants (SNVs). Using the new assembly and annotation as reference, 190 genes are identified as evolving under positive selection, including many not detected previously. The new C. parvum IOWA reference genome assembly is larger, gap free, and lacks ambiguous bases. This chromosomal assembly recovers all 16 chromosome ends, 13 of which are contiguously assembled. The three remaining chromosome ends are provisionally placed. These ends represent duplication of entire chromosome ends including subtelomeric regions revealing a new level of genome plasticity that will both inform and impact future research.
Cryptosporidium spp. are parasitic apicomplexans that cause moderate-to-severe diarrhea in humans and animals. Studies have revealed that Cryptosporidium is one of the most common causes of waterborne disease in humans and the second leading cause of diarrheal etiology in children <2 yr (Kotloff et al. 2013; GBD Diarrhoeal Diseases Collaborators 2017). In 2016, acute infections caused more than 48,000 global deaths and more than 4.2 million disability-adjusted life years lost (Khalil et al. 2018).
Currently, 38 species of Cryptosporidium are recognized (Šlapeta 2013; Feng et al. 2018). Most species have preferred hosts, and hosts range from fish to mammals. Fifteen species have an assembled genome sequence, however, only eight are annotated (Supplemental Table S1). Most genomic sequence data are from the zoonotic C. parvum and anthroponotic C. hominis, the species primarily detected in humans (Chalmers et al. 2011; Zahedi et al. 2016; Khan et al. 2018). These two species are only 3%–5% divergent at the DNA level (Mazurie et al. 2013). Cryptosporidium genome sequences are shorter than most other apicomplexans at around 9 Mbp distributed over eight chromosomes and containing less than 4000 protein-coding genes in the species examined. Most genome reduction consists of gene and intron loss, intron shortening, and very short intergenic regions (Abrahamsen et al. 2004; Xu et al. 2004; Kissinger and DeBarry 2011).
As the Cryptosporidium field is exploding with newfound interest and much needed breakthroughs in genetics and culturing (Vinayak et al. 2015; Morada et al. 2016; DeCicco RePass et al. 2017; Heo et al. 2018; Wilke et al. 2019), the limitations of existing reference genome sequences need to be addressed. The C. parvum IOWA II reference genome sequence, (CpIRef), was assembled with a limited physical map (Abrahamsen et al. 2004) and a few hundred ESTs for training gene finders. Genomic, transcriptomic, and proteomic work has been lacking owing to the obligate quasi-intracellular nature of portions of the parasite's life cycle, the historical lack of a continuous in vitro tissue culture system, the parasite's small size relative to host cells, and difficult animal models. The physical map for the CpIRef assembly was generated using a genome-wide HAPPilly anchored physical mapping technique (Piper et al. 1998; Bankier et al. 2003). Despite the cutting-edge approaches, some regions, especially chromosome ends, lacked support or were poorly resolved. Subsequent whole-genome sequencing data remain unassembled or in a large number of contigs.
In 2015, the CpIRef was reannotated using new RNA-seq evidence, and a new C. hominis sequence from a recent human isolate (UdeA01) was generated (Isaza et al. 2015). Many ambiguities in gene models were improved, but the new C. hominis UdeA01 genome sequence is fragmented. Incomplete, misassembled (i.e., gapped sequence, indels, frameshifts, compressed repetitive regions, artifactual inversions), and independently annotated reference genome sequences as discussed in Guo et al. (2015) can mislead analyses of the differences between isolates and species owing to these artifacts rather than the biology. Comparative analyses require additional assays to confirm indels and copy number variations (CNVs). Because incomplete and misassembled sequences are usually caused by repetitive and complex sequence regions, it is imperative to revisit reference genome sequences with new long-read technologies.
Long-read sequence technologies (Pacific Biosciences [PacBio] and Oxford Nanopore Technologies [ONT]) are becoming an essential tool to close full genome sequence assemblies across the tree of life (Vembar et al. 2016; Berna et al. 2018; Miga et al. 2020). They can be used to resolve complex regions such as repetitive content; structural variants (SVs) such as inversions, translocations, and duplications; or for use as scaffolding evidence for existing fragmented genome assemblies (Mahmoud et al. 2019). They are proving crucial for completing pathogen genome sequences that are often riddled with large virulence-related gene families that may have been improperly assembled in shorter-read assemblies (Xia et al. 2021). Here, we provide a new de novo hybrid long-read assembly for C. parvum strain IOWA (CpIA), and new consistent comparative genome annotations for CpIA, C. hominis 30976 (Ch30976), and C. tyzzeri UGA55 (CtUGA55). The new data were used to assess genome-level species differences and assess rapidly evolving genes.
Results
An improved long-read genome assembly for C. parvum (IOWA-ATCC)
The CpIRef genome assembly, generated in 2004, has only 10 physical gaps of unknown size, but it has 18,558 ambiguous bases and is missing six telomeres. Alignment of 54,882,187 Illumina 100-bp paired-end reads (Supplemental Fig. S1) to this reference sequence revealed many regions that had become collapsed during assembly (Supplemental Table S2). To resolve these issues, we generated a new PacBio + Illumina + Nanopore hybrid genome assembly (Table 1; Supplemental Fig. S1) for the C. parvum strain IOWA (ATCCPRA-67DQ), CpIA. To minimize strain variation differences, we performed our analysis on the IOWA strain. However, because there is a 14-yr window of propagation between these two isolates, and cryopreservation has only recently been developed (Jaskiewicz et al. 2018), we modified the strain name to IOWA-ATCC (CpIA).
Comparative Cryptosporidium genome assembly statistics
The new CpIA genome assembly is compared to the current CpIRef sequence and two closely related species with different host preferences and pathogenicity, C. hominis (Ch30976) and C. tyzzeri (CtUGA55) (Table 1; Šlapeta 2013; Nader et al. 2019; Sateriale et al. 2019). These particular assemblies were selected because they are the best available. The new CpIA long-read assembly increases the genome size by 19,939 bases (∼152 kb when including new proposed subtelomeric regions) and putatively identifies all 16 telomeres. There are no gaps and no ambiguous bases. As expected, the CpIA genome sequence has diverged slightly but shares 99.93% average pairwise identity with the 2004 assembly in regions that are comparable (Supplemental Table S3). The main Cryptosporidium subtyping marker, the 60 kDa surface protein (gp60 locus subtype IIa), shows four amino acid differences (two in the serine repeat region) between CpIA and CpIRef (Supplemental Fig. S2; Supplemental Methods).
Structural differences between the C. parvum IOWA assemblies are confirmed
The 2004 CpIRef genome assembly used Sanger reads combined with available HAPPY-map data to scaffold the contigs. We compared the CpIRef and CpIA assemblies to identify potential rearrangements. Inversions and relocations were detected in Chromosomes 2, 4, and 5 (Fig. 1A). These inversions may be previous assembly artifacts or represent genuine differences between the isolates. We investigated the synteny between CpIRef, CpIA, Ch30976, and CtUGA55 and observed that C. hominis and C. tyzzeri also share the Chr 4 and Chr 5 inversions. Examination of the inverted region boundaries in CpIRef revealed regions of ambiguous nucleotide bases or physical gaps (Fig. 1A). To further investigate, PCR primers were designed to test each possible inversion arrangement in genomic DNA from C. parvum KSU-1 strain 2006 and Bunch Grass farms IOWA (CpBGF) (Fig. 1B; Supplemental Fig. S3; Supplemental Table S4). The results support the revised assembly orientation. Long-read ONT data also support the CpIA assembly (Supplemental Fig. S4). Better assemblies for the other species will be needed to determine the true level of synteny across these species.
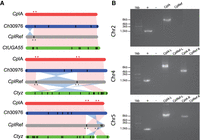
Syntenic relationships between select Cryptosporidium chromosome assemblies. (A) Synteny between Chromosomes 2, 4, and 5. Vertical black lines within a chromosome represent known physical gaps. Synteny between chromosomes is shown in pink and inversions in blue. (B) PCR validation using C. parvum KSU-1 DNA (Supplemental Table S4). Lanes 1, 2, and 3 in all gels are 1-kb ladder, positive control Dnmt2 gene, and no template control, respectively. The remaining lanes test each orientation of the left (L) and right (R) inversion boundaries. Red stars indicate the location of primers designed based on the CpIA assembly, and gray stars indicate the same on the CpIRef assembly.
Consistent structural gene annotation resolves inconsistencies and improves functional annotation
We consistently annotated and compared CpIRef, CpIA, Ch30976, and CtUGA55 which have >95% genome identity to assess differences in gene content. The new annotation for each species was generated with three de novo approaches and evidence-based manual annotation. Curation of the annotation was performed pairwise between each assembly to take full advantage of syntenic regions. Data from one species could be used to assess computational predictions in others. Using this approach, fragments of genes that were previously missing in C. hominis were identified. This approach resulted in 1926 gene alterations in CpIA when compared to CpIRef, resulting in improved functional annotation. These changes increase the overall number of predicted genes, introns (100% supported by RNA-seq data), and exons (Table 2; Supplemental Results). The average mRNA length increased. These structural fixes led to the repair of the N terminus of the methylase ortholog, Dnmt2 (Supplemental Fig. S5) as well as 523 other genes and 113 fragmented genes previously annotated as pseudogenes.
Reannotation summary statistics
Cryptosporidium has a very compact genome sequence with 76.88% covered by protein-coding sequences (CDS). As a result, RNA-seq data, which is the best evidence for annotation, contains reads that overlap adjacent genes creating false fusions of exons belonging to different genes. Available strand-specific RNA-seq was used to characterize some of these regions, but expression data were not available for all predicted genes (87% of the annotated genes were covered); thus, genes of unknown function in close proximity on the same strand remain problematic. The expression data also revealed three putative alternative spliced genes (CPATCC_0027530; CPATCC0027960; CPATCC_0035590) and 474 potential noncoding RNAs (ncRNAs), predominantly antisense lncRNAs with differential expression as reported (Li et al. 2021).
Comparative analysis reveals few gene content differences between closely related Cryptosporidium species
There is a cluster of species, C. parvum, C. hominis, C. tyzzeri, C. meleagridis, and C. ubiquitum that are highly syntenic relative to species outside of this cluster. The syntenic species are biologically distinct and largely host-adapted with the main zoonotic exceptions being C. parvum and C. ubiquitum. A synteny analysis of the clustered species and Cryptosporidium muris as an outgroup reveals high synteny (99.4%–87%) within the cluster and only 4% synteny to C. muris (Supplemental Fig. S6; Supplemental Tables S5, S14).
The consistent annotation of the species closest to CpIA (GCA_015245375.1), Ch30976 (GCA_001483515.1), and CtUGA55 (GCA_007210665.1) permitted the analysis of differences in CDS content and CNVs. Orthology analysis revealed that 94% of the genes were conserved among all species. Of the 4008 ortholog groups identified, most gene families were maintained with a similar number of paralogs (max = 6) detected in the same ortholog group, but variation was detected among singletons (Fig. 2A; Supplemental Table S6). Some of these gene differences appear to be unique to a particular species (Supplemental Table S7). Of the 224 singletons detected, we observed only 0, 1, and 1 potential truly species-specific genes in CpIA, Ch30976, and CtUGA55, respectively, following manual inspection (Fig. 2B,D). Both species-specific genes are uncharacterized proteins. The remaining 253 singletons are detected but incomplete in the fragmented assemblies of Ch30976 and CtUGA55, appearing as split genes, frameshifts, missed calls near a gap and missing subtelomeric regions, or contig break and putative false gene predictions in small contigs (Fig. 2C). The major protein-coding gene content differences between these species are gene copy number variations and not gene presence or absence.
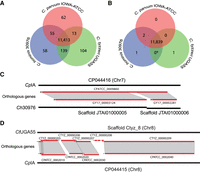
Ortholog distribution of protein-coding genes reveals few differences between the species. (A) Venn diagram of automated protein orthology assignment between CpIA, Ch30976, and CtUGA55. (B) Venn diagram of the same orthologous genes following manual investigation and removal of putative false positives. (*) The 139 genes shared between C. hominis and C. tyzzeri in A are in complex regions with repeats and gaps and do not have enough evidence to prove their uniqueness at this stage (Supplemental Table S7). (C) Example of a false positive paralog count caused by gene fragments on different scaffolds. (D) Putative unique uncharacterized gene found on Chr 8 in CtUGA55.
To identify and assess putatively overly collapsed repetitive regions within the genome assemblies analyzed in this study, that is, repetitive regions represented by only a single repeat in the assembly, we mapped Illumina reads from CpIA to the new CpIA, CpIRef, Ch30976, and CtUGA55 genome assemblies (Supplemental Table S2; Supplemental Fig. S1). Our pipeline detected 12 compressions of at least 2× read depth and >100 bp in length in the CpIRef genome assembly compared to six in the new CpIA assembly. The six compressed regions drop to four if the three putative new subtelomeric regions proposed in this study are included (see below). The Ch30976 and CtUGA55 genome assemblies contain more than 20 compressions mostly attributed to the short reads used to generate these assemblies. The CpIA collapsed regions have two hits in regions with gene annotations in Chr 1 and Chr 2. Both genic regions are composed of rRNA genes, some uncharacterized proteins, GMP synthase, aspartate-ammonia ligase, tryptophan synthase beta, and MEDLE genes, all associated with complex subtelomeric regions discussed below. The four intergenic compressions all match small simple repeat regions (Supplemental Table S8).
Functional annotation identifies new protein features
Several approaches to assess function were applied including InterProScan and I-TASSER among others (Methods). As a result, 138 new C. parvum protein annotations were generated or modified. The percentage of CpIA genes annotated as uncharacterized proteins was reduced from 40% to 33% in all reannotated sequences (Supplemental Table S9). Many new features including domain and repeat content were added to 738 previously uncharacterized proteins. In addition, 729 predicted CpIA CDSs have signal peptides, and 1990 have GO assignments (Supplemental Results). Using I-TASSER protein structure searches, 1414 CDSs were further assessed for confidence, and 1008 predicted structures were assigned as high-confidence by random forest categorization. In CpIRef, 143 previously uncharacterized proteins were assigned high-confidence GO terms.
New transporter genes are identified
We further characterized transporter genes using three different prediction methods. A total of 152 proteins in CpIA and Ch30976 were identified as transporters, including 128 confident candidates and 24 putative candidates (Supplemental Table S10). This represents an increase of 53 transporters relative to the CpIRef GO annotation (CryptoDB v36) (Heiges et al. 2006). Most identifiable transporters are related to purine metabolism, peptidoglycan biosynthesis, oxidative phosphorylation, and N-Glycan biosynthesis pathways (Fig. 3). Six translocases were also identified.

CpIA assembly and annotation reveal new transporters. The numbers of transporters correspond to the counts of genes encoding each type of transporter protein: (ABC) ATP-binding cassette transporter; (MFS) major facilitator superfamily; (DMT) divalent metal transporter; (AAAP) amino acid/auxin permease; (MC) mitochondrial carrier; (ZIP) zinc transporter protein; (CPA) cation/proton antiporter; (SulP) sulfate transporter; and (PUP) purine permeases.
Entire subtelomeric regions are duplicated
As shown in the read depth coverage analysis and in Table 1, Supplemental Table S2, and Supplemental Figure S1, the new CpIA assembly was able to recover ∼2.3 kb cumulative length in collapsed regions relative to CpIRef. One subtelomeric region on Chr 1 in CpIA, previously reported on Chr 5 in CpIRef (but not linked to Chr 5 in the HAPPY map), still shows signs of sequence compression suggesting that most of the genes present in this region have more than one copy (Fig. 4A; Supplemental Fig. S7). This region reveals at least 13 genes that vary in copy number between different Cryptosporidium species (Supplemental Fig. S8). The genes contained in this region are 18S rRNA, 5S rRNA, and 28S rRNA; uncharacterized proteins; a GMP synthase; an aspartate-ammonia ligase; tryptophan synthase beta; and a cluster of several MEDLE genes. Some of these genes, such as the tryptophan synthase beta and the MEDLE's are the focus of considerable research because they may be related to parasite survival and are potentially involved in parasite invasion, respectively (Sateriale and Striepen 2016; Li et al. 2017; Fei et al. 2018). The predicted number of copies of rRNAs and MEDLE's are underrepresented because they also have paralogs on Chr 2 and Chr 5, respectively. The Illumina pileup of ∼1350 reads on Chr 2, positions 681,607 to 686,953 (Supplemental Table S2; Supplemental Fig. S1) is the region where the 18S/28S rRNA gene(s) are located on this chromosome. The five 18S genes are identical, and 28S rRNAs have three gaps (Supplemental Fig. S9). Thus, alignment competition explains why the read coverage varies relative to the equivalent 18S/28S rRNA Chr 1 pileups. Regions with pileups on inner portions of Chr 5, 7, and 8 are low complexity regions composed by tandem repeats (Supplemental Table S8). In Chr 5, we have one uncharacterized protein (CPATCC_0023030), full of tandem repeats and good RNA-seq support for its expression. On Chr 7 and 8, these regions are smaller than 100 bp and do not contain any annotated genes.
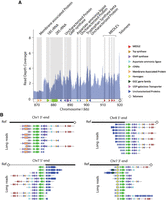
Resolution of repetitive subtelomeric regions found on Chr 1 identifies missing telomeres on Chromosomes 7 and 8. (A) Illumina reads from CpIA are mapped to the CpIA Chr 1 long-read assembly subtelomeric region to identify read pileups and estimates of sequence copy number by normalizing against the average genomics Illumina read depth. Vertical gray areas indicate regions with annotated genes. Annotated genes are represented below the shaded regions, the 5.8S rRNA is present but not indicated. (B) Subtelomeric variaton observed on different CpIA chromosomes is supported by CpBGF ONT long reads. Individual ONT long reads provide evidence of at least four different yet related subtelomeric regions that extend into the chromosomes that were missing telomeres in CpIA (Chr 7 and Chr 8) in addition to Chr 1. The white and black reference bar above each collection of annotated ONT reads identify the resolved subtelomeric regions (white) and linkage to existing assembly (black). The penultimate read on the Chr 7 3′ end panel indicates a unique region of insertion (nucleotide positions 1,191,705–1,217,462). This region contains mostly uncharacterized proteins and two transferases. Each ONT read is annotated as indicated in the key shown in A.
Because there is an apparent compression in a subtelomeric region assembly with no gaps and good PacBio long-read coverage, we hypothesized that these extra copies might derive from additional copies of this region. The CpIA assembly was only missing three telomeric regions, both ends of Chr 7 and one telomere of Chr 8. Using existing PacBio long reads we were able to identify a few reads that extended into rRNA regions on the chromosomes missing telomeres. We attempted reassembly with only PacBio reads and we could not resolve the missing regions. Thus, we generated very deep (2260×) ONT single-molecule reads from CpBGF, (ATCC DNA was not available, only 143 SNVs are detected between the strains, of which 108 are indels) (Supplemental Table S11). The ONT reads revealed related, yet unique subtelomeric regions linked to the chromosomes missing their telomeres, in addition to Chr 1 (Fig. 4B). We found good ONT long-read support for these regions (Supplemental Fig. S7). Each distinct subtelomeric region begins with chromosome-specific sequences followed by a conserved ribosomal RNA cluster that is followed by the duplicated subtelomeric region and telomere. There are many ONT and PacBio reads that link the unique chromosomal regions and the beginning of the subtelomeric gene families but only a few span the entire chromosome end. We also note that there is slight variation observed among the reads for each subtelomeric region distal to the rRNA cluster.
New positively selected genes are identified in C. parvum
The new gapless CpIA genome assembly and annotation presented an opportunity to revisit the prediction of genes evolving under positive selection in this species. We performed a single-nucleotide variant (SNV) analysis using 136 different C. parvum WGS data sets obtained from the NCBI GenBank database (https://www.ncbi.nlm.nih.gov/genbank/) (Supplemental Table S12) using the new CpIA assembly and annotation. A total of 24,407 positions were found to contain at least one high-confidence biallelic variant. Multiallelic calls were removed to guard against mixed infections. The biallelic variants reflect 3892 genes, 342 of which show a πN/πS ratio of nonsynonymous/synonymous rates of >1.5 (Supplemental Table S13). Of the 342, 17 genes were previously identified and 145 are classified as uncharacterized proteins, 105 of which are annotated as having a signal peptide or being secreted. All previously identified genes evolving under positive selection were detected, including Insulinase-like protein (CPATCC_0017080), an uncharacterized secreted protein (CPATCC_0010380), gp60 (CPATCC_0012540), and others (Strong et al. 2000; Sanderson et al. 2008; Nader et al. 2019; Zhang et al. 2019). Of the top 10 genes by πN/πS ratio, nine appear to be new to this study. Gene family members such as MEDLEs, FLGN, and SKSR were also detected, but new members of each of these families are identified as also evolving under positive selection. Because the putative new subtelomeric repeats (Fig. 4) were not included in these analyses (they were identified in a different strain), evolution of the MEDLE genes may be an overestimation. A family of WYLE proteins (Sanderson et al. 2008) is also identified as being positively selected.
Discussion
The first genome sequence assembly of C. parvum IOWA II, referred to as CpIRef here, was excellent given the technology at the time. As a result, the community has relied on this genome assembly and annotation to this day to design their experiments. However, gaps and ambiguous bases remain, and there was little available expression and orthology evidence at the time to facilitate the annotation. We used PacBio and Illumina sequencing technologies to generate a new complete genome assembly of C. parvum strain IOWA-ATCC. We then applied de novo computational and evidence-based annotation approaches with manual curation of two additional species to generate consistent annotation that can be used to detect differences between species and strains. CpIA DNA was not available for Nanopore sequencing or PCR validation of the assembly, so CpBGF DNA (which differs by fewer than 200 SNVs) was used instead. However, all results are consistent when strains can be compared; for example, compressions of CpIA Chr 1 detected with CpIA Illumina reads are the same when CpBGF is used, lending strength to the broader applicability of the findings.
The first expected finding was that the C. parvum IOWA strain is continuing to evolve (Cama et al. 2006) as it is maintained by passage through cattle in a few different locations for research use. Some natural Cryptosporidium isolates have been propagated in unnatural hosts before sequencing. Thus, potential selection during the move to a non-natural host and subsequent drift in propagated and naturally circulating parasites has led to accumulated differences. This phenomenon has been observed in other protozoan parasites (Akiyoshi et al. 2002; Cama et al. 2006; Chan et al. 2015; Isaza et al. 2015). Genomic DNA for the 2004 CpIRef and CpIA were obtained from the same source, but many years apart. We note small differences in the gp60 sequence, and an overall genome average difference of 0.07% in identity (Supplemental Table S3).
We detect chromosomal inversions in CpIA relative to CpIRef that have also been detected by others (Guo et al. 2015; Isaza et al. 2015). Chromosomal inversions are known to affect rates of adaptation, speciation, and the evolution of chromosomes (Rieseberg 2001; Guo et al. 2015), but they can also represent assembly artifacts. PCR spanning the genomic regions flanking each major inversion in each orientation using genomic DNA from an unsequenced isolate from 2006, C. parvum KSU-1 and CpBGF from 2019 validated the long-read CpIA assembly. Because the other species still lack physical evidence for their chromosomal structures, further long-read sequencing or chromosome conformation capture sequencing, such as Hi-C, will be needed to detect and validate species-specific genomic structural variations for the other Cryptosporidium species.
C. hominis and C. tyzzeri, which are 95%–97% identical at the nucleotide level to C. parvum, show incongruences in annotated genes with respect to the new CpIA genome assembly. The differences result in part from numerous sequence gaps and a lack of experimental evidence (e.g., RNA-seq data) to facilitate annotation. Assembly gaps can lead to frameshift artifacts, fragments of genes split on different contigs, and missing genes. These differences affect similarity-based analyses such as ortholog detection, giving the impression that some of these partially annotated genes are unique to a species when they are not. These misinterpretations can sabotage some experimental designs and analysis (Baptista and Kissinger 2019). The reannotation of the original assembly had 114 pseudogenes, now reduced to only one. This improvement facilitated ortholog and functional identification of the genes involved. Assembly gap regions are usually complex (with repetitive sequence patterns) or hypervariable regions in the population analyzed, and some have high polymorphism rates. False assumptions regarding species-specific genes can affect many downstream analyses including the detection of highly polymorphic loci.
In this study we were able to improve the structural and functional annotation for three Cryptosporidium species using two different approaches: (1) inclusion of seven full-length stranded cDNA libraries derived from three time points (0 h, 24 h, and 48 h post infection) (Tandel et al. 2019), covering ∼90% of the protein-coding genes in C. parvum; and (2) by using synteny information to construct a consistent genome annotation between three different closely related species. This approach facilitated a new comparative analysis of genome content between species. Our analyses reveal that C. parvum, C. hominis, and C. tyzzeri show few differences in gene content for the regions that can be compared. Most differences are related to slight structural variation, such as small translocations and inversions, and copy number variation as revealed by read depth coverage analysis.
Apicomplexans have streamlined genome sequences that approximately range from 8.5 to 125 Mbp (Woo et al. 2015) relative to the only sequenced free-living ancestor, Chromera velia at 194 Mbp (Kissinger and DeBarry 2011). Cryptosporidium species have among the most compact apicomplexan genomes with about 3900 protein-coding genes and ∼77% of the genome sequence being protein coding. They also lack a mitochondrial genome and apicoplast organelle (Keeling 2004), a finding that holds with our deep sequencing. Thus, the higher number of transporters found in our reanalysis makes biological sense and adds to growing work in this area; for example, Cryptosporidium may have adapted a novel type of nucleotide transporter for ATP uptake from the host (Striepen et al. 2004; Pawlowic et al. 2019). The new CpIA assembly and annotation reveals a complete ortholog of the Dnmt2 methylase family. The C. parvum Dnmt2 sequence was previously annotated as truncated and lacking a DNMT-specific motif containing a prolyl-cysteinyl dipeptide (Abrahamsen et al. 2004; Ponts et al. 2013; Isaza et al. 2015). DNMT2 proteins share high sequence and structural similarity with DNA methyltransferases; however, they appear to function primarily as RNA methyltransferases in plants and animals (Goll et al. 2006). Substrates for DNMT2 in protozoa remain unclear.
The lack of three telomeres in the new CpIA long-read assembly was an intriguing result that could be explained by the detection of three putative similar but not identical copies of subtelomeric regions containing genes including tryptophan synthase beta; the MEDLE genes; and 18S, 5.8S, and 28S rRNAs, among others. This finding raises the possibility that Cryptosporidium has recombination between telomeres by break-induced replication like some yeasts (McEachern and Iyer 2001; McEachern and Haber 2006) or telomere maintenance by recombination as is observed in human cancers (Natarajan et al. 2006). Some genes in this region may be essential for parasite survival (Sateriale and Striepen 2016). It is possible (but remains to be proven) that extra or altered copies of these genes may confer an advantage to individual parasites or the population as a whole. In fact, we have not shown that all four subtelomeric regions coexist in the same cell, but 4× coverage of these sequences are present in the population sequenced. We have support from single ONT reads indicating that this region is detected on four different chromosome ends. The ONT reads also prove that these structures are varying within the sequenced CpBGF population (Fig. 4), raising the possibility of recombination or gene conversion during sexual reproduction. This subtelomeric plasticity in which transfer or duplication of important gene sequences between homologous and nonhomologous chromosome ends may affect genetic manipulations of the parasite and their resulting phenotype. Currently, cloning does not exist for Cryptosporidium, and oocysts, which can be sequenced (Troell et al. 2016), must still be considered a population of four haploid meiotic progeny (sporozoites). Single-cell sporozoite sequencing will facilitate recombination and subtelomeric plasticity studies but currently is still impossible in the absence of genome amplification.
Cryptosporidium species are usually typed and characterized by a small number of genetic markers: 18S, cowp, hsp70, and gp60 (Ghaffari et al. 2014). As shown in this study, gp60, which is a gene evolving under positive selection used for Cryptosporidium subtyping characterization, had small differences between CpIRef and CpIA. Using a single marker to characterize an obligately sexual organism with eight chromosomes is problematic. In this study, we confirm an existing group of genes evolving under positive selection and identify 325 additional potential candidates distributed across all eight chromosomes. Some of these genes belong to gene families; to avoid artifacts, only uniquely mapped reads were used for the SNV analysis. The genes identified here can be used to help the community develop additional markers for typing parasite isolates. However, the global diversity of Cryptosporidium is yet to be characterized. Only 136 isolates from a small geographic region have been sampled here. Newer techniques such as hybrid capture bait set techniques (Mamanova et al. 2010) are a powerful future alternative to characterize and select Cryptosporidium population variants and better characterize genetic diversity.
The new C. parvum long-read assembly combined with a consistent comparative annotation has proven powerful. The species analyzed here have different host preferences and pathogenicity. Comparisons of previous sequences and annotation suggested numerous gene content differences. However, this systematic study reveals that the primary differences between the zoonotic C. parvum, the anthroponotic C. hominis, and the rodent-infecting C. tyzzeri are SNVs and CNVs rather than differences in unique gene content. Finally, new findings related to within parasite and/or within population subtelomeric amplification and variation events in C. parvum reveal a new level of genome plasticity that will complicate some genetic manipulations and may affect the organisms’ phenotype.
Methods
Sample DNA sources
C. parvum IOWA-ATCC (CpIA) DNA from oocysts/sporozoites was purchased from the ATCC. The source was the University of Arizona, Sterling Parasitology Laboratory. It is a GP60 subtype (IIa) like the current C. parvum IOWA II reference (CpIRef) genome sequence. C. parvum IOWA DNA was also prepared from oocysts obtained in 2018 from Bunch Grass Farms, Deary, Idaho, referred to as CpBGF in this study. C. parvum KSU-1 genomic DNA was also prepared in 2006 from oocysts obtained from Steve Upton. Public sequence data were accessed from the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJNA252787, PRJEB3213, PRJNA388495, and PRJEB10000. Accession numbers for the 136 C. parvum sequences used for evolutionary analysis are detailed in Supplemental Table S12.
C. parvum IOWA-ATCC sequencing and genome assembly
PacBio RSII and Illumina HiSeq 2000 sequencing were performed at the Wellcome Sanger Institute, United Kingdom. The CpIA reads were first assembled using the PacBio open source SMRTlink v6.0 from nine PacBio SMRT cells, with ∼75× mean genome coverage. The resulting assembly was then submitted to the accuracy improver tool Sprai 0.9.9.23 (https://sprai-doc.readthedocs.io/en/latest/index.html), and then gaps were filled using PBJelly 15.24.8 (English et al. 2014) with PacBio reads and IMAGE 2.4.1 (Swain et al. 2012) with Illumina reads. A manual inspection and improvement using GAP5 (Bonfield and Whitwham 2010) was applied, and the final scaffolded genome assembly was polished with Illumina reads using iCORN2 0.95 (Otto et al. 2010) and Pilon 1.22 (Walker et al. 2014).
ONT single-molecule long-read sequencing was performed on DNA from CpBGF (ATCCPRA-67DQ was out of stock) following the recommended R9.4.1 flow cell protocol. MinION ONT sequencing was performed at the Georgia Genomics Bioinformatics Core at the University of Georgia, using an R.9.4 flow cell and the Ligation Sequencing kit (SQK-LSK109). The ONT long reads generated >2000× coverage of the C. parvum genome. This high coverage complemented the PacBio data to confirm and resolve several complex regions (Supplemental Methods). The final assembly was submitted along with CpIRef, Ch30976, and CtUGA55 to QUAST v.5.02 (Gurevich et al. 2013) to compare and evaluate the quality of CpIA. All sequencing statistics are in Supplemental Table S12.
Cryptosporidium genome reannotation
Genome annotation was generated with ab initio prediction using GeneMark-ES 4.57 (Lomsadze et al. 2005); evidence-trained predictions were made using SNAP/MAKER (Cantarel et al. 2008; Johnson et al. 2008) and AUGUSTUS (Stanke and Morgenstern 2005). For training, we used publicly available data from each respective species: RNA-seq, ESTs, previously predicted proteins, and MassSpec proteomics data when available. In parallel we also generated transcriptome assemblies using HISAT2 v.2.1.0 (Kim et al. 2015) and StringTie v.1.3.4 (Pertea et al. 2015), and noncoding RNA predictions were generated for C. parvum as described (Li et al. 2021). Manual curation of all genes in the context of existing molecular evidence was performed using WebApollo2 (Lee et al. 2013).
We performed comparative genome annotation using the Artemis Comparison tool 17.0.1 (Carver et al. 2005). OrthoFinder v.2.3.7 (Emms and Kelly 2015) was used to detect paralogs, orthologs, and singletons. All singletons were then manually compared using MCScanX 0.8 (Wang et al. 2012) and JBrowse (Buels et al. 2016) to verify their uniqueness and assess the contribution of sequence gaps or misassembly to the findings. We considered the following error types: split genes caused by frameshifts or early stop codons, lack of stranded RNA-seq to confirm the gene model, and the presence of a gapped region in the genome assembly. All genes that did not fall into one of these categories were identified as unique.
Functional annotation
Following structural annotation, the predicted protein sequences were used to search the Swiss-Prot, Trembl, and the NCBI nonredundant protein database with BLASTP using an e-value threshold at the superfamily level of 1 × 10−6. Protein structure similarity was explored using I-TASSER (Roy et al. 2010) as in Ansell et al. (2019) and Supplemental Methods. Blast2GO (Conesa et al. 2005) version 4.1.9 was used to assign Enzyme Code (EC) and Gene Ontology (GO) terms. We compared the existing protein product names to the new functional results. Some structural information, such as protein domain and repeat pattern content were added to some uncharacterized proteins, and nomenclature errors were corrected according to the NCBI guidelines.
Transporter prediction
Predicted proteins were submitted to four different transporter prediction methods: (1) BLASTP against TCDB (Saier et al. 2009) with a threshold e-value of 1 × 10−5 cutoff; (2) TMHMM (Server v. 2.0) (Krogh et al. 2001) and SignalP (Server 4.1) (Bendtsen et al. 2004) to reduce false positives from the TCDB BLASTP results. Transporter candidates with no transmembrane domains or candidates with only one transmembrane prediction while having signal peptides predicted were removed. (3) TransAAP (Ren et al. 2007), a Transporter Classification tool at TransportDB v2.0 (http://www.membranetransport.org/transportDB2/index.html), was used to provide information about potential transporter identity and substrate; and (4) a structural proof for candidate transporters using Phyre2.0 (Kelley et al. 2015). Final candidate transporters were checked according to the preceding results as well as annotations obtained from InterProScan 5.44 (Jones et al. 2014).
Comparative analyses
Structural variation sites were calculated using NucDiff v2.0.3 (Khelik et al. 2017), and the major inversions observed between CpIRef and CpIA were verified by PCR. Primers to test both ends of each orientation were designed using Primer3 v0.4.0 (Supplemental Table S4; Untergasser et al. 2012). Primers designed to be specific and conserved among the species were tested using an in silico PCR amplification tool (San Millán et al. 2013). These regions contain repeats, so the amplicons range from 2 to 9 kb to avoid them. PrimeSTAR GXL DNA polymerase (TAKARA) was used with Long-PCR conditions: initial 3 min hot start at 98°C, 35 cycles of 10 sec denaturation at 98°C; 15 sec primer annealing at 55.4°C; and 10 min elongation at 68°C; followed by 10 min elongation at 72°C. PCR products were separated in a 0.8% agarose gels and stained with ethidium bromide.
The consistency of annotation and potential gene family CNVs were determined with OrthoFinder v.2.2.7. CNVs were also determined by aligning Illumina sequence reads from each species studied to the new CpIA genome sequence to check for variations in read depth coverage. Alignment was performed using BWA-MEM 0.7.17 (Li and Durbin 2009) with default options, and the alignment depth per base was calculated using BEDTools genomecov 2.29.2 (Quinlan and Hall 2010) and SAMtools depth 1.6 (Li et al. 2009). Read depth coverage plots were generated using the reshape R package (Wickham 2007; R Core Team 2011). To avoid the interference of multiply-mapped regions, only mapped reads were kept for this analysis. For plotting purposes, the Ch30976 genome was scaffolded using the CpIA chromosomal genomic structure using RagTag v.2.0.1 (Alonge et al. 2019).
Resolving the structure of repetitive subtelomeric regions
Following the CNV analysis, the sequence content of the subtelomeric compressed regions and their CpIA assembly noncompressed chromosomal sequence boundaries containing at least 10 genes of Chromosomes 1, 7, and 8 were used to build a BLAST database. We then used this database and BLASTN 2.10.0 (Camacho et al. 2009) to detect CpBGF ONT reads capable of aligning to both subtelomeric and chromosomal boundary regions. The few reads meeting these criteria were evaluated and visualized by aligning all subtelomeric ONT reads to the unique pre-subtelomeric regions of Chromosomes 1, 7, and 8 using the Geneious mapper 2019.1.3 (https://www.geneious.com) with medium sensitivity and minimap2 v.2.22 (Li 2018). Finally, the longest ONT reads were polished with Illumina reads and annotated as previously described for gene content analysis.
Variant analysis, selection prediction, and populational analysis
Illumina sequence reads from 136 different isolates of C. parvum from different geographical locations (Supplemental Table S8) as well as CpBGF were aligned against the CpIA reference genome sequence using BWA-MEM. The BAM files were parsed to select uniquely mapped reads using Picard (http://broadinstitute.github.io/picard/) and then submitted to the GATK 3.8 Haplotypecaller (McKenna et al. 2010). The results were filtered by mapping quality greater than 40 and depth coverage greater than 10. Because mixed infections exist, we restricted analysis to biallelic sites. The individual VCF files were combined into one GVCF file using the GATK tool GenotypeGVCF. After selecting only SNVs from this data, the combined GVCF file was annotated using SnpEff v.4.3 (Cingolani et al. 2012). The SNV variants from the combined annotated GVCF file had their πN/πS ratio (Nei and Gojobori 1986) estimated using SNPGenie 1.0 (Nelson et al. 2015). To avoid noise in the data and identify top candidates, genes with ratios >1.5 were detected and denoted as evolving under positive selection in the C. parvum population analyzed. The higher threshold of 1.5 was chosen based on known genes evolving under positive selection, such as gp60 (Strong et al. 2000) and Insulinase-like (Zhang et al. 2019).
Data access
The sequence data and annotation generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession numbers PRJNA573722 and PRJEB3213. Subtelomeric sequences from Chr 7 and 8 have GenBank accession numbers MZ892386, MZ892387, and MZ892388.
Competing interest statement
J.C.K. has a financial interest in PacBio.
Acknowledgments
We thank Dr. Lihua Xiao for sharing C. hominis 30976 and permitting us to update the annotation. This work was supported by Bill and Melinda Gates Foundation grant OPP1151701 to J.C.K., The Wellcome Trust via its core funding of the Wellcome Sanger Institute (grant WT206194), The National Health and Medical Research Council Investigator Grant (APP1194330) to A.R.J., and National Institutes of Health (NIH) R01AI127798 and R01AI112427 to B.S. J.E.D. was supported by NIH T32AI007532 and A.S. by NIH K99AI137442.
Author contributions: R.P.B. and J.C.K. designed research; R.P.B. and J.C.K. performed research; A.S., J.E.D., and B.S. contributed new reagents and samples; B.R.E.A., A.R.J., B.J.P., and P.G. contributed analytical tools; M.J.S., K.L.B., A.T., M.B., and J.A.C. contributed Illumina and PacBio sequencing; R.P.B., Y.L., K.L.B., A.T., R.X., E.D.S., G.W.C., and J.C.K. analyzed data; R.P.B. and J.C.K. wrote the paper; and A.R.J., B.R.E.A., B.S., A.S., and J.A.C. provided feedback.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.275325.121.
-
Freely available online through the Genome Research Open Access option.
- Received February 11, 2021.
- Accepted November 10, 2021.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








