
Diploid genome architecture revealed by multi-omic data of hybrid mice
- Zhijun Han1,2,
- Kairong Cui3,
- Katarzyna Placek3,
- Ni Hong1,
- Chengqi Lin2,
- Wei Chen1,
- Keji Zhao3 and
- Wenfei Jin1
- 1Department of Biology, Southern University of Science and Technology, Shenzhen, Guangdong 518055, China;
- 2Institute of Life Sciences, Southeast University, Nanjing, Jiangsu 210096, China;
- 3Systems Biology Center, National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, Maryland 20892, USA
Abstract
Although mammalian genomes are diploid, previous studies extensively investigated the average chromatin architectures without considering the differences between homologous chromosomes. We generated Hi-C, ChIP-seq, and RNA-seq data sets from CD4 T cells of B6, Cast, and hybrid mice, to investigate the diploid chromatin organization and epigenetic regulation. Our data indicate that inter-chromosomal interaction patterns between homologous chromosomes are similar, and the similarity is highly correlated with their allelic coexpression levels. Reconstruction of the 3D nucleus revealed that distances of the homologous chromosomes to the center of nucleus are almost the same. The inter-chromosomal interactions at centromere ends are significantly weaker than those at telomere ends, suggesting that they are located in different regions within the chromosome territories. The majority of A|B compartments or topologically associated domains (TADs) are consistent between B6 and Cast. We found 58% of the haploids in hybrids maintain their parental compartment status at B6/Cast divergent compartments owing to cis effect. About 95% of the trans-effected B6/Cast divergent compartments converge to the same compartment status potentially because of a shared cellular environment. We showed the differentially expressed genes between the two haploids in hybrid were associated with either genetic or epigenetic effects. In summary, our multi-omics data from the hybrid mice provided haploid-specific information on the 3D nuclear architecture and a rich resource for further understanding the epigenetic regulation of haploid-specific gene expression.
Chromatin is well organized within the 3D nucleus, which renders efficient packaging of DNA while simultaneously allowing precise gene regulation and genome replication. Studies based on high-throughput chromosome conformation capture assay (Hi-C) showed that the chromatin is segregated into A and B compartments, which correspond to euchromatin and heterochromatin, respectively (Lieberman-Aiden et al. 2009). Furthermore, the mammalian genome is organized into a large number of topologically associated domains (TADs) (Dixon et al. 2012), and active TADs appear to be segregated from inactive TADs (Simonis et al. 2006; de Wit et al. 2013; Gibcus and Dekker 2013; Meuleman et al. 2013). The TAD structure is relatively stable across different cell types, which requires the function of CTCF and cohesin (Dixon et al. 2012; Zuin et al. 2014; Nora et al. 2017; Ren et al. 2017; Wang et al. 2019).
Most mammalian genomes are diploid, with one paternal haploid and one maternal haploid. Previous studies have provided rich information about the dynamics of chromatin status and histone modification during the early gamete and embryo development (Liu et al. 2016; Lu et al. 2016; Jung et al. 2017; Nagano et al. 2017; Stevens et al. 2017). However, almost all the studies only inferred the average chromatin interactions and average epigenetic status across the diploid genomes, partially because genetic variations in general individuals could not provide sufficient segregating sites for investigating allele-specific interactome. Although Tan et al. (2018) recently investigated the diploid genome at single cell resolution, characterizing the 3D genome structures of diploid mammalian cells remains challenging, and the principle of 3D nucleus organization is a mystery to some extent (Carstens et al. 2016).
Here, we introduce a hybrid mouse model that contains a large number of segregating sites for distinguishing paternal and maternal haploids, which serves as an ideal model for investigating the allele-specific 3D nucleus organization and epigenetic regulation. Using the multi-omics data from this model system, we systematically investigated the inter-chromosomal interaction pattern for each of the haploid genomes, constructed the diploid 3D nucleus, and identified allele-specific A|B compartments and TADs.
Results
Constructing the hybrid mouse system and generating multi-omics data sets
To study diploid genome architecture, we generated hybrid mice by crossbreeding male C57BL/6J (B6) and female CAST/EiJ (Cast) (Fig. 1A). Naive CD4 T cells were isolated from the hybrid mice and their parents. We applied the 3e Hi-C protocol (Ren et al. 2017) to capture the global chromatin interaction information, ChIP-seq to profile genome-wide histone modification (or transcription factor binding), and RNA-seq to profile gene expression in these cells (Supplemental Table S1). We assigned a read to B6 or Cast if there was a strain-specific allele on that read (Methods). For convenience, haploid genomes in hybrid mice inherited from B6 and Cast were called Fb6 and Fcast, respectively. The length distribution of paired-end tags (PETs) in Fb6 and Fcast are similar to that in B6 or Cast Hi-C libraries (Supplemental Fig. S1; Supplemental Table S2).
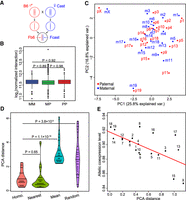
Homologous chromosomes show similar interaction patterns and the similarity correlates with allelic coexpression level. (A) Schematic representation of the hybrid mouse model, in which male B6 and female Cast were crossbred to generate hybrid progeny. Haploid genome in the hybrid originating from B6 and Cast were called Fb6 and Fcast, respectively. (B) Box plot of chromosomal interactions between maternal–maternal (MM), maternal–paternal (MP), and paternal–paternal (PP) based on allelic specific PETs. (C) PCA analysis of chromosomal interactions. The chromosomes are projected on a 2D plot, in which short distance indicated high similarity of interaction patterns: (p) paternal chromosomes; (m) maternal chromosomes. (D) Distances between homologous chromosomes on PCA projection compared with other situations: (Homo) distance between homologous chromosome pair; (nearest) distance between a chromosome and its nearest nonhomologous chromosome; (mean) mean distance between a chromosome and the other chromosomes; (random) distance between two randomly picked chromosomes. P-values were calculated using t-test. (E) Biallelic coexpression level is correlated with the similarity of chromosomal interaction pattern between homologous chromosomes. (R = 0.7, P-value = 0.0006.)
Homologous chromosomes show similar interaction patterns
We built an inter-chromosomal interaction heatmap among all chromosomes using the Hi-C PETs with both ends having strain-specific alleles (Supplemental Fig S2A). Then we classified the inter-chromosomal interactions into three groups: maternal–maternal (MM), maternal–paternal (MP), and paternal–paternal (PP). There are no significant differences among the normalized inter-chromosomal interaction for MM, MP, and PP (Student's t-test) (Fig. 1B), indicating that the data are not parentally biased or the chromosomes’ paternal or maternal origins do not bias inter-chromosomal interactions.
We conducted principal component analysis (PCA) on the interaction matrix to capture the major inter-chromosomal interaction patterns. The homologous chromosomes are in close proximity on the PCA projection compared to nonhomologous chromosomes (Fig. 1C), indicating that the homologous chromosomes have similar interaction patterns. The distances between two chromosomes on PCA projection were called PCA distance to indicate the similarity of interaction patterns. The PCA distance between homologous chromosomes is comparable to that between two nearest nonhomologous chromosomes, significantly shorter than the mean distance between a chromosome to the other chromosomes, as well as significantly shorter than the distance between two random chromosomes (Fig. 1D). The conclusions were not affected after we masked the interactions between homologous chromosomes (Supplemental Fig. S2B), suggesting that the similarity of interaction patterns between homologous chromosomes are not caused by PETs directly linking them.
It is well known that females have two copies of the X Chromosomes, with one being randomly inactivated owing to the dosage compensation effects (Schulz and Heard 2013; Crane et al. 2015). Both paternal and maternal X Chromosomes are far from the autosomes in the PCA plot (Fig. 1C), indicating that the interaction patterns of X Chromosomes are quite different from that of autosomes. The result is consistent with a recent report showing that the imprinted X Chromosome was highly compacted and had few interactions with other chromosomes (Wang et al. 2016). Recent studies showed that the inactivated X Chromosome consisted of a distinct active region (X-a) and inactive region (X-i) (Deng et al. 2015; Marks et al. 2015). We also found the similarity of interaction pattern between homologous X-a was higher than that between homologous X-i (Supplemental Fig. S2C), consistent with our expectation.
Similarity of interaction patterns between homologous chromosomes is correlated with the allelic coexpression level
The overall expression level of a gene depends on the coordinated regulation between the two alleles. We measured the allelic coexpression level of homologous chromosomes by calculating the correlation coefficient of allele-specific expressions between homologous chromosomes. We found that the similarity of interaction patterns between homologous chromosomes is highly correlated with the allelic coexpression level (Fig. 1E) (R = 0.7, P = 0.0006). The X-a shows an autosome-like pattern, whereas the X-i showed low interaction similarity and low allelic coexpressions and thus was far separated from autosomes (Supplemental Fig. S2D).
Spatial organization of chromosomes in the nucleus
Chromosome painting showed that each chromosome occupies a roughly elliptical domain termed as chromosome territory in interphase (Cremer et al. 2003; Bolzer et al. 2005; Hua and Mikawa 2018). To resolve the relative positioning of chromosome territories in the nucleus, we built an iteratively weighted adjusting model to fit each chromosome into the 3D nuclear space based on the inter-chromosomal allele-specific interaction matrix (Supplemental Fig. S3A; Methods). In our constructed 3D nucleus, there are no significant differences among the 3D distances between MM, MP, and PP (Supplemental Fig. S3B). The large chromosomes tended to locate in one polar region and the small chromosomes locate in the other polar region (Fig. 2A). The paternal and maternal X Chromosomes are far away from each other, consistent with a recent study (Giorgetti et al. 2016). We found that Chromosomes 14 and X are located toward the periphery of the nucleus (Fig 2A), which is supported by FISH analyses of lymphocytes (Mayer et al. 2005).
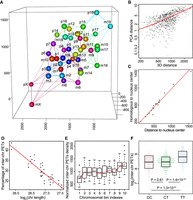
Spatial organization of chromosomes in the 3D nucleus and principles of nucleus organization. (A) Constructed 3D nucleus at chromosomal resolution. Each chromosome and its five closest chromosomes are connected by lines: (p) paternal chromosomes; (m) maternal chromosomes. (B) Chromosomal interaction pattern is positively correlated with chromosomal distance in the 3D nucleus (R = 0.67, P-value < 2.2 × 10−16). (C) Distances to the center of the 3D nucleus of paternal chromosomes are highly correlated with that of their maternal homologous counterparts. Center of the 3D nucleus is calculated as the mean coordinate of all chromosomes (R = 0.98, P-value = 1.2 × 10−14). (D) The lengths of chromosomes are negatively correlated with the percentages of inter-chromosomal interactions (R = 0.9, P-value = 1.5 × 10−14). (E) Inter-chromosomal interactions are increasing from centromere to telomere along the chromosomes. Each chromosome is evenly divided into 10 bins. The red line plots the mean value for each bin. (F) Telomere ends have much stronger inter-chromosomal interactions than that of centromere ends. Each chromosome is evenly binned into three parts, namely centromere end (C), middle (ignored), and telomere end (T). P-values were calculated using unpaired t-test.
We further found the inter-chromosomal distance in the 3D nucleus correlated with PCA distance that represents interaction patterns (Fig. 2B) (R = 0.67, P-value < 2.2 × 10−16), which is consistent with the notion that the positioning of a chromosome in the 3D nucleus contributes critically to chromatin interaction patterns. We inferred the center of the 3D nucleus according to the coordinates of all chromosomes. We found the distance of a chromosome to the center of the 3D nucleus is highly correlated with that of its homologous counterpart (Fig. 2C) (R = 0.98, P-value = 1.2 × 10−14). We also discovered that the distance of a chromosome to each of the two homologous chromosomes is highly correlated (Supplemental Fig. S3C) (R = 0.87, P-value < 2.2 × 10−16).
Chromatin organization in chromosome territories
The length of a chromosome is negatively correlated with the percentage of inter-chromosomal PETs detected on the chromosome (Fig. 2D) (R = 0.9, P-value = 1.5 × 10−14), potentially implying that the longer the chromosome is, the bigger the chromosome territory it occupies, thus the smaller the surface-area-to-volume ratio and lower fraction of inter-chromosomal interactions it has. We separated each chromosome from centromere to telomere into 10 equal bins and found that the inter-chromosomal interactions are increasing from centromere to telomere along the chromosome (Fig. 2E). Further, inter-chromosomal interactions between telomere ends are significantly higher than that between centromere ends (Fig. 2F) (P = 1.3 × 10−51). The features of centromeres or telomeres are unlikely significant impacts on the results because they only account for a tiny fraction of chromosome (Supplemental Materials; Supplemental Table S3). These results suggest that the telomere ends are located in an environment that has a higher chance to contact other chromosomes than do the centromere ends (Discussion).
Parental divergent compartments transit into the same status in hybrid mice
Global analysis showed the A|B compartment scores of haploids in hybrid mice are highly correlated with its parent of origin (Fig. 3A).We further found that the correlation coefficient between Fb6 and Fcast is higher than that between B6 and Cast (Fig. 3A). We identified A|B compartments in each genome and found that ∼88% of the compartment bins showed exactly the same status among all the four genomes (Supplemental Fig. S4A). In total, 870 A|B compartment bins (12% of the genome) showed a different status between B6 and Cast. Gene Ontology (GO) terms including olfactory transduction, neuroactive ligand-receptor interaction, and G-protein coupled receptor signaling pathway were significantly enriched in B6/Cast compartment divergent bins (Fig. 3B). The significant GO terms did not change when compartments with low score (−0.004 < score < 0.004) were removed. We found that the majority of olfactory clusters were in the same TADs that displayed divergent A|B compartments between B6 and Cast. The compartment status in Fb6 and Fcast was the same as that of their parents, respectively (Supplemental Fig. S4B).
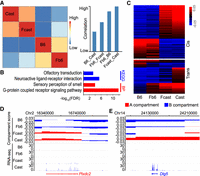
Both cis and trans effects of hybrid genomes influence the status of chromatin compartment and gene expression. The compartment analyses were conducted at 100-kb resolution. (A) Correlation of genome-wide A|B compartment scores among parents and the haploids from hybrid. Heatmap (left) and bar plot (right) of correlation coefficient between different genomes. (B) GO analysis of genes in B6 and Cast divergent A|B compartments. (C) A|B compartment status of Fb6, Fcast, B6, and Cast at B6/Cast divergent A|B compartments: (Cis) compartment status is consistent between haploid and parent of origin; (Trans) compartment status is different between haploid and parent of origin. (D) A|B compartments and gene expression in B6, Cast, Fb6, and Fcast around cis-regulated Plxdc2. (E) A|B compartments and gene expression in B6, Cast, Fb6, and Fcast around trans-regulated Dlg5.
For all 870 A|B compartment divergent bins between B6 and Cast, 58% of the haploid compartments in the hybrid mice retained the same status as did the parent of origin. For example, Plxdc2 was in an A compartment and expressed in Cast/Fcast, and it was in a B compartment and not expressed in B6/Fb6 (Fig. 3D). However, we also found that Fb6 and Fcast display different compartment status compared to their parents of origin in 42% of A|B compartment divergent bins. Among the compartments with status changes, 95% converged into same compartment status in the hybrid mice (Fig. 3C). For instance, Dlg5 was in an A compartment and highly expressed in Cast, and it did not maintain the A compartment status and became silent in Fcast (Fig. 3E).
TAD boundary shift is associated with gene expression changes
Although many methods were developed to identify TADs (Dixon et al. 2012; Filippova et al. 2014; Crane et al. 2015; Durand et al. 2016; Shin et al. 2016; Wolff et al. 2018), it is still troublesome to quantitatively compare the TADs between samples. Here, we introduce the local boundary score (LBS; available in Supplemental Code), a quantitative value along the genome with peaks representing TAD boundaries (Fig. 4A). Further analyses showed that LBSs were statistically robust and not sensitive to sequencing depth (Supplemental Fig. S5A,B). TAD boundaries and the size of a TAD inferred by LBS were essentially consistent with that inferred by HiCExplorer (Supplemental Fig. S5C–E; Wolff et al. 2018). Thus, differential LBS peaks could reveal the TAD boundary shifts between different samples (Fig. 4B). Using this approach, we only identified 648 TAD boundaries that have showed a shift between B6 and Cast owing to the conservation of TAD (Fig. 4C), among which we identified 2460 CTCF binding sites with CTCF motif. There are only 97 of these 2460 CTCF motifs that contain strain-specific SNPs, with only eight motifs potentially impacting the binding of CTCF. Therefore, genetic changes of CTCF sites may play a limited role in the TAD boundary shift between B6 and Cast. Therefore, the boundary shift should associate with chromatin state and gene expression change. Indeed, genes near the strain-specific boundaries showed much higher expression changes than those near the conserved boundaries (Fig. 4D).
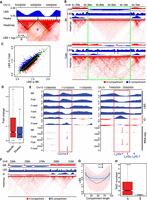
Strain-specific TAD boundaries and relationships between TAD and A|B compartment. Resolution: LBS (5 kb); ID (5 kb); Heatmap (20 kb); compartment (100 kb). (A) Definition and calculation of LBS. (B) LBSs could accurately detect the TAD boundaries. The green box outlines a region with several strain-specific TAD boundaries (black arrows) and divergent A|B compartments. (C) Significantly different TAD boundaries between B6 and Cast inferred by LBS. (D) Genes near strain-specific TAD boundaries showed stronger strain-specific expression than these near strain-shared TAD boundaries: (Specific) strain-specific TAD boundaries; (Shared) strain-shared TAD boundaries. (E) Strain-specific boundaries are associated with strain-specific gene expressions near Gzma and Ly6e, respectively. The red box highlights strain-specific boundary. (F) LBSs in B compartments are flat and smooth, whereas LBSs in A compartment fluctuate. (G) Comparison of mean and SD of LBSs in A and B compartments, showing a high variability of LBS on A compartments. (H) Box plot of number of TADs in A compartments and B compartments (P-value < 2.2 × 10−16).
A low LBS means high density of local interactions, hence nearby genes tend to have high gene expression. For instance, Gzma and Ly6 family genes are located at shifted TAD boundaries. Gzma shows a high LBS and is not expressed in B6/Fb6, and it shows a low LBS and is expressed in Cast/Fcast (Fig. 4E). In contrast, Ly6e shows a low LBS and is expressed in B6/Fb6, and it shows a high LBS and is not expressed in Cast/Fcast (Fig. 4E). Interestingly, we found that LBS values within A compartments showed substantial fluctuations, whereas LBS values within B compartments were flat and showed low variation (Fig. 4F). Global analysis showed that LBS values of A compartments show much higher inter-compartment variations than that of B compartments (Fig. 4G), indicating heterogeneities of A compartments are much higher than that of B compartments. Further analysis showed that one A compartment contains multiple TADs, and one B compartment usually contains only one TAD, further indicating the complexity of A compartments (Fig. 4H). The multiple TADs within a single A compartment may explain the high variation of LBS values within the A compartment.
Both genetic and epigenetic factors influence gene expression in the hybrid mice
We identified 721 B6-specific and 1157 Cast-specific genes in naive CD4 T cells by analyzing RNA-seq data of the B6 and Cast samples. GO analyses showed that genes associated with immune function are significantly enriched in these strain-specific genes (Fig. 5A). We further identified 158 Fb6-specific and 178 Fcast-specific genes in the hybrid mice, with 72% and 66% of them overlapped with B6- and Cast-specific genes, respectively (Supplemental Fig. S6A). The number of differentially expressed genes identified between Fb6 and Fcast is much less compared with that identified between B6 and Cast, largely owing to limited allelic reads coverage in Fb6 and Fcast.
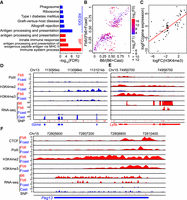
Both genetic and epigenetic regulations shape the gene expression in hybrid mouse. (A) GO enrichment for differentially expressed genes between B6 and Cast. (B) Relative expressions in parents and haploids of differentially expressed genes between Fb6 and Fcast. Dot size represents average expression level of Fb6 + Fcast. Blue to red represents average expression level of B6 + Cast. (C) Allele-specific H3K4me3 is positively correlated with allele-specific gene expression in the hybrid. Each point represents a biased ChIP-seq peak and its regulated gene (R = 0.85, P-value = 1.2 × 10−13). (D) Cast/Fcast-specific epigenetic modifications and expression near Gzma. (E) B6/Fb6-specific epigenetic modification and expression near Ly6e. (F) Maternal imprinted gene Peg13 only showed paternal-specific active epigenetic modifications in the hybrid.
We showed the relative expression of differentially expressed genes between Fb6 and Fcast in the two haploids and two parental mice in one plot. The genes distributed along the diagonal were differentially expressed in both parents and two haploids (Fig. 5B), which can be mainly attributed to cis effects. The genes distributed near the vertical line were differentially expressed in two haploids but not differentially expressed in two parents (Fig. 5B), which may be attributed to trans effects. The allele-specific H3K4me3 signals (Fig. 5C, R = 0.85, P-value = 1.2 × 10−13) and H3K4me2 signals (Supplemental Fig. S6B, R = 0.82, P-value < 2.2 × 10−16) are positively correlated with allele-specific gene expression, respectively. Gzma and Ly6e loci showed accordant allele-specific epigenetic status and gene expression in the haploids (Fig. 5D,E), potentially owing to cis effects. Paternally expressed 13 (Peg13), a maternal imprinted gene (Smith et al. 2003), was identified as a trans-effect gene in the hybrid mice. Although Peg13 was expressed in both parents, it is silent in Fcast/maternal and has Fb6/paternal-specific active histone modifications, CTCF binding, and expression (Fig. 5F).
Discussion
Most Hi-C studies lacked sufficient allele-specific information to distinguish between the paternal haploid and maternal haploid in the diploid genomes and thus only investigated the average chromatin architectures. A recent study identified interesting differences between the maternal haloid and paternal haloid using Dip-C (Tan et al. 2018). In this study, we used nearly 20 million single-nucleotide polymorphism (SNPs) existing between B6 and Cast (Keane et al. 2011; Yalcin et al. 2012), making it possible to assign the Hi-C, ChIP-seq, and RNA-seq reads to strain-specific haploids at high-resolution. To some extent, this study was the first endeavor that focused on investigation of the 3D nucleus organization of haploid chromosomes. Our analyses revealed that homologous chromosomes have a similar interaction pattern and similar distances to the center of the 3D nucleus, which has many implications for understanding gene regulation and nuclear architecture. For instance, although many studies inferred the TADs, A|B compartments, and chromatin loops using averaged interaction matrix from different cells and from different homologous chromosomes without considering the differences between the two haploids, the conclusions from these studies hold true to some extent because the homologous chromosomes have similar interaction patterns, as we have shown here.
Mouse autosomes and the X Chromosome are telocentric chromosomes that are rodlike with one centromere end and one telomere end. Our inter-chromosomal interaction analysis showed that the telomere ends have stronger inter-chromosomal interaction than the centromere ends. This observation could be explained by two nonexclusive models: (1) The centromere ends are more likely located near the center of a 3D chromosomal territories, and the telomere ends are more likely toward the surface of the 3D chromatin territories; and (2) the centromere ends are positioned toward the nuclear periphery, and the telomere ends are toward the center of the nucleus. In the second model, the positioning of a centromere end at the nuclear periphery may effectively reduce its chance of interaction with other chromosomes and thus lead to decreased inter-chromosomal interaction at the centromere end. The second model is also supported by the previous observation that the pericentromeric heterochromatin is positioned at the nuclear periphery (Holla et al. 2020).
B6 and Cast hybrid mice had been used to investigate cis and trans effects of gene regulation (Goncalves et al. 2012). The allele-specific epigenetic regulation is rarely studied, and the dynamics of epigenetic status in hybrid mice is unknown. We found parental divergent compartments converged into the same compartment status in hybrid mice owing to shared cellular environment. We found gene expression changes in hybrid owing to trans effects, with transition of histone modifications and transcription factor binding. These results confirm that microenvironment plays a critical role in the epigenetic regulation on gene expression. In summary, this study provides a landscape of diploid genome architecture and allele-specific epigenetic profiles that are important for understanding the regulation of differential gene expression between homologous chromosomes.
Methods
Mice and CD4 T cell isolation
Both C57BL/6J (B6) mice and CAST/EiJ (Cast) mice were purchased from Jackson Laboratories. The study was reviewed and approved by the Animal Care and Use Committee of the National Heart Lung and Blood Institute. All mice received humane treatment according to “Guiding Principles for Research Involving Animals and Human Beings.” Male B6 and female Cast mice were crossbred to generate the F1 hybrid mice. Naive CD4 T cells were purified from spleen and lymph nodes of B6, Cast, and hybrid mice by magnetic selection using CD4 microbeads (Miltenyi Biotech, CD4+CD62L+ T cell isolation kit II, mouse).
3e Hi-C, ChIP-seq, and RNA-seq
The multiple-enzyme Hi-C (3e Hi-C) was performed according to our previously described protocol (Ren et al. 2017; Hu et al. 2018). Different from conventional Hi-C, the fixed cells were digested with CviQI, CviAII, and BfaI. ChIP-seq experiments were performed as described previously (Barski et al. 2007). We conducted ChIP-seq using antibodies against CTCF (Millipore 07-729), RNA Polymerase II (Abcam ab5408), H3K4me3 (Abcam ab8580), and H3K4me2 (Abcam ab32356). RNA-seq was prepared as described previously (Hu et al. 2018).
SNP calling and cross validation
We obtained the mouse SNPs from the Mouse Genomes Project (Keane et al. 2011) and extracted the SNPs between C57BL/6J and CAST/EiJ using SNPsplit (Krueger and Andrews 2016). Meanwhile, we built the mm10 index with SNP positions being masked by “N.” We mapped reads from B6 and Cast Hi-C to the N-masked reference using Bowtie 2 (Langmead and Salzberg 2012). We called SNPs using mpileup (MAPQ ≥ 30) (Li et al. 2009). The called SNPs showing identical states with that downloaded from mouse genome project were kept for further analyses. In total, we obtained 13 million highly confident SNPs for distinguishing Fcast and Fb6 genome in the hybrid mice.
Hi-C, ChIP-seq, and RNA-seq data processing
For the reads mapped to N-masked mm10, redundant PETs and reads with MAPQ < 30 were filtered out. Intra-chromosomal PETs within 10 kb were filtered out because of potential self-ligations. The interaction matrices were normalized using iterative correction algorithm ICE (Imakaev et al. 2012) and further optimized in HiC-Pro (Servant et al. 2015). A|B compartments were calculated as described in Lieberman-Aiden et al. (2009). PETs from hybrid mice were split into haploid using SNPsplit with highly confident SNPs.
ChIP-seq data processing such as mapping reads, filtering reads, and constructing haploid was similar to the process for Hi-C. Peaks were called using MACS2 (Zhang et al. 2008). We counted the allele-specific reads located in the peaks using intersect in BEDTools (Quinlan 2014). The significantly different loci between Fb6 and Fcast were identified by DESeq (Love et al. 2014). ChIP-seq reads were extended to 150 bp and normalized by library size, then converted to bedGraph for visualization.
RNA-seq reads were mapped to N-masked mm10 by TopHat2 (Kim et al. 2013), and reads with MAPQ ≥ 30 were kept. The reads were split into haploid using SNPsplit. The batch effect of replicates was adjusted by ComBat from R package sva (Leek et al. 2012), and differentially expressed genes were identified by DESeq2 (Love et al. 2014) with FDR < 0.05 and fold change > 1.8.
PCA analysis of inter-chromosomal interaction matrix
We obtained all inter-chromosomal PETs with both ends containing confident SNPs in hybrid to construct an inter-chromosomal
interaction matrix. To eliminate the bias induced by chromosome size, the PETs number was normalized by the following (Imakaev et al. 2012):
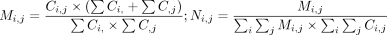 (1)
where Ci,j is the raw interaction count for each chromosome pair i and j; Mi,j is the intermediate count after normalization; and Ni,j is the final normalized count, which is scaled to the original library size.
(1)
where Ci,j is the raw interaction count for each chromosome pair i and j; Mi,j is the intermediate count after normalization; and Ni,j is the final normalized count, which is scaled to the original library size.
Construction of 3D nucleus
We developed an iteratively weighted adjusting algorithm to infer the relative positioning of each chromosome in 3D space,
in which the iterative process will continuously minimize the sum of errors between coordinate-based distance and the “real”
distance which was converted from the allele-specific interaction matrix (Supplemental Fig. S3A). In brief, we first initialized random xyz values for each chromosome, then iteratively adjusted the xyz values based on the distance errors between these chromosomes by the following formula:
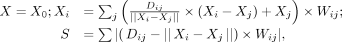 (2)
where X0 is randomly initialized 3D coordinates (xyz values) for each chromosome; Dij is the “real” distance converted from the allele-specific interaction matrix based on the fitted PETs count to distance function
(Supplemental Fig. S1A); and Wij represents the weight matrix for each chromosome pair converted from the allele-specific interaction matrix. The aim of the
iteration is to achieve the smallest S.
(2)
where X0 is randomly initialized 3D coordinates (xyz values) for each chromosome; Dij is the “real” distance converted from the allele-specific interaction matrix based on the fitted PETs count to distance function
(Supplemental Fig. S1A); and Wij represents the weight matrix for each chromosome pair converted from the allele-specific interaction matrix. The aim of the
iteration is to achieve the smallest S.
Because the interaction between homologous chromosomes may introduce bias to the 3D model, we reset the interactions between homologous chromosome pairs to the average interaction density of the corresponding chromosome to all nonhomologous chromosomes. We also constructed the 3D model with separated X Chromosome (X-a and X-i) (Supplemental Fig. S3C) and without X Chromosome (Supplemental Fig. S3D).
Local boundary score (LBS)
LBS is defined as logarithm of the ratio of local interactions to cross-local interactions in a given window (Fig. 4A). In short, interactions within left side (A1) and right side (A2) of the window were defined as local interaction, whereas interactions between the left side and right side was defined as cross-local interaction (B) (Fig 4A); thus LBS of a bin was calculated by LBS = log2(A1 + A2)/B. The peak of LBS indicated the presence of the TAD boundary and Peakdet (http://billauer.co.il/peakdet.html) was used to call peaks. ROSE, initially for identifying super enhancers (Whyte et al. 2013), was used to calculate LBS biases for identifying TAD boundary shifts between samples.
Software availability
All data were analyzed using Perl (https://www.perl.org) and R version 3.5.3 (2019-03-11) (R Core Team 2019). The scripts for processing Hi-C data including LBS are maintained in the GitHub code repository (https://github.com/hangeneral/hybridMiceHiC) and are also available as Supplemental Code.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE132898.
Competing interest statement
The authors declare no competing interests.
Acknowledgments
The study was supported by National Key Research and Development Program of China (2018YFC1004500) to W.J.; Shenzhen Science and Technology Program (JCYJ20170817111841427, KQTD20180411143432337) to W.J.; and Division of Intramural Research, National Heart, Lung and Blood Institute to K.Z. This study was supported by the Center for Computational Science and Engineering of Southern University of Science and Technology.
Author contributions: K.Z. and W.J. conceived this project. K.C. and K.P. did the experiments. Z.H. constructed the computational model and performed data analysis. W.J., K.Z., W.C., N.H., and C.L. supervised this study. Z.H., W.J., and K.Z. wrote the manuscript with input from all authors.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.257568.119.
- Received September 26, 2019.
- Accepted July 23, 2020.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








