
CRISPR-based modular assembly of a UAS-cDNA/ORF plasmid library for more than 5500 Drosophila genes conserved in humans
- 1Shanghai Diabetes Institute, Shanghai Key Laboratory of Diabetes Mellitus, Shanghai Clinical Center for Diabetes, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai 200233, China;
- 2Cyrus Tang Hematology Center, Soochow University, Suzhou 215123, China;
- 3Shanghai Institute of Endocrine and Metabolic Diseases, Shanghai 200025, China;
- 4Shanghai National Clinical Research Center for Endocrine and Metabolic Diseases, Key Laboratory for Endocrine and Metabolic Diseases of the National Health Commission of the PR China, Shanghai Institute of Endocrine and Metabolic Diseases, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China;
- 5Department of Anatomy and Physiology, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China
Abstract
Construction of a genome-wide transgenic UAS-cDNA/ORF library in Drosophila based on the binary GAL4/UAS system has been severely hampered by technical difficulties, although genome-wide cDNA or ORF resources of Drosophila, human, and mouse have been publicly available for more than a decade. Here, we developed a new method named CRISPR-based modular assembly (CRISPRmass) for the high-throughput construction of a genome-wide UAS-cDNA/ORF library from publicly available cDNA/ORF resources. Through cleavage of shared vector sequences of cDNA/ORF plasmids by CRISPR/Cas9 and subsequent insertion of UAS modules by Gibson assembly, the procedure of construction of such a library by CRISPRmass is standardized as massively parallel two-step test tube reactions before bacterial transformation. Using CRISPRmass, we generated 5551 UAS-cDNA/ORF constructs covering 83% of the Drosophila genes conserved in humans in the Drosophila Genomics Resource Center (DGRC) Gold Collection, and among them, 5518 were generated within 3 mo by three people. Our results show that CRISPRmass allows modulization, simplicity, efficiency, and adaptability in the generation of a genome-wide UAS-cDNA/ORF plasmid library by using publicly available cDNA/ORF resources. CRISPRmass can be applied to editing various genome-wide libraries in general and is an alternative to Gateway technology in high-throughput plasmid library editing. Furthermore, the more than 5500 UAS-cDNA/ORF plasmids of Drosophila genes serve as a powerful resource for gain-of-function (GOF) screening in cultured cells and for generation of a transgenic UAS-cDNA/ORF library in Drosophila.
Genome-wide genetic screens are powerful tools to explore gene function in complex biological processes. In Drosophila, genome-wide genetic screens have been relying on loss-of-function (LOF) screens. Based on the binary GAL4/upstream activation sequence (UAS) system (Brand and Perrimon 1993), genome-wide transgenic UAS-RNAi libraries for LOF screens have been constructed for targeted knockdown of most of the Drosophila genes (Dietzl et al. 2007; Perkins et al. 2015). However, the majority of Drosophila genes show no obvious LOF phenotypes (Miklos and Rubin 1996), therefore the function of such genes cannot be determined by LOF studies. Thus, gain-of-function (GOF) screens may provide a valuable tool to uncover function of such genes (Miklos and Rubin 1996; St Johnston 2002).
In contrast to construction of genome-wide transgenic UAS-RNAi libraries for LOF screens, construction of a genome-wide transgenic UAS-cDNA/ORF library for GOF screens in Drosophila has been challenging. Genome-wide cDNA/ORF resources of Drosophila (Rubin et al. 2000; Stapleton et al. 2002), human (Yang et al. 2011), and mouse (https://genecollections.nci.nih.gov/MGC) have been publicly available for more than a decade, but the efforts by different research groups to create such a genome-wide cDNA/ORF library using these resources have never ceased. In 2008, a collection of UAS-ORF plasmids of 236 human genes and the corresponding transgenic fly lines have been created based on the conventional cloning technique (Xu et al. 2008). In 2013, a platform of the recombinational (Gateway) cloning technique was established and used successfully in constructing the UAS-ORF plasmids for 655 Drosophila genes (Bischof et al. 2013); based on this platform, to date, the UAS-ORF plasmids and transgenic flies mapping to about 2850 Drosophila genes have been generated (FlyORF, http://www.FlyORF.ch). In 2018, the human ORF collection containing 365 UAS-ORF plasmids for Drosophila transgenesis was constructed and deposited to the Drosophila Genomics Resource Center (DGRC, https://dgrc.bio.indiana.edu). Despite these efforts, there is still a long way to go before a genome-wide transgenic Drosophila UAS-cDNA/ORF library is created. The rate-limiting step of construction of a genome-wide transgenic Drosophila UAS-cDNA/ORF library is the technical difficulty of constructing a library of UAS-cDNA/ORF plasmids. Currently, construction of such a plasmid library has almost exclusively relied on the recombinational (Gateway) cloning technique (Bischof et al. 2013). The recombinational (Gateway) cloning technique requires handling tens of thousands of cDNAs or ORFs by time-consuming individualized manipulations of every single gene including primer design and synthesis, PCR, gel purification, and sequencing. Particularly, construction of UAS-cDNA/ORF plasmids for long cDNAs or ORFs by the recombinational (Gateway) cloning technique is truly problematic. Therefore, the technical difficulties of construction of a library of UAS-cDNA/ORF plasmids have severely hampered construction of such a genome-wide transgenic UAS-cDNA/ORF library in Drosophila.
To achieve the goal of performing genome-wide GOF screens, some research groups established alternative approaches that circumvented construction of UAS-cDNA/ORF plasmids. Through random insertion of the P-element transposon bearing a UAS enhancer and an Hsp70 minimal promoter (EP) into the Drosophila genome, about 8500 EP lines have been created (Rørth 1996; Rørth et al. 1998; Mata et al. 2000). Because more than one EP line may overexpress or misexpress the same gene in the presence of GAL4, it is unclear how many genes were covered by these lines, probably because it is very laborious to identify genomic locations of UAS insertions (Mata et al. 2000). Currently a representative set of these EP lines containing about 1700 EP lines which maps to fewer than 1700 genes is available in the Bloomington Drosophila Stock Center and covers <12.38% of 13,733 Drosophila euchromatic protein-coding genes (Lin et al. 2007). However, this approach has intrinsic drawbacks. If an EP element inserts into a gene in antisense orientation, an antisense transcript may be generated and therefore causes loss of function of the gene. Furthermore, EP lines do not allow expression of genes from other organisms including human. The P-element transposon shows inherent insertional hot spots and orientation bias, and therefore it is impossible to construct a saturated genome-wide GOF library through generation of EP lines (St Johnston 2002). Nevertheless, GOF screens by EP lines have made great contributions to biological research over the past two decades.
CRISPRa is another approach to construct a genome-wide GOF library. During the past two years, two CRISPRa-based methods for construction of a genome-wide GOF library in Drosophila were developed (Ewen-Campen et al. 2017; Jia et al. 2018). These two methods serve as useful strategies for GOF screening, especially for genes of which the cDNA/ORFs are not available. However, for every single gene, DNA oligos for at least one sgRNA have to be individually well designed in order to improve success rates of sgRNAs and to reduce their off-target effects, followed by synthesis of DNA oligos and sequencing of sgRNA constructs (Jia et al. 2018). Furthermore, these two CRISPRa-based methods have some other drawbacks due to the intrinsic properties of CRISPRa. For example, CRISPRa-based GOF is sometimes toxic in vivo even in the absence of sgRNAs when crossed with some GAL4 drivers (Jia et al. 2018); sometimes two or more genes share a common promoter region in the Drosophila genome, which may cause the expression of more than one gene to be controlled by a single sgRNA, and Drosophila CRISPRa does not allow expression of genes from other organisms, including human.
Ideally, the best genome-wide GOF library in Drosophila is still the standard cDNA/ORF-based transgenic UAS-cDNA/ORF library, which can overcome all the aforementioned drawbacks. Therefore, there is an urgent need to develop a simple and efficient method of creating a genome-wide UAS-cDNA/ORF plasmid library using cDNA/ORF resources of different species. Here, we developed a new method named CRISPR-based modular assembly (CRISPRmass) for high-throughput construction of a genome-wide UAS-cDNA/ORF plasmid library from publicly available cDNA/ORF resources, and using CRISPRmass, we created more than 5500 UAS-cDNA/ORF plasmids for Drosophila genes conserved in humans.
Results
The CRISPRmass pipeline for construction of a UAS-cDNA/ORF plasmid library
The procedure of construction of a genome-wide UAS-cDNA/ORF plasmid library from publicly available cDNA/ORF resources by CRISPRmass was standardized as massively parallel two-step test tube reactions before bacterial transformation (Fig. 1). The first step is that the identical vector backbones of the cDNA/ORF constructs are cleaved by Cas9/sgRNA. The cleavage site is adjacent to the 5′ end of cDNA/ORF. The backbones of some vectors are cleaved twice by Cas9/sgRNA in order to remove unnecessary DNA sequence. The cleavage products, without purification, are directly subjected to the subsequent step. The second step is that a vector-specific UAS module is joined into the Cas9-linearized cDNA/ORF constructs through Gibson assembly, resulting in UAS-cDNA/ORF constructs. The vector-specific UAS module comprises a core UAS module and 28–40 bps of 5′- and 3′-terminal sequences overlapping with the backbone cleavage site 5′ and 3′ ends, respectively. The 5′ and 3′ overlapping terminal sequences enable Gibson assembly. A core UAS module comprises 10 copies of UAS, an Hsp70 minimal promoter, an attB sequence for phiC31-mediated genomic integration, a mini-white transformation marker for Drosophila, and a positively selectable antibiotic resistance gene distinct from that of the vector. A vector-specific UAS module can be built up by PCR modifying a core UAS module according to the vector backbone cleavage site. After the second step of the two test tube reactions, Gibson assembly products are directly subjected to Escherichia coli transformation. Transformants are selected on LB plates containing antibiotic corresponding to the antibiotic resistance gene of the vector-specific UAS module. Theoretically, only the desired UAS-cDNA/ORF colonies can grow.
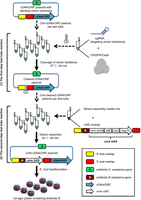
The CRISPRmass pipeline for construction of a UAS-cDNA/ORF library. (1) The first step test tube reaction. The identical vector backbones of cDNA/ORF plasmids are cleaved by Cas9/sgRNA. The cleavage site is located in vector backbones adjacent upstream of the cDNA/ORF 5′ end. The cleavage products, without purification, are directly subjected to the second step test tube reaction. The sgRNAs used in cleavage reactions are prepared by in vitro transcription and can be used directly without purification. Then, 28–40 bp of the 5′ end of the cleavage site (yellow box) is defined as “5′ end overlap,” and 28–40 bp of the 3′ end of the cleavage site (red box) is defined as “3′ end overlap.” The vector backbone carries the antibiotic A resistance gene (green box). (2) The second step test tube reaction. A vector-specific UAS module is joined into the Cas9-linearized cDNA/ORF plasmids right upstream of cDNA/ORF 5′ end through Gibson assembly, resulting in UAS-cDNA/ORF constructs. Gibson assembly products are directly subjected to E. coli transformation. Transformants are selected on LB agar plates containing antibiotic B corresponding to the antibiotic B resistance gene (brown box) of the vector-specific UAS module. Only the desired UAS-cDNA/ORF colonies can grow. A UAS module comprises 10 copies of UAS, a Hsp70 minimal promoter, an attB sequence for phiC31-mediated genomic integration, a mini-white transformation marker for Drosophila transgenesis, a selectable antibiotic B resistance gene for positive selection, and the 5′ end overlap and 3′ end overlap enabling Gibson assembly. Gibson assembly filters out any potential off-target DNA cleavages caused by CRISPR/Cas9.
Proof of principle for construction of UAS-cDNA/ORF plasmids by CRISPRmass
To show the feasibility of using CRISPRmass to construct a UAS-cDNA/ORF plasmid library from publicly available cDNA or ORF resources such as the DGRC Gold Collection, we first attempted to create a UAS-cDNA/ORF plasmid sublibrary, UAS-DUBs, by CRISPRmass for all the 41 deubiquitinases (DUBs) of Drosophila from the DGRC Gold Collection (Tsou et al. 2012). There are three reasons for selecting DUBs to create a UAS-cDNA/ORF plasmid sublibrary as proof of principle. First, DUBs play important roles in diverse biological processes, and altered DUB activity is associated with a number of diseases. However, a collection of UAS-DUBs has not yet been reported. Second, of all the 41 Drosophila DUBs, the cDNA/ORF clones for 31 Drosophila DUBs are available in the DGRC Gold Collection, which were constructed in four of six total vectors, except pBS SK (−) and pCR2.1 vectors. To build a complete CRISPRmass platform for construction of the UAS-cDNA/ORF library from the DGRC Gold Collection, we only need to further show CRISPRmass also works for cDNA/ORF clones constructed in pBS SK (−) and pCR2.1; therefore, we randomly picked up a clone in which CG7747 cDNA was cloned in pBS SK (−) and a clone in which Prdm13 ORF was cloned in pCR2.1. Third, for the rest of the 10 Drosophila DUBs, six Drosophila DUB ortholog clones are available from the human CCSB-Broad Lentiviral Expression Library (Yang et al. 2011), and one Drosophila DUB ortholog clone is available from the mouse Mammalian Gene Collection (MGC). These human and mouse DUB clones allow us to examine whether CRISPRmass can apply to cDNA/ORF libraries of other species. The remaining three Drosophila DUBs do not have publicly available cDNA/ORF clones of human or mouse orthologs. Thus, in total, 40 Drosophila, human, and mouse cDNA/ORF clones correspond to eight distinct vectors (Supplemental Table S1). All the six human DUB ORFs from the human CCSB-Broad Lentiviral Expression Library were fused with a C-terminal V5 tag in pLX304 vector. To facilitate detection of MYC expression of Drosophila and mouse cDNA/ORFs by immunostaining and western blotting, we picked up one cDNA/ORF clone for each of the remaining seven vectors and inserted two or three copies of the MYC epitope tag right before the stop codon (Supplemental Table S1).
To generate UAS-CG7747, UAS-Prdm13, and 38 UAS-DUB constructs by CRISPRmass, we first determined the optimal Cas9 cleavage sites upstream of cDNA/ORF for all eight vectors. The vector backbones of cDNA/ORF clones in pOT2, pOTB7, pFLC-I, pBS SK(−), pCR2.1, and pDNR-Dual vectors are cleaved once by Cas9 together with a single sgRNA, whereas the vector backbones of cDNA/ORF clones in pLX304 and pCMV-SPORT6 vectors are cleaved twice by Cas9 together with two different sgRNAs to remove unnecessary sequences (Supplemental Fig. S1). The vector backbone of pOTB7_DraIII is identical to that of pOTB7, so these two vectors can be treated as one in terms of sgRNA design. We designed 3–20 sgRNA target sequences for each cleavage site of the eight vectors and synthesized 53 sgRNAs by in vitro transcription (Supplemental Table S2). Then we examined in vitro cleavage efficiency of all these sgRNAs and found most of them can be used for CRISPRmass (Supplemental Figs. S2, S3; Supplemental Table S2). We selected one or two sgRNAs for each vector (Supplemental Table S3). We also searched the genome of the corresponding species for sequences with 14 consecutive nucleotides identical to the optimal sgRNA target sequences, and we did not find any potential off-target sites in Drosophila, human, or mouse cDNA sequences. Based on the cleavage sites determined by the optimal sgRNAs, we generated a UAS module for each of the eight vectors. After confirmation by DNA sequencing, the vector-specific UAS modules were released by EcoRI digestion and joined into the corresponding Cas9/sgRNA linearized cDNA/ORF plasmids by Gibson assembly. The Gibson assembly products were directly used to transform E. coli competent cells, and transformants were selected on LB plates containing ampicillin or chloramphenicol depending on the antibiotic resistance gene of the UAS modules. Subsequent isolation and restriction analysis of plasmids showed that we successfully generated UAS-CG7747, UAS-Prdm13, and 38 UAS-DUB constructs by CRISPRmass (Fig. 2; Supplemental Table S1). Similar to UAS-cDNA/ORF constructs created by conventional cloning techniques, the UAS-cDNA/ORF constructs generated on eight distinct vectors by CRISPRmass retain the key features of a classic UAS-cDNA/ORF construct of GAL4/UAS system, particularly the UAS and the Hsp70 minimal promoter. Therefore, we reason that UAS-cDNA/ORF constructs generated by CRISPRmass should have no difference in terms of cDNA/ORF expression with those generated by conventional cloning techniques.
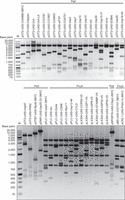
Restriction analysis of 40 UAS-cDNA/ORF constructs generated by CRISPRmass. UAS-CG7747, UAS-Prdm13, and 38 UAS-DUB constructs were generated by CRISPRmass from the DGRC Gold Collection and analyzed by PstI or PvuII digestion, and the expected restriction patterns for all the UAS-cDNA/ORF constructs were observed (Supplemental Table S1). (M) DNA marker.
The UAS-cDNA/ORF constructs generated by CRISPRmass were expressed in the presence of GAL4 in Drosophila
We next generated transgenic UAS-CG7747, UAS-Prdm13, and 38 UAS-DUB lines in Drosophila using these 40 constructs created in eight distinct vectors by CRISPRmass. To test whether these transgenes can be expressed in the presence of GAL4 in vivo, we selected one tagged transgene for each of eight vectors and crossed to motor neuron-specific OK6-GAL4 and eye-specific GMR-GAL4 drivers, respectively. Immunostaining showed these transgenes were expressed in the larval motor neurons (Fig. 3A,B), and western blotting showed these transgenes were expressed in adult fly eyes (Fig. 3C–E). The levels of expressed proteins vary among different transgenes, probably due to differences in translation regulation and protein degradation, but not distinct vectors. Together, these results showed that the UAS-cDNA/ORF constructs in eight distinct vectors generated by CRISPRmass were expressed in the presence of GAL4 in Drosophila; therefore, we conclude that CRISPRmass can be used for construction of a genome-wide UAS-cDNA/ORF library using the publicly available cDNA/ORF resources of Drosophila, human, and mouse.
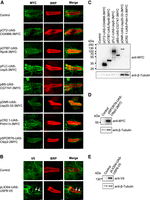
The UAS-cDNA/ORF constructs generated by CRISPRmass were expressed in the presence of GAL4 in Drosophila. Transgenic UAS-CG7747, UAS-Prdm13, and 38 UAS-DUB lines were generated by CRISPRmass in Drosophila using the 40 cDNA/ORF plasmids constructed in eight distinct vectors. An epitope-tagged transgene for each of the eight vectors was chosen and crossed to motor neuron–specific OK6-GAL4 and eye-specific GMR-GAL4 drivers, respectively. (DUB) Deubiquitinase. (A,B) Immunostaining showed these transgenes were expressed in the larval motor neurons, and V5 signals are indicated by arrows. Presynaptic active zones are labeled with anti-BRP (nc82) (red). (C–E) Western blotting showed these transgenes were expressed in adult fly eyes. The epitope-tagged proteins and their expected molecular weights are as follows: CG4968-3MYC, 33.9 kD; Rpn8-3MYC, 41.6 kD; Usp5-3MYC, 95.6 kD; CG7747-3MYC, 62.5 kD; Usp20-33-3MYC, 113.2 kD; Prdm13-3MYC, 54.1 kD; Ctbp2-2MYC, 48.3 kD; USP8-V5, 130.0 kD.
The UAS-cDNA/ORF constructs generated by CRISPRmass were functional in the presence of GAL4 in Drosophila
To further validate whether transgenes generated by CRISPRmass are functional, we screened the UAS-CG7747, UAS-Prdm13, and 38 UAS-DUB transgenes for phenotypes by crossing to eye-specific GMR-GAL4, wing-specific A9-GAL4, and muscle-specific MEF2-GAL4 drivers, respectively. Activation of the UAS-Prdm13 and several UAS-DUB transgenes elicited distinct phenotypes (Fig. 4A–C), indicating the UAS-CG7747, UAS-Prdm13, and 38 UAS-DUB transgenes generated by CRISPRmass are functional.
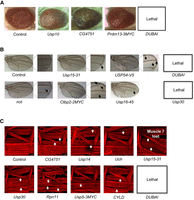
The UAS-cDNA/ORF constructs generated by CRISPRmass were functional in the presence of GAL4 in Drosophila. UAS-CG7747, UAS-Prdm13, and 38 UAS-DUB transgenes were screened for phenotypes by crossing to eye-specific GMR-GAL4 (A), wing-specific A9-GAL4 (B), and muscle-specific MEF2-GAL4 (C) drivers, respectively. Activation of the UAS-Prdm13 and several UAS-DUB transgenes elicited distinct phenotypes (arrows).
Construction of a UAS-cDNA/ORF plasmid library for more than 5500 Drosophila genes conserved in humans by CRISPRmass
We applied CRISPRmass to generate a genome-wide UAS-cDNA/ORF plasmid library using conserved cDNA/ORF clones from the DGRC Gold Collection (Fig. 5). As far as we know, until now the most comprehensive Drosophila cDNA/ORF library is the DGRC Gold Collection, which comprises 12,192 clones mapping to 10,066 Drosophila genes, and among them, 6687 Drosophila genes conserved in humans are available based on OrthoDB v9.1 (Zdobnov et al. 2017). Given the general interest of researchers, we focused on the 6687 Drosophila genes conserved in humans to generate a genome-scale UAS-cDNA/ORF library. For the 6687 conserved genes, excluding the clones of which vectors are not clearly specified in the DGRC list and the 33 clones for which UAS-cDNA/ORF plasmids have been previously constructed successfully, 6515 cDNA/ORF clones mapping to 6515 genes were subjected to CRISPRmass. At least one colony grew on LB plates with appropriate antibiotics for 5987 CRISPRmass reactions after transformation. We randomly picked up only one colony from each plate for subsequent restriction analysis. We performed restriction analysis of UAS-cDNA/ORF constructs with PstI- or PvuII-dependent specific vectors, because UAS modules bear at least two recognition sites for either of the two restriction enzymes, and meanwhile all these vectors bear at least one recognition site for either of the two restriction enzymes. Thus, restriction digestions of UAS-cDNA/ORF constructs with PstI or PvuII produce the UAS module-specific fragments and the UAS module-vector assembly-specific fragments. Indeed, we observed both UAS module-specific fragments and UAS module-vector assembly-specific fragments for 5903 constructs, demonstrating that UAS modules were inserted into 98.6% of the 5987 constructs successfully under the condition of randomly analyzing only one colony for each UAS-cDNA/ORF construct, resulting in 5903 UAS-cDNA/ORF constructs (Supplemental Fig. S4; Supplemental Table S4). Using an Hsp70 minimal promoter-specific primer, we then sequenced the rest of 84 constructs for which no UAS module-specific fragments or UAS module-vector assembly-specific fragments were observed, and we obtained no sequencing signals.
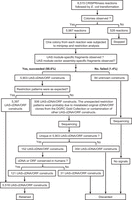
Flow chart of generation of 5518 UAS-cDNA/ORF constructs within 3 mo, in which 6515 DGRC cDNA/ORF plasmids covering 6515 Drosophila genes conserved in humans were subjected to CRISPRmass reactions. At least one colony grew on LB agar plates with appropriate antibiotics for 5987 reactions after transformation. Only one colony from each plate was randomly picked up for subsequent restriction analysis. UAS module-specific fragments and UAS module-vector assembly-specific fragments were observed in 5903 constructs, demonstrating that UAS modules were inserted into 98.6% of the 5987 constructs successfully, resulting in 5903 UAS-cDNA/ORF constructs. After a series of analyses of 5903 UAS-cDNA/ORF constructs were conducted, 5518 UAS-cDNA/ORF constructs covering 5518 Drosophila genes conserved in humans were retained as a resource.
We next analyzed the restriction patterns of the 5903 UAS-cDNA/ORF constructs and found the restriction patterns were as expected for 5397 UAS-cDNA/ORF constructs mapping to 5397 genes. The unexpected restriction patterns for the remaining 506 UAS-cDNA/ORF constructs were probably caused by mislabeled original cDNA/ORF clones from the DGRC Gold Collection or contamination of other UAS-cDNA/ORF constructs. Subsequent sequencing results of the 506 UAS-cDNA/ORF constructs indicated that 354 constructs were not unique in the 5923 UAS-cDNA/ORF constructs and were therefore discarded. The remaining 152 constructs were unique in the 5923 UAS-cDNA/ORF constructs; among them, 121 constructs of which cDNAs or ORFs are conserved in humans were retained and the remaining 31 constructs were discarded. In summary, we created 5518 UAS-cDNA/ORF constructs mapping to 5518 Drosophila genes conserved in humans. All 5518 UAS-cDNA/ORF constructs were generated by CRISPRmass successfully within 3 mo by three people (Fig. 5).
We further evaluated the quality of the more than 5500 UAS-cDNA/ORF constructs generated by CRISPRmass. We fully Sanger sequenced the cDNA/ORFs of the first six UAS-cDNA/ORF constructs cloned in each of the four vectors on the list (Supplemental Tables S4–S6) and found that the entire sequences of cDNA/ORFs of the 24 UAS-cDNA/ORF constructs were all as expected, demonstrating that the accuracy rate of the cDNA/ORFs of those 24 constructs is 100%. To examine whether the cDNA/ORFs of those 24 constructs can be expressed in the presence of GAL4, we fused the 24 cDNA/ORFs with a 3xFLAG tag to the N terminus or C terminus and transfected HEK293T cells with each UAS-cDNA/ORF construct together with a GAL4-expressing plasmid, pCMV-GAL4 (Potter et al. 2010). The subsequent western blotting experiments detected specific signals for all the 24 cDNA/ORFs (Supplemental Fig. S5), demonstrating that all the 24 UAS-cDNA/ORFs generated by CRISPRmass were expressed in the presence of GAL4.
Together with the previously generated UAS-cDNA/ORF constructs for 31 Drosophila DUBs and two functionally unknown genes, we generated a total of 5551 UAS-cDNA/ORF constructs mapping to 5551 genes, which cover 83% of the 6687 Drosophila genes conserved in humans (Supplemental Table S4).
Thus, CRISPRmass may be even more suitable for high-throughput UAS-cDNA/ORF library construction than the recombinational (Gateway) cloning technique, specifically when a suitable Gateway entry clone set is not available, but a clone set of any other sort does exist. Compared to the Gateway cloning technique, CRISPRmass does not need to generate any entry constructs and thus circumvent all the individualized manipulation for each entry construct, such as primer design and synthesis, PCR, sequencing, gel purification, and so forth. For example, for construction of a UAS-cDNA/ORF library containing 5551 genes from the DGRC Gold Collection, which were cloned in six distinct vectors, the Gateway cloning technique requires at least 5551 individualized manipulations, including primer design and synthesis, PCR, gel purification, and sequencing. In contrast, CRISPRmass allows us to manipulate shared vector sequences; therefore, we only need to design and synthesize primers for preparation of only six sgRNAs and to build up only six UAS modules by PCR modifying the core module according to the six vectors. The remaining manipulations are uniformed test tube reactions followed by E. coli transformation (Table 1). Moreover, of 6687 conserved genes in the DGRC Gold Collection, we observed that for the total 17 cDNAs of >7000 bps (the longest one is 9337 bps), the UAS-cDNA constructs of 12 cDNAs were successfully created, whereas for the remaining five cDNAs, two original cDNA bacterial clones failed to grow and the other three original cDNA bacterial clones were not as expected probably due to mislabeling of the original clones. The size limit for CRISPRmass is not known, because we do not have cDNAs larger than 9337 bps at hand. We infer that the size limit for CRISPRmass depends on the second step reaction, insertion of modules by Gibson assembly, because the first step reaction of CRISPRmass, CRISPR/Cas9-mediated cleavage, is not affected by DNA size. It has been reported that Gibson assembly products as large as 300 kb have been cloned in E. coli successfully (Gibson et al. 2009). We therefore conclude that CRISPRmass is also suitable for generating UAS-cDNA/ORF constructs from long cDNAs or ORFs, which could be because CRISPRmass does not manipulate the cDNAs or ORFs themselves but vector backbones and the high cloning capacity of Gibson assembly. Furthermore, even if a suitable Gateway entry clone set is available, the cost of key commercially available kits for construction of the UAS-cDNA/ORF library by CRISPRmass is merely ∼40% of that by the Gateway LR reaction (Table 1). In summary, CRISPRmass is faster and cheaper than Gateway for high-throughput construction of desired destination plasmids, specifically when a starting plasmid library is not Gateway compatible.
Comparison of construction of a UAS-cDNA/ORF library (5551 plasmids, six vector backbones) from the Drosophila DGC Gold Collection by recombinational cloning and CRISPRmass
Discussion
Here, we developed a method named CRISPRmass, and to our knowledge, this is the first time that CRISPR/Cas9 has been used in high-throughput plasmid library construction. In addition, by using CRISPRmass, we generated a total of 5551 UAS-cDNA/ORF constructs, covering 83% of the 6687 conserved genes in humans in the DGRC Gold Collection, and among them, 5518 were generated within 3 mo by three people. The 5551 UAS-cDNA/ORF plasmids of Drosophila genes serve as a resource for GOF screening in cultured cells and for generation of transgenic UAS-cDNA/ORF library in Drosophila.
Compared to the conventional or recombinational (Gateway) cloning technique, CRISPRmass offers several advantages in generation of a genome-wide UAS-cDNA/ORF plasmid library. First, it offers modularization. We introduced a new concept of modular assembly design and manipulation of vector backbone for construction of a genome-wide library. A combinatory use of CRISPR/Cas9 and Gibson assembly allows editing shared sequence in vector backbone, therefore it modulizes library construction. Once it is constructed for a given vector, a UAS module can fit any cDNA or ORF clones as long as these clones are in the same vector, which avoids individualized design and manipulation for every single cDNA or ORF clone. Another benefit of modulization is that CRISPRmass is also suitable for generating UAS-cDNA/ORF constructs from long cDNAs or ORFs, because CRISPRmass does not manipulate the cDNAs or ORFs themselves but vector backbones. In contrast, construction of UAS-cDNA/ORF plasmids of long cDNAs or ORFs by the conventional or recombinational (Gateway) cloning techniques is difficult. Second, it offers simplicity. The construction of a UAS-cDNA/ORF library from existing cDNA/ORF resources is uniformed and simplified as two steps of test tube reactions followed by bacterial transformation and analysis of only one colony by restriction digestion. For hundreds of thousands of genes, these procedures can be carried out in parallel. The remaining manipulations do not require primer design and synthesis, PCR, gel purification, sequencing, or any advanced experimental techniques (Table 1). Third, it offers efficiency. Introduction of an antibiotic resistance gene into a UAS module enables positive selection of UAS-cDNA/ORF clones. Moreover, Gibson assembly filters out any potential off-target DNA cleavages caused by CRISPR/Cas9, because it requires that both 5′ and 3′ ends of a Cas9-cleaved vector overlap with those of a UAS module respectively. Fourth, it offers adaptability. A UAS module can fit any vector by modifying a core UAS module, and the expression levels of genes under the control of UAS can be easily modulated by adjusting the UAS copy number.
The core concept of CRISPRmass is to manipulate shared vector sequences for high-throughput plasmid library editing. Theoretically, the first step of CRISPRmass manipulation, which is cleavage of shared vector sequences of library plasmids by CRISPR/Cas9, could be altered to other methods such as zinc-finger nuclease (ZFN) (Kim et al. 1996; Smith et al. 2000) and transcription activator-like effector nuclease (TALEN) (Christian et al. 2010). The second step of CRISPRmass manipulation, which is insertion of modules by Gibson assembly, could be altered to other seamless cloning methods such as cotransformation cloning (Bubeck et al. 1993), seamless ligation cloning extract (SLiCE) (Zhang et al. 2012), ligation-independent cloning (LIC) (Aslanidis and de Jong 1990), sequence- and ligation-independent cloning (SLIC) (Li and Elledge 2007), In-Fusion cloning (Zhu et al. 2007), and T5 exonuclease-dependent DNA assembly (TEDA) (Xia et al. 2019).
In addition to generating UAS-cDNA/ORF plasmid libraries, CRISPRmass has broader applicability to constructing some other genome-scale plasmid libraries, for example, constructing mammalian cDNA/ORF expression libraries by CRISPRmass-assisted insertion of a CMV promoter into upstream of cDNA/ORFs, which is equivalent to transferring cDNA/ORFs into a CMV promoter–containing expression vector. Likewise, CRISPRmass can also be used for high-throughput transferring clone collections that are not compatible with Gateway into the Gateway entry vectors, followed by LR reactions to produce desired destination constructs when multiple different expression vectors are likely to be desirable. Thus, CRISPRmass can be applied to editing various genome-wide plasmid libraries in general and is an alternative to Gateway technology in high-throughput plasmid library editing, paving the way for global studies of the biological networks.
Methods
cDNA and ORF clones
The Drosophila, mouse, and human cDNA and ORF clones used in this study are from the Drosophila Genome Resource Center (https://dgrc.bio.indiana.edu) Gold Collection, the mouse Mammalian Gene Collection (MGC, https://genecollections.nci.nih.gov/MGC/), and the human CCSB-Broad Lentiviral Expression Library (Yang et al. 2011).
Drosophila strains
GAL4 lines used in this study are the motor neuron driver OK6-GAL4 (Aberle et al. 2002), eye driver GMR-GAL4 (Freeman 1996), wing driver A9-GAL4 (BDSC Stock 8761), and muscle driver MEF2-GAL4 (BDSC Stock 27390). The UAS-cDNA/ORF transgenic flies were generated using phiC31-mediated integration into the attP2 landing site, and the UAS-cDNA/ORF constructs used for transgenes were described in Supplemental Table S1.
sgRNA preparation
To design the sgRNAs used for cleavage of vector backbones of cDNA/ORF plasmids, we used Optimized CRISPR Design (http://crispr.mit.edu) to search vector backbone sequences for sgRNA targets. The vector backbone sequences used for sgRNA design are adjacent to the 5′ end of cDNAs or ORFs, thus all cDNA/ORF plasmids with an identical vector backbone can be uniformly digested by Streptococcus pyogenes Cas9 together with the same sgRNA. The target candidates of sgRNAs for vector backbones of cDNA/ORF clones are listed in Supplemental Table S2.
To prepare a DNA template for in vitro transcription (IVT) of sgRNA, we synthesize two PAGE-purified oligos, forward primer [5′-TAATACGACTCACTATAGG(N)20GTTTTAGAGCTAGAAATAG-3′], where (N)20 is the target sequence of a sgRNA, and a sgRNA scaffold reverse primer sgRNA-REV (5′-AAAAGCACCGACTCGGTGCCACTT-3′). Next a 100-μL PCR reaction is set up by mixing 5 μL template pX330 (Addgene plasmid 42230, a gift from Feng Zhang) (Bischof et al. 2013) (1 ng/μL), 5 μL forward primer (10 μM), 5 μL sgRNA-REV (10 μM), 50 μL Q5 Hot Start High-Fidelity 2× Master Mix (NEB), and 35 μL DEPC-treated water (Invitrogen) in a PCR tube. A PCR program on an ABI 2720 PCR Thermal Cycler is run as follows: 30 sec at 98°C; five cycles of 10 sec at 98°C, 10 sec at 51°C, and 5 sec at 72°C; 30 cycles of 10 sec at 98°C and 15 sec at 72°C; and a final extension of 2 min at 72°C. The PCR product is separated by 0.8% agarose gel electrophoresis and a ∼120-bp DNA fragment should be visible on the gel. The ∼120-bp DNA fragment is then purified by QIAquick Gel Extraction Kit (Qiagen). The gel-purified ∼120-bp DNA fragment serves as a template for IVT of sgRNA.
IVT of sgRNA is performed using HiScribe T7 Quick High Yield RNA Synthesis Kit (NEB). A 10-μL IVT reaction is set up as follows: 330 ng ∼120-bp gel-purified DNA fragment, 3.33 μL NTP Buffer Mix, 0.67 μL T7 RNA Polymerase Mix, and DEPC-treated water. The 10-μL IVT reaction is incubated for 4 h at 37°C, then the in vitro transcribed sgRNA, without purification, is quantified, diluted to 20 ng/μL with DEPC-treated water, aliquoted, stored at −80°C, and is ready for CRISPRmass.
sgRNA evaluation
To evaluate sgRNAs targeting vector backbone sequences that are adjacent to the 5′ end of cDNAs or ORFs in cDNA/ORF plasmids, we performed in vitro CRISPR/Cas9 cleavage analysis of DNA substrates. A 5-μL CRISPR/Cas9 cleavage reaction contains 0.2 μL 1.22 μM S. pyogenes Cas9 (GenScript Z03386-50), 0.5 μL 20 ng/μL sgRNA, 0.015 pmol DNA substrate, 0.5 μL 10× Cas9 Buffer, and DEPC-treated water. After the 5-μL cleavage reaction is incubated for 1 h at 37°C, cleavage fragments are separated by 0.8% agarose gel electrophoresis and analyzed by Tanon 2500 Gel Imaging System (Tanon). A DNA fragment containing the sgRNA targeting sequences of a vector backbone is released by restriction digestion of a cDNA/ORF plasmid and used as a DNA substrate. DNA substrates and the sizes of substrate cleavage fragments are listed in Supplemental Table S2. The sgRNAs selected for CRISPRmass and their targets on vector backbones of cDNA/ORF clones are listed in Supplemental Table S3.
Construction of UAS modules
UAS module for pOT2
A 1141-bp EcoRI/HindIII fragment carrying ampicillin resistance gene (Amp) was PCR amplified from pBluescript KS (+) using EcoRI-pOT2G4-Amp-F and HindIII-AscI-Amp-R primers, digested with EcoRI/HindIII, inserted into the EcoRI/HindIII sites of pMartini vector (Wang et al. 2012), resulting in pMartini-Amp. pMartini-Amp was verified by sequencing.
A 938-bp HindIII/EcoRI fragment carrying attB-UAS-Hsp70 was PCR amplified from DopR-Tango (a gift from David Anderson) (Inagaki et al. 2012) using HindIII-attB-F and EcoRI-pOT2G4-Hsp70-R primers, digested with EcoRI/HindIII, inserted into the EcoRI/HindIII sites of pBluescript KS (+), resulting in pBS-attB-UAS-Hsp70. pBS-attB-UAS-Hsp70 was verified by sequencing.
A 1127-bp EcoRI/HindIII fragment of pMartini-Amp and a 924-bp attB-UAS-Hsp70 EcoRI/HindIII fragment of pBS-attB-UAS-Hsp70 were ligated together into the EcoRI site of pCR8GW-TOPO vector (Invitrogen), resulting in pCR8GW-Amp-attB-UAS-Hsp70.
A 4127-bp mini-white AscI fragment of pJFRC150-20XUAS-IVS-Flp1::PEST (Addgene plasmid 32132, a gift from Gerald Rubin) (Nern et al. 2011) was inserted into the AscI site of pCR8GW-Amp-attB-UAS-Hsp70, giving rise to pCR8GW-Amp-W-attB-UAS-Hsp70. The 6178-bp Amp-W-attB-UAS-Hsp70 fragment was released by EcoRI from pCR8GW-Amp-W-attB-UAS-Hsp70, and this fragment serves as a UAS module for construction of UAS-cDNA/ORF plasmids from pOT2 vector-based cDNA/ORF clones.
UAS module for pFLC-I
A 866-bp EcoRI/HindIII fragment carrying chloramphenicol resistance gene (Cam) was PCR amplified from pOT2-CG4968 using EcoRI-pFLCG3-Cam-F and HindIII-AscI-Cam-R2 primers, digested with EcoRI/HindIII, inserted into the EcoRI/HindIII sites of pMartini vector, resulting in pMartini-Cam. pMartini-Cam was verified by sequencing.
A 940-bp HindIII/EcoRI fragment carrying attB-UAS-Hsp70 was PCR amplified from DopR-Tango using HindIII-attB-F and EcoRI-pFLCG3-Hsp70-R primers, digested with EcoRI/HindIII, inserted into the EcoRI/HindIII sites of pBluescript KS (+), resulting in pBS-attB-UAS-Hsp70-2. pBS-attB-UAS-Hsp70-2 was verified by sequencing.
A 852-bp EcoRI/HindIII fragment of pMartini-Cam and a 926-bp attB-UAS-Hsp70 EcoRI/HindIII fragment of pBS-attB-UAS-Hsp70-2 were ligated together into the EcoRI site of pCR8GW-TOPO vector, resulting in pCR8GW-Cam-attB-UAS-Hsp70.
A 4127-bp mini-white AscI fragment of pJFRC150-20XUAS-IVS-Flp1::PEST was inserted into the AscI site of pCR8GW-Cam-attB-UAS-Hsp70, giving rise to pCR8GW-Cam-W-attB-UAS-Hsp70. The 5905-bp Cam-W-attB-UAS-Hsp70 fragment was released by EcoRI from pCR8GW-Cam-W-attB-UAS-Hsp70, and this fragment serves as a UAS module for construction of UAS-cDNA/ORF plasmids from pFLC-I vector-based cDNA/ORF clones.
UAS module for pBS SK(−)
A 1805-bp Cam-attB-UAS-Hsp70 EcoRI fragment was PCR amplified from pCR8GW-Cam-attB-UAS-Hsp70 using EcoRI-pBSG3-Cam-F and EcoRI-pBSG3-hsp70-R primers, digested with EcoRI, inserted into the EcoRI site of pCR8GW-TOPO vector, resulting in pCR8GW-Cam-attB-UAS-Hsp70-2. pCR8GW-Cam-attB-UAS-Hsp70-2 was verified by sequencing.
A 4127-bp mini-white AscI fragment of pJFRC150-20XUAS-IVS-Flp1::PEST was inserted into the AscI site of pCR8GW-Cam-attB-UAS-Hsp70-2, giving rise to pCR8GW-Cam-W-attB-UAS-Hsp70-2. The 5918-bp Cam-W-attB-UAS-Hsp70-2 fragment was released by EcoRI from pCR8GW-Cam-W-attB-UAS-Hsp70-2, and this fragment serves as a UAS module for construction of UAS-cDNA/ORF plasmids from pBS SK(−) vector-based cDNA/ORF clones.
UAS module for pOTB7 and pOTB7_DraIII
A 2070-bp Amp-attB-UAS-Hsp70 EcoRI fragment was PCR amplified from pCR8GW-Amp-attB-UAS-Hsp70 using EcoRI-pOTB7G9-Amp-F and EcoRI-pOTB7G9-hsp70-R primers, digested with EcoRI, inserted into the EcoRI site of pCR8GW-TOPO vector (Wang et al. 2012), resulting in pCR8GW-Amp-attB-UAS-Hsp70-2. pCR8GW-Amp-attB-UAS-Hsp70-2 was verified by sequencing.
A 4127-bp mini-white AscI fragment of pJFRC150-20XUAS-IVS-Flp1::PEST was inserted into the AscI site of pCR8GW-Amp-attB-UAS-Hsp70-2, giving rise to pCR8GW-Amp-W-attB-UAS-Hsp70-2. The 6183-bp Amp-W-attB-UAS-Hsp70 fragment was released by EcoRI from pCR8GW-Amp-W-attB-UAS-Hsp70-2, and this fragment serves as a UAS module for construction of UAS-cDNA/ORF plasmids from pOTB7 and pOTB7_DraIII vectors-based cDNA/ORF clones.
UAS module for pCR2.1
A 1805-bp Cam-attB-UAS-Hsp70 EcoRI fragment was PCR amplified from pCR8GW-Cam-attB-UAS-Hsp70 using EcoRI-pCR2.1G1-Cam-F and EcoRI-pCR2.1G1-hsp70-R primers, digested with EcoRI, inserted into the EcoRI site of pCR8GW-TOPO vector, resulting in pCR8GW-Cam-attB-UAS-Hsp70-3. pCR8GW-Cam-attB-UAS-Hsp70-3 was verified by sequencing.
A 4127-bp mini-white AscI fragment of pJFRC150-20XUAS-IVS-Flp1::PEST was inserted into the AscI site of pCR8GW-Cam-attB-UAS-Hsp70-3, giving rise to pCR8GW-Cam-W-attB-UAS-Hsp70-3. The 5918-bp Cam-W-attB-UAS-Hsp70-3 fragment was released by EcoRI from pCR8GW-Cam-W-attB-UAS-Hsp70-3, and this fragment serves as a UAS module for construction of UAS-cDNA/ORF plasmids from pCR2.1 vector-based cDNA/ORF clones.
UAS module for pDNR-Dual
A 1805-bp Cam-attB-UAS-Hsp70 EcoRI fragment was PCR amplified from pCR8GW-Cam-attB-UAS-Hsp70 using EcoRI-pLX304OriG11-Cam-F and EcoRI-pLX304OriG11-hsp70-R primers, digested with EcoRI, inserted into the EcoRI site of pCR8GW-TOPO vector, resulting in pCR8GW-Cam-attB-UAS-Hsp70-4. pCR8GW-Cam-attB-UAS-Hsp70-4 was verified by sequencing.
A 4127-bp mini-white AscI fragment of pJFRC150-20XUAS-IVS-Flp1::PEST was inserted into the AscI site of pCR8GW-Cam-attB-UAS-Hsp70-4, giving rise to pCR8GW-Cam-W-attB-UAS-Hsp70-4. The 5918-bp Cam-W-attB-UAS-Hsp70-4 fragment was released by EcoRI from pCR8GW-Cam-W-attB-UAS-Hsp70-4, and this fragment serves as a UAS module for construction of UAS-cDNA/ORF plasmids from pDNR-Dual vector-based cDNA/ORF clones.
UAS module for pLX304
A 1800-bp Cam-attB-UAS-Hsp70 EcoRI fragment was PCR amplified from pCR8GW-Cam-attB-UAS-Hsp70 using EcoRI-pLX304OriG13-Cam-F and EcoRI- pLX304CMVG7-hsp70-R primers, digested with EcoRI, inserted into the EcoRI site of pCR8GW-TOPO vector, resulting in pCR8GW-Cam-attB-UAS-Hsp70-5. pCR8GW-Cam-attB-UAS-Hsp70-5 was verified by sequencing.
A 4127-bp mini-white AscI fragment of pJFRC150-20XUAS-IVS-Flp1::PEST was inserted into the AscI site of pCR8GW-Cam-attB-UAS-Hsp70-5, giving rise to pCR8GW-Cam-W-attB-UAS-Hsp70-5. The 5913-bp Cam-W-attB-UAS-Hsp70-5 fragment was released by EcoRI from pCR8GW-Cam-W-attB-UAS-Hsp70-5, and this fragment serves as a UAS module for construction of UAS-cDNA/ORF plasmids from pLX304 vector-based cDNA/ORF clones.
UAS module for pCMV-SPORT6
A 1800-bp Cam-attB-UAS-Hsp70 EcoRI fragment was PCR amplified from pCR8GW-Cam-attB-UAS-Hsp70 using EcoRI-pLX304OriG13-Cam-F and EcoRI-pCMVST6G15-hsp70-R primers, digested with EcoRI, inserted into the EcoRI site of pCR8GW-TOPO vector, resulting in pCR8GW-Cam-attB-UAS-Hsp70-6. pCR8GW-Cam-attB-UAS-Hsp70-6 was verified by sequencing.
A 4127-bp mini-white AscI fragment of pJFRC150-20XUAS-IVS-Flp1::PEST was inserted into the AscI site of pCR8GW-Cam-attB-UAS-Hsp70-5, giving rise to pCR8GW-Cam-W-attB-UAS-Hsp70-6. The 5918-bp Cam-W-attB-UAS-Hsp70-6 fragment was released by EcoRI from pCR8GW-Cam-W-attB-UAS-Hsp70-6, and this fragment serves as a UAS module for construction of UAS-cDNA/ORF plasmids from pCMV-SPORT6 vector-based cDNA/ORF clones.
Primers used for constructing and sequencing UAS modules are listed in Supplemental Table S7.
Construction of UAS-cDNA/ORF plasmids by CRISPRmass
Cas9 cleavage of cDNA or ORF clones in pOT2, pOTB7, pOTB7_DraIII, pFLC-I, pBS SK(−), pCR2.1, and pDNR-Dual vectors is performed as follows: 0.4 μL of 0.019 μM cDNA or ORF plasmid is cleaved for 1 h at 37°C in a 2.4 μL of reaction containing 0.16 μL of 1.22 μM of Cas9 (GenScript), 0.4 μL of 20 ng/μL sgRNA, and 0.24 μL 10× Cas9 Buffer. cDNA/ORF clones in pLX304 and pCMV-SPORT6 are cleaved twice by Cas9 together with two different sgRNAs to remove unnecessary sequences (Supplemental Fig. S1). Then 0.7 μL Cas9-cleaved cDNA/ORF plasmid, without purification, is directly subjected to Gibson assembly together with 0.07 μL of 0.006 μM UAS module and 0.7 μL of NEBuilder HiFi DNA Assembly Master Mix (NEB). Gibson assembly is performed for 1 h at 50°C, then 0.7 μL of Gibson assembly product is directly used to transform E. coli. Transformants are selected on LB plates containing antibiotic corresponding to the antibiotic resistance gene on the UAS module.
Construction of 2×MYC or 3×MYC-tagged UAS-cDNA/ORF plasmids
A 3×MYC epitope sequence (EQKLISEEDLEQKLISEEDLEQKLISEEDL) was inserted right before the stop codon of a representative Drosophila cDNA/ORF clone for each of the six vectors of the DGRC Gold Collection using Q5 Site-Directed Mutagenesis Kit (NEB). Briefly, a 5-μL PCR reaction was set up as follows: 0.2 ng cDNA/ORF plasmid, 0.5 μM forward primer (Supplemental Table S8), 0.5 μM reverse primer (Supplemental Table S8), and 2.5 μL Q5 Hot Start High-Fidelity 2× Master Mix (NEB). A PCR program on an ABI 2720 PCR Thermal Cycler was performed run as follows: 30 sec at 98°C; five cycles of 10 sec at 98°C, 10 sec at 51°C, and 8 min at 72°C; 30 cycles of 10 sec at 98°C and 8 min at 72°C; and a final extension of 5 min at 72°C. Then 0.2 μL of the PCR product is subjected to a KLD reaction together with 0.2 μL of 10× KLD Enzyme Mix, 1 μL of 2× KLD Reaction Buffer, and 0.6 μL ddH2O. KLD reactions were performed for 10 min at room temperature, and the KLD reaction products were used to transform E. coli. The resulting plasmids were confirmed by sequencing (Supplemental Table S8). Similarly, a 3×MYC epitope sequence was inserted right before the stop codon of the mouse Ctbp2 cDNA on pCMV-SPORT6 using Q5 Site-Directed Mutagenesis Kit, but subsequent sequencing indicated that a copy of MYC tag was removed from the 3×MYC epitope, resulting in a 2×MYC-tagged Ctbp2 plasmid (Supplemental Table S8). 2×MYC or 3×MYC-tagged cDNA/ORF plasmids and all the primers used for 2×MYC or 3×MYC epitope tagging are listed in Supplemental Table S8.
Construction of 3xFLAG-tagged UAS-cDNA plasmids
A 3xFLAG epitope sequence (DYKDHDGDYKDHDIDYKDDDDK) was inserted right before the stop codon of the 23 cDNA clones of the DGRC Gold Collection using Q5 Site-Directed Mutagenesis Kit (NEB). Briefly, a 5-μL PCR reaction was set up as follows: 0.2 ng cDNA plasmid, 0.5 μM forward primer, 0.5 μM reverse primer, and 2.5 μL Q5 Hot Start High-Fidelity 2× Master Mix (NEB). A PCR program on an ABI 2720 PCR Thermal Cycler was performed run as follows: 30 sec at 98°C; five cycles of 10 sec at 98°C, 10 sec at 51°C or 56°C and 8 min at 72°C; 20 cycles of 10 sec at 98°C, 10 sec at 70°C and 8 min at 72°C; and a final extension of 5 min at 72°C. Then 0.2 μL of the PCR product is subjected to a KLD reaction together with 0.2 μL of 10× KLD Enzyme Mix, 1 μL of 2× KLD Reaction Buffer, and 0.6 μL ddH2O. KLD reactions were performed for 10 min at room temperature, and the KLD reaction products were used to transform E. coli. The resulting plasmids were confirmed by sequencing. The Nhe3 gene was fused with a 3xFLAG tag at its N terminus by Gibson assembly. Briefly, because the SphI site overlaps the Nhe3 start codon, the Nhe3 cDNA clone was linearized by SphI, resulting in a 6792-bp fragment. The overlapping 3FLAG-Nhe3-F and 3FLAG-Nhe3-R primers were annealed for 1 min at 50°C and then extended for 2 min at 72°C, resulting in a 102-bp fragment containing a 3xFLAG sequence and partial sequence of Nhe3 cDNA. The 6792-bp and 102-bp fragments were then joined through Gibson assembly using NEBuilder HiFi DNA Assembly Master Mix (NEB), and the resulting 3xFLAG-tagged Nhe3 plasmid, pFLC-3FLAG-Nhe3, was confirmed by sequencing. All the 3xFLAG-tagged UAS-cDNA plasmids were constructed by CRISPRmass based on the 3xFLAG-tagged cDNA plasmids, and all the 3xFLAG-tagged cDNA and UAS-cDNA plasmids and the primers used for 3xFLAG epitope tagging and sequencing are listed in Supplemental Tables S5 and S6.
Western blotting and immunohistochemistry
These two methods are available in Supplemental Methods.
Data access
All processed sequencing data generated in this study have been submitted to the NCBI GenBank (https://www.ncbi.nlm.nih.gov/genbank/) under accession numbers MN503478–MN503501 and MN518567–MN518687. The GenBank accession numbers MN503478–MN503501 are for the cDNA/ORFs of 24 UAS-cDNA/ORF constructs from the more than 5500 UAS-cDNA/ORF constructs generated by CRISPRmass. The GenBank accession numbers MN518567–MN518687 are for the 121 UAS-cDNA/ORF constructs that were verified by sequencing from the more than 5500 UAS-cDNA/ORF constructs. The plasmids are available upon request.
Competing interest statement
Two patents have been filed on this study (China patent application Nos. 2019101104852 and 2019101110552).
Acknowledgments
We thank the Drosophila Genomics Resource Center (DGRC), supported by National Institutes of Health grant 2P40OD010949-10A1, for the DGRC Gold Collection. We thank the Developmental Studies Hybridoma Bank for antibodies and the Bloomington Stock Center for the Drosophila stocks. We thank the Core Facility of Drosophila Resource and Technology, Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences for Drosophila transgenesis. We thank Jihui Guo, Yan Wan, Xueyan Song, and Hongsuo Wang for their assistance on the project. This work was sponsored by the National Natural Science Foundation of China (31471010 and 31401013), Shanghai Pujiang Program (14PJ1405900), and Natural Science Foundation of Shanghai (19ZR1446400).
Author contributions: J.W. conceived and supervised this project. J.W. and P.W. designed the experiments. J.W., P.W., and W.X. performed the experiments. Y.Z. and G.N. provided suggestions. J.W. and P.W. analyzed the data and wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.250811.119.
- Received March 23, 2019.
- Accepted November 8, 2019.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








