
Blood-derived mitochondrial DNA copy number is associated with gene expression across multiple tissues and is predictive for incident neurodegenerative disease
- Stephanie Y. Yang1,
- Christina A. Castellani1,
- Ryan J. Longchamps1,
- Vamsee K. Pillalamarri1,
- Brian O'Rourke2,
- Eliseo Guallar3 and
- Dan E. Arking1,2
- 1McKusick-Nathans Department of Genetic Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA;
- 2Division of Cardiology, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205, USA;
- 3Departments of Epidemiology and Medicine, and Welch Center for Prevention, Epidemiology, and Clinical Research, Johns Hopkins University Bloomberg School of Public Health, Baltimore, Maryland 21205, USA
Abstract
Mitochondrial DNA copy number (mtDNA-CN) is a proxy for mitochondrial function and is associated with aging-related diseases. However, it is unclear how mtDNA-CN measured in blood can reflect diseases that primarily manifest in other tissues. Using the Genotype-Tissue Expression Project, we interrogated relationships between mtDNA-CN measured in whole blood and gene expression from whole blood and 47 additional tissues in 419 individuals. mtDNA-CN was significantly associated with expression of 700 genes in whole blood, including nuclear genes required for mtDNA replication. Significant enrichment was observed for splicing and ubiquitin-mediated proteolysis pathways, as well as target genes for the mitochondrial transcription factor NRF1. In nonblood tissues, there were more significantly associated genes than expected in 30 tissues, suggesting that global gene expression in those tissues is correlated with blood-derived mtDNA-CN. Neurodegenerative disease pathways were significantly associated in multiple tissues, and in an independent data set, the UK Biobank, we observed that higher mtDNA-CN was significantly associated with lower rates of both prevalent (OR = 0.89, CI = 0.83; 0.96) and incident neurodegenerative disease (HR = 0.95, 95% CI = 0.91;0.98). The observation that mtDNA-CN measured in blood is associated with gene expression in other tissues suggests that blood-derived mtDNA-CN can reflect metabolic health across multiple tissues. Identification of key pathways including splicing, RNA binding, and catalysis reinforces the importance of mitochondria in maintaining cellular homeostasis. Finally, validation of the role of mtDNA CN in neurodegenerative disease in a large independent cohort study solidifies the link between blood-derived mtDNA-CN, altered gene expression in multiple tissues, and aging-related disease.
Mitochondria perform multiple essential metabolic functions including energy production, lipid metabolism, and signaling for apoptosis. Mitochondria possess circular genomes (mtDNA) that are distinct from the nuclear genome. Although cells typically only possess two copies of the nuclear genome, they contain 100s to 1000s of mitochondria, and each individual mitochondrion can hold 2–10 copies of mtDNA, resulting in wide variation in mtDNA copy number (mtDNA-CN) (Wai et al. 2010). The amount of mtDNA-CN also varies widely across cell types, with higher energy demand cell types typically possessing higher levels of mtDNA-CN (Chabi et al. 2003; Miller et al. 2003; Clay Montier et al. 2009; Kelly et al. 2012). Due to the importance of mitochondria in metabolism and energy production, mitochondrial dysfunction plays a role in the etiology of many human diseases (Herst et al. 2017). mtDNA-CN has been shown to be a proxy for mitochondrial function and is consequently an attractive biomarker due to its ease of measurement (Malik and Czajka 2013; Castellani et al. 2020b). Indeed, low levels of mtDNA-CN in peripheral blood have been associated with an increased risk for a number of chronic aging-related diseases including frailty, kidney disease, cardiovascular disease, heart failure, and overall mortality (Ashar et al. 2015, 2017; Huang et al. 2016; Tin et al. 2016).
Crosstalk between the mitochondrial and nuclear genomes is essential for maintaining cellular homeostasis. Many essential mitochondrial proteins are encoded by the nuclear genome, and expression of these nuclear genes must be modified to match mitochondrial activity. Likewise, mitochondrial activity must respond to cellular energy demands. Polymorphisms in the nuclear genome have been associated with changes in mitochondrial gene expression, and mitochondrial genome variation has been associated with changes in nuclear gene expression, suggesting interplay between the two genomes (Lee et al. 2017b; Ali et al. 2019).
In cancer cells, mtDNA-CN alters gene expression through modifying DNA methylation (Reznik et al. 2016; Sun and St John 2018). Recent work from our lab has shown that mtDNA-CN is also associated with nuclear DNA methylation in noncancer settings (Castellani et al. 2020a). Given that DNA methylation can modify gene expression, the current study seeks to explore the potential association between blood-derived mtDNA-CN and gene expression. Past work has shown that mtDNA-CN is associated with gene expression of nuclear-encoded genes in lymphoblast cell lines, but this may not reflect biological processes occurring in other tissues, especially after an extended culturing period (Gibbons et al. 2014). Therefore, we leveraged data from the Genotype-Tissue Expression Project (GTEx), a cross-sectional study with gene expression data from multiple nondiseased postmortem tissues, to examine associations between mtDNA-CN and expression of both nuclear and mitochondrially encoded genes (The GTEx Consortium 2013). This study aimed to evaluate associations between blood-derived mtDNA-CN and gene expression across multiple tissues and to follow up on a novel association between neurodegenerative disease and blood-derived mtDNA-CN.
Results
Determination and validation of mtDNA-CN metric
mtDNA-CN estimates were generated from whole genome sequences performed on DNA derived from whole blood using the ratio of mitochondrial reads to total aligned reads. As mtDNA-CN is known to be affected by cell type composition, cell counts for samples with available RNA-sequencing data were deconvoluted using gene expression measured in whole blood (Aran et al. 2017; Zhang et al. 2017). We identified a batch effect that resulted in significantly altered mtDNA-CN for individuals sequenced prior to January 2013. Therefore, only individuals sequenced after January 2013 were retained for analysis (Supplemental Fig. S1). After quality control, outlier filtering, and normalization of the RNA-sequencing data, 419 individuals remained for analyses (see Methods).
To validate mtDNA-CN measurements in the filtered GTEx data, we determined the association between mtDNA-CN and known correlated measures, including age, sex, and neutrophil count (Mengel-From et al. 2014; Zhang et al. 2017; Moore et al. 2018). We observed a significant association with neutrophil count (P = 8.4 × 10−5), with higher neutrophil count associated with lower mtDNA-CN. Although not statistically significant, effect size estimates between mtDNA-CN and age (P = 0.18) and sex (P = 0.14) were also in the expected direction, with older individuals and males having lower mtDNA-CN (Supplemental Fig. S2). Effect size estimates for age and neutrophils were also consistent with prior literature (Supplemental Table S1; Longchamps et al. 2020). Based on variance explained from previous studies, the current study was only powered to detect a significant effect for neutrophil count. For all downstream analyses, mtDNA-CN was defined as the standardized residual from a linear regression model adjusted for age, sex, cell counts estimated from RNA-seq deconvolution, ischemic time, and cohort (see Methods).
Association of mtDNA-CN derived from whole blood with gene expression in blood
A priori, we expect that mitochondrially encoded gene expression would be positively correlated with mtDNA-CN. Likewise, multiple nuclear-encoded genes are involved in the regulation of mtDNA replication, and thus, expression levels of these genes are expected to be correlated with mtDNA-CN (Garcia et al. 2017; Rusecka et al. 2018). We therefore evaluated the associations between mtDNA-CN and expression of these two classes of genes, correcting for cohort, sample ischemic time, genotyping PCs, age, race, and surrogate variables derived from RNA-sequencing data to capture known and hidden confounders (Supplemental Fig. S3; Leek and Storey 2007).
To minimize the potential impact of outliers, we performed an inverse normal transformation on both the mtDNA-CN metric and the gene expression values. To evaluate the association between mtDNA-CN and mitochondrial RNA (mtRNA) levels, we used the median gene expression value calculated from scaled expression values across 36 mtDNA-encoded genes that passed expression thresholds (see Methods).
We observed a highly significant association between mtDNA-CN and overall mtRNA expression (P = 9.10 × 10−9) (Table 1), with 33 out of 36 individual mtDNA-encoded genes nominally significant (P < 0.05) (Supplemental Fig. S4).
Blood-derived mtDNA-CN is positively associated with gene expression for mitochondrially encoded genes and nuclear-encoded genes required for mtDNA replication
In addition to genes coding directly for mtDNA replication machinery, genes involved in mtDNA transcription and nucleotide metabolism are also required for mtDNA replication. The mtDNA transcription machinery provides the RNA primers used in mtDNA replication, and nucleotides are needed to synthesize new mtDNA molecules. Of the 17 mtDNA major replication genes tested (Rusecka et al. 2018), all were positively associated with mtDNA-CN, as would be expected based on gene function; eight of them were nominally significant (P < 0.05), and four were significant after Bonferroni correction (P < 2.94 × 10−3) for multiple testing (Table 1).
To identify additional genes and pathways associated with mtDNA-CN, we performed a transcriptome-wide analysis. There was an overall inflation of test statistics, which we quantified using the genomic inflation factor (lambda = 4.71) (Devlin and Roeder 1999). Two-stage permutation testing with 1000 permutations demonstrated no inflation in null data sets, suggesting that this inflation represents a true global association between blood-derived mtDNA-CN and gene expression (Supplemental Fig. S5).
When stratified by gene functional categories (Harrow et al. 2012), all categories showed elevated test statistics, but protein-coding genes were the most enriched (lambda = 7.44) (Fig. 1).
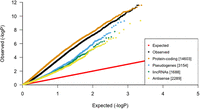
Global inflation of test statistics from linear regressions between blood-derived mtDNA-CN and gene expression in blood. After stratification by gene category, protein-coding genes have the most inflation, suggesting that mtDNA-CN is strongly associated with genes that code for proteins.
Gene expression levels of most of the nominally significant genes were positively correlated with mtDNA-CN (7769 genes with positive t-values vs. 285 genes with negative t-values). Although much of this positive skewing is likely due to correlated gene expression, permuted data sets demonstrate that this positive shift is significant (P < 0.001) (Supplemental Fig. S6), perhaps reflecting a more active transcriptional state associated with higher mtDNA-CN. Whereas 698 of the significantly associated genes were positively associated, only two negatively associated genes passed the permutation cutoff (P < 2.38 × 10−6), CAMP (P = 1.58 × 10−8), and PGLYRP1 (P = 1.78 × 10−7), both of which are involved in innate immunity.
Gene set enrichment analysis uncovers gene regulatory networks in whole blood
To identify specific molecular pathways, transcription factors, and gene ontologies associated with mtDNA-CN in whole blood, we performed gene set enrichment analyses (Irizarry et al. 2009) using gene sets obtained from the Molecular Signatures Database (MSigDB) (Ashburner et al. 2000; Xie et al. 2005; Liberzon et al. 2011; Meng et al. 2019; The Gene Ontology Consortium 2019). Previous studies have shown that cross-mappability can lead to false pseudogene positives in eQTL association studies (Saha and Battle 2019); we therefore excluded pseudogenes from subsequent analyses. Significantly associated KEGG pathways included “Spliceosome” (P = 1.03 × 10−8) and “Ubiquitin-mediated proteolysis” (P = 2.4 × 10−10) (Table 2).
Top five genes that were most significantly associated with mtDNA-CN within the “Spliceosome” and “Ubiquitin-mediated proteolysis” KEGG pathways
A number of transcription factor target sequences were also significantly enriched, including those for ELK1 (P = 8.58 × 10−66), NRF1 (P = 1.76 × 10−35), GABPB (P = 3.54 × 10−21), YY1 (P = 3.14 × 10−19), and E4F1 (P = 3.98 × 10−15). All of these transcription factors regulate genes that play a role in mitochondrial function (Barrett et al. 2006; Blesa et al. 2008; Yang et al. 2014; Rodier et al. 2015; Chen et al. 2019). Gene expression levels of these transcription factors were all positively correlated with mtDNA-CN, with five out of six nominally significant, and three remaining significant after Bonferroni correction (P < 8.33 × 10−3) (Table 3).
Gene expression for transcription factors whose targets are enriched for association with blood-derived mtDNA-CN is nearly all nominally significantly associated with blood-derived mtDNA-CN
Many mitochondrially related cellular component Gene Ontology (GO) terms were significant, including “Mitochondrion” (P = 7.77 × 10−23), “Mitochondrial part” (P = 2.79 × 10−15), and “Mitochondrion organization” (P = 2.87 × 10−14) (Fig. 2; Supek et al. 2011).
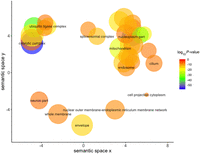
REVIGO visualization of GO cellular component terms significantly associated with mtDNA-CN after removal of redundant GO terms. Size of the circle represents the relative number of genes in each gene set, color represents significance. Axes represent semantic similarities between GO terms; GO terms that are more similar will cluster with one another.
Additional significantly associated GO terms included “ubiquitin ligase complex” (P = 6.6 × 10−18) and “spliceosomal complex” (P = 4.46 × 10−14), supporting the KEGG pathway findings. Genes with substantial evidence of mitochondrial localization, determined through integration of several genome-scale data sets, were obtained from MitoCarta2.0 and demonstrated significant enrichment (P = 8.22 × 10−21) (Calvo et al. 2016).
Cross-tissue analysis reveals associations between gene expression in multiple tissues and blood-derived mtDNA-CN
mtDNA-CN measured in blood has been associated with a number of aging-related diseases, including chronic kidney disease, heart failure, and diabetes (Huang et al. 2016; Tin et al. 2016; Al-Kafaji et al. 2018). Given that these diseases primarily manifest in nonblood tissues, we evaluated associations between blood-derived mtDNA-CN and gene expression measured from 47 additional tissues that had greater than 50 samples after filtering.
Though blood-derived mtDNA-CN appears to be associated with gene expression in other tissues, we did not observe a significant association between blood-derived mtDNA-CN and scaled mtRNA gene expression in any tissue other than blood, and only two out of 47 tested tissues had nominally significant associations between tissue-specific scaled mtRNA expression and blood-derived mtDNA-CN (uterus [P = 0.004], left ventricle of the heart [P = 0.017]) (Supplemental Table S2). However, mtRNA expression for 35/47 nonblood tissues was positively associated with blood mtDNA-CN, which is more than what would be expected by chance (P < 0.001). This suggests that, although our study may be underpowered to detect a significant association in individual tissues due to small sample sizes, mtDNA-CN measured in blood is broadly correlated with mtDNA-CN in other tissues. As expected, mtRNA expression varies widely across tissues, with brain tissues having notably more expression than other tissues (Supplemental Fig. S7).
We calculated genomic inflation factors for each tissue to quantify test statistic inflation. Genomic inflation factors were elevated across multiple nonblood tissues, suggesting that blood-derived mtDNA-CN was broadly associated with gene expression in other tissues (Fig. 3).
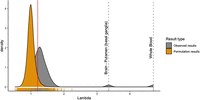
Observed genomic inflation factors are significantly different from permuted genomic inflation factors for certain tissues. A higher genomic inflation factor represents increased global associations between blood-derived mtDNA-CN and gene expression in a specific tissue. One thousand permuted genomic inflation factors were obtained using two-stage permutation testing. Red line represents permuted lambda cutoff of 1.20.
To determine true signal from noise, we performed 1000 two-stage permutations for each tissue and obtained a genomic inflation factor lambda cutoff of >1.20 representing a significant elevation of lambda (study-wide P < 0.05). Using this cutoff, we identified 30 nonblood tissues with a global inflation of test statistics (Supplemental Table S3). Other than blood, the most strongly enriched tissue was the putamen region of the brain, with a lambda of 3.27. Principal components analysis revealed that the putamen region of the brain was not significantly different from other brain regions, and we are uncertain what is biologically causing the strong enrichment (Supplemental Fig. S8). We note that the two cell lines, EBV transformed lymphocytes (lambda = 0.84) and cultured fibroblasts (lambda = 0.84), showed no global inflation of test statistics, suggesting that blood-derived mtDNA-CN loses its association with gene expression after the cell culturing process.
To examine the similarity of associations of mtDNA-CN observed in blood with other tissues, we calculated Spearman's rank correlation coefficients between effect estimates for blood-derived mtDNA-CN on blood gene expression (βblood) and effect estimates for blood-derived mtDNA-CN on gene expression in other tissues (βtissue). All genes that passed a permutation cutoff for significance in blood (P = 2.38 × 10−6, 700 genes total) were included. To distinguish tissues with correlations more extreme than baseline, we calculated reference correlations between blood and other tissues for 1000 randomly selected sets of 700 genes. Twenty-six tissues had observed values that were significantly more extreme than baseline (Supplemental Table S4), with 22 tissues showing greater correlation and four tissues showing less correlation. Of these 26 tissues, 20 were among the 30 tissues with significantly inflated lambdas.
To identify pathways associated with mtDNA-CN across multiple tissues, we performed gene set enrichment analysis in each of the 30 tissues with a significant genomic inflation factor. Multiple terms were significant in greater than one tissue (Table 4), including terms related to oxidative phosphorylation and mitochondria, suggesting that mtDNA-CN derived from blood can reflect mitochondrial function occurring in other tissues.
Pathways, transcription factor targets, and GO terms significantly enriched in multiple tissues (excluding blood)
ELK1 transcription factor binding sites were significantly enriched in 19 of the 30 significant tissues and were also significant in whole blood, suggesting that mtDNA-CN may regulate ELK1 or vice versa. We note that gene expression for ELK1 was nominally significantly associated (P < 0.05) with blood-derived mtDNA-CN in four of the 18 tissues for which ELK1 targets were significantly enriched (Supplemental Fig. S9). Effect estimates for ELK1 targets were generally consistent with the directionality of ELK1 effect estimates. For example, in blood, where ELK1 expression is positively associated with mtDNA-CN, 747/750 (99.6%) nominally significant ELK1 target genes were positively associated. On the other hand, mtDNA-CN was negatively associated with nerve ELK1 gene expression, and 204/306 (66.67%) nominally significant ELK1 target genes were also negatively associated. Of note, nearly all the noted transcription factors are ubiquitously expressed throughout the body, except for ELK1, which is not expressed in brain putamen or spinal cord (Supplemental Fig. S10).
To identify genes driving enrichment of significant pathways in multiple tissues, we performed a random effects meta-analysis for all expressed genes using effect size estimates from all 47 nonblood tissues. Genes encoding both the large and small ribosomal subunits were negatively associated with blood-derived mtDNA-CN across all tested tissues, implying an inverse relationship between ribosomal abundance and mitochondrial DNA quantity (Table 5).
Random-effects meta-analysis for genes driving the enrichment of pathways in multiple tissues
Huntington's disease (HD), Parkinson's disease (PD), and Alzheimer's disease (AD) were among the most significantly associated KEGG pathways that appear in multiple tissues (Table 4). This is an intriguing finding, given the known role of mitochondria in neurodegenerative disease (Reddy 2009).
Although neurodegenerative disorders primarily manifest in nervous tissues (Wood et al. 2015), we observed significant enrichment of disease pathways in colon, pancreas, and testis tissues. When limiting our query to brain tissues, HD and PD were nominally significantly enriched in cerebellum, caudate (basal ganglia), and cortex, whereas AD was nominally significantly enriched in cerebellum and spinal cord (Supplemental Fig. S11).
mtDNA-CN is associated with incident and prevalent neurodegenerative disease in the UK Biobank
To examine the association between mtDNA-CN and neurodegenerative disease risk, we used the UK Biobank (UKB) (Bycroft et al. 2018), a prospective cohort study with whole exome sequencing (WES) for ∼50,000 individuals and genotyping for ∼500,000 individuals. mtDNA-CN was estimated from a combination of WES data and mitochondrial SNP probe intensities from genotyping arrays (see Methods) and was significantly associated with age and sex in the expected directions (Supplemental Fig. S12). Analyses were restricted to unrelated individuals of self-identified European descent, and individuals with blood cell type count outliers were excluded, followed by adjustment of mtDNA-CN for age and sex. Associations between mtDNA-CN and prevalent and incident neurodegenerative disease were evaluated using logistic regression models and Cox proportional-hazards models, respectively. mtDNA-CN was significantly associated with prevalent Parkinson's disease and prevalent dementia (Table 6). As there were only 12 individuals with prevalent Alzheimer's disease, we did not test for an association with prevalent Alzheimer's disease. For incident disease, median follow-up time was ∼10 yr. mtDNA-CN was significantly associated with incident Parkinson's disease and incident dementia (Table 6). Consistent with other aging-related diseases (Ashar et al. 2015, 2017), higher mtDNA-CN was associated with lower risk for developing incident neurodegenerative disease (Table 6). A combined analysis for all individuals with neurodegenerative disease revealed a consistent strongly significant association for both prevalent (OR = 0.89, CI = 0.83;0.96) and incident (HR = 0.95, CI = 0.91;0.98) disease.
mtDNA-CN is associated with incident and prevalent neurodegenerative disease
Discussion
In this study, blood-derived mtDNA-CN was significantly associated with a host of blood-expressed genes. As expected, nearly all genes involved in mtDNA replication were significantly associated with mtDNA-CN in a positive direction. There was also a clear overall shift toward significant positive estimates, possibly indicating that increased mtDNA-CN is reflective of a more active transcriptional state. This finding is consistent with previous literature demonstrating that higher mitochondrial content is correlated with increased transcriptional activity (Guantes et al. 2015; Márquez-Jurado et al. 2018). The two negatively associated genes both play roles in innate immune function (Gombart et al. 2005; Osanai et al. 2011), suggesting that higher mtDNA-CN levels are correlated with decreased immune response. Mitochondria play a role in immune responses to pathogens in several ways; for example, mitochondrial DNA release from compromised mitochondria can trigger an intracellular antiviral response through the cGAS–STING pathway (West et al. 2015), binding of viral dsRNA to the mitochondrial antiviral signaling complex (MAVS) can trigger an interferon responses through STAT6 activation (Chen et al. 2011), and release of mitochondrial components from cells can bind to damage-associated molecular pattern (DAMP) receptors to trigger innate immune responses (Nakahira et al. 2015). These novel findings correlating expression of mtDNA-CN with specific immune response genes in tissues represent an area for further investigation.
Gene set enrichment analyses revealed pathways potentially involved in mitochondrial DNA control, including ubiquitin-mediated proteolysis and splicing. Supporting this finding, Guantes et al. demonstrated that mitochondrial content modulates alternative splicing (Guantes et al. 2015). Additionally, we found that genes expressed in whole blood that were associated with blood-derived mtDNA-CN were enriched for target sequences for the ELK1, NRF1, YY1, GABPB, and E4F1 transcription factors. All of these transcription factors have been implicated in mitochondrial pathways, as ELK1 is associated with the mitochondrial permeability transition pore complex in neurons, NRF1 regulates expression of the mitochondrial translocase TOMM34, YY1 binds to and represses mitochondrial gene expression in skeletal muscle, GABPB is required for mitochondrial biogenesis, and E4F1 controls mitochondrial homeostasis (Barrett et al. 2006; Blesa et al. 2008; Yang et al. 2014; Rodier et al. 2015; Chen et al. 2019). Additionally, we found significant enrichment of signal for genes implicated in ubiquitin-mediated proteolysis and splicing. Given that mitochondrial quality control is regulated through ubiquitination, and that nuclear-encoded spliceosomes are involved in mtRNA splicing, our results likely implicate processes involved in mitochondrial DNA regulatory networks (Bragoszewski et al. 2017; Herai et al. 2017).
mtDNA-CN measured in one tissue has previously been found to be uncorrelated with mtDNA-CN in another tissue from the same individual (Wachsmuth et al. 2016). We found that, although mtRNA transcription in individual tissues was not significantly correlated with blood-derived mtDNA-CN, across all tissues, there was a significant enrichment for positive associations, suggesting a weak positive correlation between blood-derived mtDNA-CN and mtDNA-CN in other tissues. Moreover, we found that blood-derived mtDNA-CN was associated with various biological pathways in nonblood tissues (including mitochondrial function), providing a possible explanation as to why blood-derived mtDNA-CN is associated with aging-related diseases that primarily manifest in nonblood tissues. Further examination of pathways significant in multiple tissues revealed that ribosomal subunit genes were significantly negatively associated with mtDNA-CN. Although there has been conflicting evidence on the relationship between mtDNA-CN and ribosomal content, our study revealed a strong inverse relationship between ribosomal DNA dosage and mtDNA-CN (Gibbons et al. 2014; Guantes et al. 2015). Importantly, because these are statistical associations, causal directionality cannot be determined between gene expression and blood-derived mtDNA-CN. Future follow-up studies are needed to determine functional causality for mtDNA-CN and gene expression.
KEGG pathways that were significantly enriched in multiple tissues included Huntington's disease, Alzheimer's disease, and Parkinson's disease. These aging-related neurodegenerative diseases all have underlying mitochondrial pathologies (Coskun et al. 2010; Petersen et al. 2014; Wei et al. 2017; Park et al. 2018) and dysregulated ubiquitination pathways (Atkin and Paulson 2014). In particular, mtDNA-CN has been implicated in Alzheimer's disease (Rice et al. 2014; Delbarba et al. 2016; Lv et al. 2019) and cognitive function (Lee et al. 2010, 2017a). Further, the ELK1 transcription factor, whose target sequences were significantly enriched in 19 tissues, plays a role in multiple neurodegenerative diseases (Besnard et al. 2011). Finally, after finding that blood-derived mtDNA-CN was associated with expression of neurodegenerative disease genes, we used an independent data set, the UK Biobank, and found that mtDNA-CN was significantly associated with both prevalent and incident neurodegenerative disease risk. In conclusion, our findings show that blood-derived mtDNA-CN is significantly associated with gene expression from tissues across the body and that higher mtDNA-CN is associated with decreased incident neurodegenerative disease risk.
Methods
GTEx sample acquisition
Whole genome sequences were downloaded from the GTEx version 8 cloud repository on 11/18/2020. RNA-sequencing data used for analyses were downloaded from the GTEx portal (http://gtexportal.org/home/datasets) on 06/18/2019, and phenotypes were obtained from the database of Genotypes and Phenotypes (dbGaP; https://www.ncbi.nlm.nih.gov/gap/) (phs000424.v8.p2).
Estimation of mtDNA-CN
SAMtools version 1.9 (Li et al. 2009) was used to count the number of mitochondrial, unaligned, and total reads for each whole genome sequence. mtDNA-CN was estimated as the number of mitochondrial reads divided by the difference between the number of total reads and the number of unaligned reads to obtain a ratio of mtDNA to nuclear DNA. Whole genome is a highly accurate method for estimation of mtDNA-CN (Wachsmuth et al. 2016; Longchamps et al. 2020).
Correcting mtDNA-CN for covariates
All statistical analyses were performed with R version 3.6.1 (R Core Team 2019). Cell type composition for whole blood samples was determined from RNA sequencing using xCell (Aran et al. 2017), only allowing for deconvolution of cell types found in blood. A stepwise regression in both directions was used to select appropriate cell types to correct mtDNA-CN. To avoid model overfitting, correlated cell types (R > 0.8) were removed. The final model used to adjust mtDNA-CN included neutrophils, hematopoietic stem cells, megakaryocytes, subject cohort, ischemic time, age, and sex. Residuals were standardized after adjusting for covariates. Power calculations were performed using R2 values from previous studies using the pwr package (Longchamps et al. 2020).
Filtering pipeline
A batch effect due to sample collection and/or sequencing methods resulted in significantly altered mtDNA-CN for individuals who were sequenced prior to January 2013. To keep this from confounding the analysis, we excluded subjects with whole genome sequencing prior to January 2013 (Supplemental Fig. S1). Individuals who had greater than 5 × 107 unaligned whole genome sequence reads were also omitted from the analysis. Cell type outliers who were greater than three standard deviations (SDs) from the mean were excluded as well. Only one individual remained from the surgical cohort after filtering and therefore was also removed (Supplemental Fig. S13).
RNA-sequencing pipeline
GTEx version 8 RNA-sequencing data was downloaded from the GTEx website in read counts and normalized using the trimmed mean of M-values method prior to analyses (Robinson and Oshlack 2010; Robinson et al. 2010). For each separate tissue, only genes with expression greater than 0.1 counts for at least 20% of samples for that tissue were retained for analysis. To identify potential hidden confounders, we used surrogate variable analysis (SVA), protecting mtDNA-CN from SV generation (Leek and Storey 2007). SVs were associated with known covariates in the data, such as whether individuals were in the postmortem or the organ donor cohorts (Supplemental Fig. S3). Individuals who were greater than three standard deviations from the mean for the first 10 SVs were omitted from analysis. SV generation was performed iteratively three times.
Linear model for evaluating associations
To reduce the influence of outliers, both the gene expression metric and the mtDNA-CN metric were inverse normal transformed prior to linear regression. We then tested for association using multiple linear regression, with mtDNA-CN as the predictor and gene expression as the outcome, correcting for SVs, sex, cohort, race, ischemic time, and the first three genotyping principal components.
Genomic inflation factor calculation
Genomic inflation factors were calculated by squaring z-scores to obtain χ2 values. The median observed χ2 value was divided by the expected median to obtain lambda (Devlin and Roeder 1999).
Two-stage permutations
To determine an appropriate P-value cutoff, we created null data sets for permutation testing. First, a multiple linear regression model for the alternate hypothesis was used to obtain gene expression residuals. Second, a multiple linear regression model for the null hypothesis was used to obtain estimates for each gene. Residuals from the alternate model were then permuted and added to effect estimates from the null model to create null data sets. Permuted gene expression data were then tested for association with mtDNA-CN. Permutations were performed 1000 times. Minimum P-values from each permuted data set were obtained, and the fifth lowest P-value was utilized as a permutation cutoff.
Annotation of gene categories
Gene annotations were downloaded from GENCODE (Harrow et al. 2012). Test statistics were then stratified by gene type, and observed and expected distributions were generated for each category.
Overrepresentation of positive beta estimates
Percentage of positive effect estimates was calculated using all nominally significant genes in blood, dividing the number of nominally significant genes with positive effect estimates by the total number of nominally significant genes. Percentages for null distributions were calculated using 1000 permutations, generated using the two-stage permutation method described above.
Gene set enrichment analysis
To examine enrichment for genes in specific pathways, gene sets for KEGG pathways, transcription factor target sequences, and Gene Ontologies were downloaded from the Molecular Signatures Database (Kanehisa and Goto 2000; Liberzon et al. 2011; The Gene Ontology Consortium 2019). Then, using the absolute value of the t-scores from the regression model with mtDNA-CN, we performed a t-test of t-scores for genes in a specific pathway versus genes that were not contained in the pathway. For each tissue, only genes with greater than 0.1 counts in at least 20% of the tissue samples were used for gene set enrichment analyses. Based on which genes passed expression thresholds, different lists of genes were used for enrichment analyses for different tissues. We also performed 1000 permutations using randomized t-scores to determine appropriate cutoffs for significance. To confirm that results were not driven by individual genes in a pathway with very large t-scores, we also performed t-tests using ranked t-scores as opposed to absolute value t-scores.
REVIGO trimming and visualization of GO terms
For visualization of significantly enriched GO terms and elimination of redundant GO terms, REVIGO (http://revigo.irb.hr/) was used with the default settings except for the allowed similarity, which was set to medium (0.7) (Supek et al. 2011).
Testing for associations between blood-derived mtDNA-CN and gene expression in other tissues
Filtering parameters and models for testing the association of blood-derived mtDNA-CN with gene expression in other tissues were identical to the pipeline used in whole blood. Only tissues with greater than 50 observations after filtering were tested. For tissues that had no variation in covariates, covariates were dropped from the linear model (i.e., sex was not used in the model for testing gene expression in reproductive organs, and cohort was not used in the model for brain tissues).
Spearman's correlations for effect estimates with whole blood
All significant genes in whole blood that passed the permutation cutoff (P = 2.38 × 10−6) were used for testing. Spearman's correlations between effect estimates in blood and effect estimates in other tissues were calculated. To compare correlations for genes significant in blood with baseline correlation, we randomly selected 100 random genes and calculated correlations between blood estimates and specific tissue estimates for those genes. We repeated this random selection 1000 times to generate multiple baseline correlation measures.
Meta-analysis of genes driving specific ontologies
To calculate meta-analysis effect estimates and P-values, the R “meta” package (Balduzzi et al. 2019) was used to perform a random-effects meta-analysis using all effect estimates and P-values for all tissues, excluding results from whole blood.
Association of mtDNA-CN with neurodegenerative disease in UKB
UKB mtDNA-CN derivation
We started with 49,997 Exome SPB CRAM files (version Jul 2018) downloaded from the UKB data repository and used SAMtools (ver1.9)
to extract read summary statistics (“idxstats” command). A custom Perl script was used to aggregate the summary statistics
from each individual file into the following categories (see Perl script and example stats file): (1) Total Reads (sum of
columns 3 and 4, across all rows); (2) Mapped Reads (sum of column 3, across all rows); (3) Unmapped Reads (sum of column
4 across all rows); (4) Autosomal Reads (sum of column 3, rows 1–22); (5) Chr X; (6) Chr Y; (7) Chr MT; (8) “Random” Reads
(sum of column 3, across rows 26–67); (9) “Unknown” Reads (sum of column 3 across rows 68–194); (10) EBV Reads; (11) “Decoy1”
Reads (sum of column 3 across rows 196–582); (12) “Decoy2” Reads (sum of column 3 across rows 583–2580). Linear regression
models were used to adjust for total DNA and potential technical artifacts. Specifically, we used 10-fold cross-validation
for variable selection, using the “leaps” R package (version 3.0), with an initial model with chrMT read count as the dependent
variable, and “Total”, “Mapped”, “unknown”, “random”, “decoy1”, and “decoy2” read counts as the independent variables. For
each of the independent variables, we included a natural spline with df = 4 to allow for nonlinear effects. The independent
variables “Total”, “unknown”, “decoy1”, and “decoy2” read counts were selected. We then increased the natural spline df to
15 and then used backward selection to reduction model complexity, requiring P < 0.005 to keep a term in the model. The final regression model residuals were generated with the following R (version 3.6.0)
code:
 Mitochondrial SNP probe intensities were obtained from the “ukb_chrMT_l2r.txt” file downloaded from the UK Biobank, and samples
were stratified by array type (UKBelieve, Axiom). To correct for potential artifacts and/or batch effects, we generated 250
principal components (PCs) using the “rpca” command from the “rsvd” package (version 1.0.3) from autosomal nuclear probes
by randomly sampling 5% of probes from either even or odd chromosomes that were required to be present on both array types
(n∼19,500 probes). Note that we generated the two independent sets of PCs so that we could ensure that probe selection for
PCA did not bias results. Prior to PCA, all probe intensities were rank-transformed to reduce the impact of any outliers.
For each array type, all mitochondrial SNP probes (UKBelieve, n = 181; Axiom, n = 244) along with the 250 PCs were regressed on the “WES.mtDNA” metric derived as described above. Beta estimates from these
analyses were then used to generate fitted values in the full UK Biobank data set using the “predict” function (“array.mtDNA”).
Mitochondrial SNP probe intensities were obtained from the “ukb_chrMT_l2r.txt” file downloaded from the UK Biobank, and samples
were stratified by array type (UKBelieve, Axiom). To correct for potential artifacts and/or batch effects, we generated 250
principal components (PCs) using the “rpca” command from the “rsvd” package (version 1.0.3) from autosomal nuclear probes
by randomly sampling 5% of probes from either even or odd chromosomes that were required to be present on both array types
(n∼19,500 probes). Note that we generated the two independent sets of PCs so that we could ensure that probe selection for
PCA did not bias results. Prior to PCA, all probe intensities were rank-transformed to reduce the impact of any outliers.
For each array type, all mitochondrial SNP probes (UKBelieve, n = 181; Axiom, n = 244) along with the 250 PCs were regressed on the “WES.mtDNA” metric derived as described above. Beta estimates from these
analyses were then used to generate fitted values in the full UK Biobank data set using the “predict” function (“array.mtDNA”).
Given the known impact of age, sex, and cell counts on mtDNA-CN, we first used visual inspection to identify outliers for cell counts:
-
Log(WBC) ≤1.25 or ≥3
-
Log(RBC) ≤1.4 or ≥2s
-
Platelet ≤10 or ≥500
-
Log(Lymphocyte) ≤0.10 or ≥2
-
Log(Mono) ≥0.9
-
Log(Neutrophil) ≤0.75 or ≥2.75
-
Log(Eos) ≥0.75
-
Log(Baso) ≥0.45
We then excluded self-identified non-white individuals due to insufficient WES data, related individuals (used.in.pca.calculation==0),
and cell count outliers and then adjusted for age and sex using the following linear regression model:
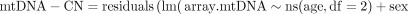 Beta estimates from these analyses were then used to generate fitted values in the full UK Biobank data set using the “predict”
function.
Beta estimates from these analyses were then used to generate fitted values in the full UK Biobank data set using the “predict”
function.
For all analyses, mtDNA-CN was standardized by subtracting the mean and dividing by the standard deviation.
A Cox proportional-hazards model was used to evaluate the association between mtDNA-CN and time to incident neurodegenerative disease, adjusting for age and sex, whereas a logistic regression model was used to evaluate associations between mtDNA-CN and prevalent neurodegenerative disease. Individuals with prevalent neurodegenerative disease were omitted from the incident analysis.
Software availability
All in-house scripts are available as Supplemental Code and at GitHub (https://github.com/syyang93/mtDNA_GE_scripts).
Competing interest statement
The authors declare no competing interests.
Acknowledgments
This work was supported by National Institutes of Health grants R01HL13573 and R01HL144569. This research was conducted using data from the Genotype-Tissue Expression (GTEx) project (dbGaP accession: phs000424.v8.p2). The GTEx project was supported by the Common Fund of the Office of the Director of the National Institutes of Health, and by the National Cancer Institute, the National Human Genome Research Institute, the National Heart, Lung, and Blood Institute, the National Institute on Drug Abuse, the National Institute of Mental Health, and the National Institute of Neurological Disorders and Stroke. This research was also conducted using the UK Biobank Resource under Application Number 17731.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at https://www.genome.org/cgi/doi/10.1101/gr.269381.120.
- Received July 27, 2020.
- Accepted January 6, 2021.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see https://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








