
Abstract
Missense variant interpretation is challenging. Essential regions for protein function are conserved among gene-family members, and genetic variants within these regions are potentially more likely to confer risk to disease. Here, we generated 2871 gene-family protein sequence alignments involving 9990 genes and performed missense variant burden analyses to identify novel essential protein regions. We mapped 2,219,811 variants from the general population into these alignments and compared their distribution with 76,153 missense variants from patients. With this gene-family approach, we identified 465 regions enriched for patient variants spanning 41,463 amino acids in 1252 genes. As a comparison, by testing the same genes individually, we identified fewer patient variant enriched regions, involving only 2639 amino acids and 215 genes. Next, we selected de novo variants from 6753 patients with neurodevelopmental disorders and 1911 unaffected siblings and observed an 8.33-fold enrichment of patient variants in our identified regions (95% C.I. = 3.90-Inf, P-value = 2.72 × 10−11). By using the complete ClinVar variant set, we found that missense variants inside the identified regions are 106-fold more likely to be classified as pathogenic in comparison to benign classification (OR = 106.15, 95% C.I = 70.66-Inf, P-value < 2.2 × 10−16). All pathogenic variant enriched regions (PERs) identified are available online through “PER viewer,” a user-friendly online platform for interactive data mining, visualization, and download. In summary, our gene-family burden analysis approach identified novel PERs in protein sequences. This annotation can empower variant interpretation.
Sequencing technologies are becoming routinely applied in clinical diagnostics (den Dunnen et al. 2016). The number of genetic variants derived from patients has increased exponentially (Lek et al. 2016), demanding scalable and accurate methods for variant interpretation. Particularly, the ability to accurately predict variants associated with rare and complex Mendelian disorders becomes crucial in the development of personalized medicine (Xue et al. 2015). Up to 85% of disease traits are explained by variation within the coding region of the genome, thereby making whole-exome and gene-panel sequencing the standard of care (Choi et al. 2009; Bamshad et al. 2011). Still, variant interpretation remains challenging (Gilissen et al. 2012) particularly for missense variants—the most prevalent genomic alteration, with 10,000 to 12,000 events per individual (The 1000 Genomes Project Consortium 2015). Protein truncating variants (PTVs) and large deletions are generally assumed to cause disease by loss-of-function mechanisms in haploinsufficient genes. In contrast, missense variants can have a variety of functional outcomes depending on the amino acid substitution and protein domain affected (Miosge et al. 2015), further complicating interpretation. Many computational tools have been developed for missense variant interpretation (Itan and Casanova 2015; Liu et al. 2016). These tools are based on a combination of criteria, including the physicochemical properties of the amino acids change (e.g., Grantham) (Grantham 1974), structural features (e.g., PolyPhen-2) (Adzhubei et al. 2010), amino acid conservation across different species (e.g., GERP++, SIFT) (Cooper et al. 2005; Kumar et al. 2009), or combined machine learning consensus approaches (e.g., CADD, FATHMM, REVEL) (Shihab et al. 2013; Kircher et al. 2014; Ioannidis et al. 2016).
Repositories of variants from the general population have been used as a resource to calculate gene constraint or to identify coding regions “intolerant to variation” (Lek et al. 2016). Constraint metrics are extensively used for the identification of potential disease genes and for individual variant interpretation (Samocha et al. 2017). Missense variants are not randomly distributed across the exome, and functionally essential genes are constrained from variation (Petrovski et al. 2013; Bartha et al. 2018). Thus, variants within genes that are intolerant to loss of function or missense variation in the general population are more likely to be pathogenic. Methods and scores that incorporate variant tolerance have been developed. For example, evaluation of the rate of missense against synonymous variants from the general population allowed the identification of missense depleted regions (MDRs) (Ge et al. 2016). Similarly, the missense tolerance ratio (MTR) score evaluates constraint over a 31-amino-acid window (Traynelis et al. 2017), and the measure of deleterious effect of missense badness, PolyPhen-2, and constraint score (MPC) combines constraint with exchange and structural scores to report a missense-specific score (Samocha et al. 2017).
From an evolutionary perspective, it has been shown that ∼80% of the genes causing Mendelian disorders have functionally redundant paralogs (Chen et al. 2013b) expressed across different cell types. Gene duplication events from ancestral genes have produced large sets of well-established paralog gene families in the human genome, with different degrees of amino acid conservation and functional redundancy. Conserved amino acids across gene-family members are more likely to hold essential functional domains. Thus, amino acid conservation across gene paralogs can be used at scale for variant interpretation (Parazscore) (Lal et al. 2017). Functional redundancy can help explain disease etiology via the accumulation of pathogenic variants in analogous domains within the tissues and organs, corresponding to the paralogous genes expressed (Ware et al. 2012; Chen et al. 2013b; Walsh et al. 2014; Barshir et al. 2018). Therefore, protein alignments of gene-family members could significantly cluster independent pathogenic variants in the same analogous domain. Variant aggregation over protein domain homologs without distinction of gene-family members has been reported for variant interpretation (Gussow et al. 2016; Wiel et al. 2017), including the identification of cancer-driver variants (Melloni et al. 2016).
Similar to genetic constraint, patient variant clustering along the linear protein sequence can also be expected in functionally essential regions. Thousands of pathogenic variants have been used to train variant interpretation tools such as the Variant Effect Scoring Tool (VEST) (Carter et al. 2013) and the Combined Annotation Dependent Depletion (CADD) (Kircher et al. 2014). However, patient variant enrichment analysis to detect disease-sensitive regions has not been conducted on an exome-wide level.
Here, we compared the distribution of patient missense variants against population missense variants within gene-family alignments and gene sequences. We developed a novel statistical framework that, based on the observed mutational distribution, can identify pathogenic variant enriched regions (PERs) across protein sequences. We show that the family-wise approach is able to identify more and larger PERs than gene-wise analyses. Our identified family-wise and gene-wise PERs are high in resolution and can be used for variant interpretation. We developed the “PER viewer” (http://per.broadinstitute.org) to facilitate the exploration of all data generated in this study, including gene-family alignments, PERs, variants, and paralog conservation scores in a user-friendly web application.
Results
Missense variant mapping in genes and gene families
Our goal was to generate an annotation for protein regions vulnerable to disease. We found that protein residues near or within clusters of pathogenic variants are more likely to be disease associated. We compared the distribution of missense variants from patients and individuals from the general population across protein sequences to identify regions enriched for patient variants. To increase our statistical power, we performed a “family-wise” approach analyzing the missense variant burden along aligned protein sequences of gene-family members (i.e., paralogs) as a single unit. First, we extracted a total of 2,219,811 missense variants from the Genome Aggregation Database (gnomAD) (Lek et al. 2016) to serve as our “population” variant data set. Patient missense variants were retrieved from two sources: the ClinVar database (Landrum et al. 2016) and the Human Gene Mutation Database (HGMD) (Stenson et al. 2003). After variant filtering, the union of ClinVar and HGMD yielded a total of 76,153 unique high-confidence pathogenic/likely-pathogenic missense variants, which were subsequently used as our “patient” data set. A detailed description of the applied filtering criteria can be found in the Methods section. Patient and population missense variant sets are available in the Supplemental Code database folder (/db) and at our GitHub repository (https://github.com/edoper/PERs/tree/master/db). The workflow designed for PER detection is summarized in Figure 1.
Study workflow and the PER viewer. (A) Starting from protein alignments of paralogous genes (gene-family approach) or all genes (gene-wise approach), missense variants from gnomAD (population; green) and ClinVar/HGMD (patient; purple) were mapped independently to the corresponding amino acid positions. (B) The mapping follows a binary notation. For sites with at least one missense variant reported, a “1” state was assigned. Alternatively, if no mutation was found, a “0” state was annotated instead. Amino acid sliding window (bin) counting over the alignment/sequence was used to calculate the corresponding missense burden. (C) The ratio between the number of sites with missense variants inside and outside the bin defines the burden area (population burden = green; patient burden = purple). Statistical comparison between the population and patient variant burden across aligned sequences allowed the identification of significant pathogenic variant enriched regions (PERs; red area).
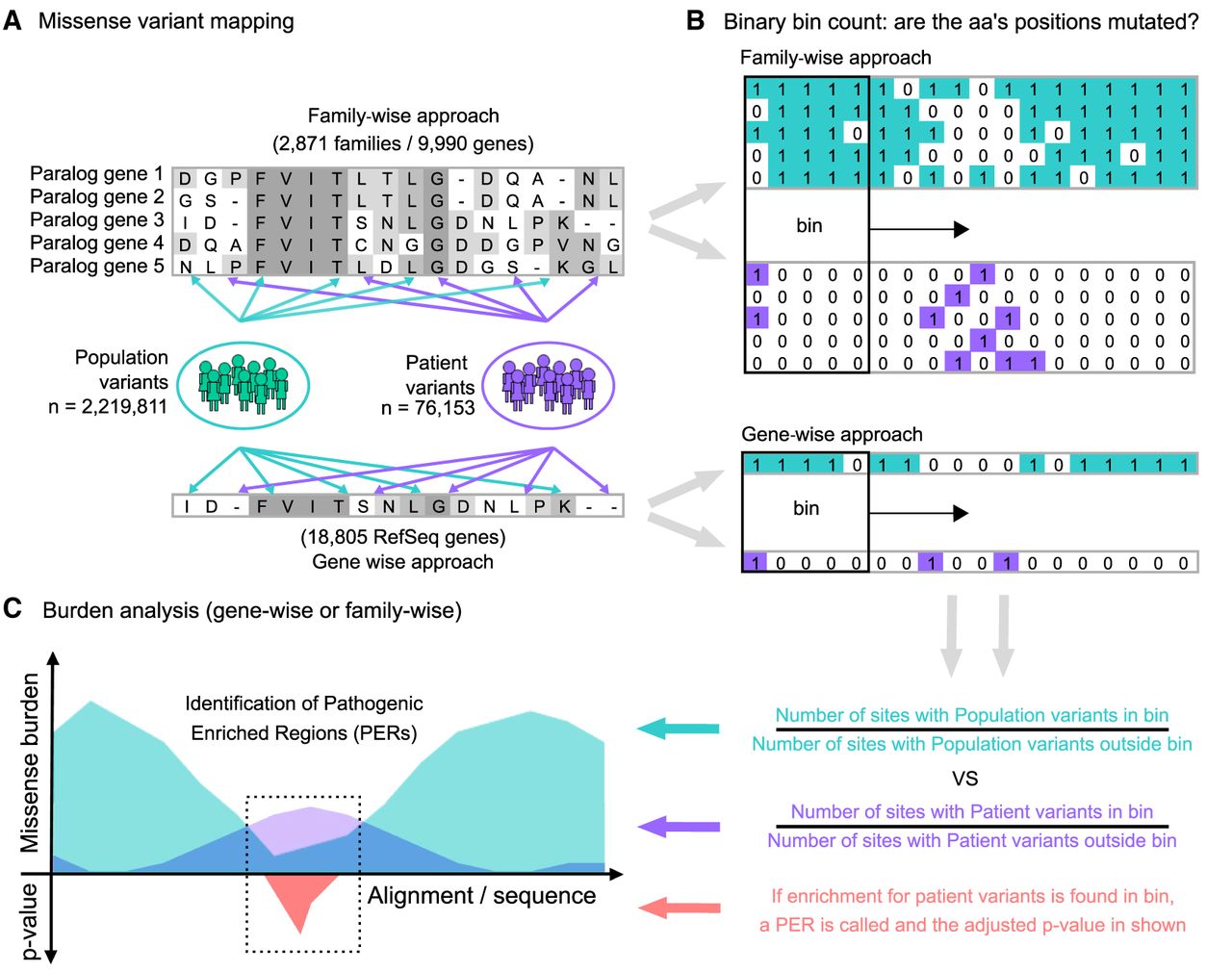
Missense burden analysis
To investigate mutational burden across paralog-conserved amino acids, we mapped all missense variants from the population and patient data sets onto a set of 2871 gene-family protein alignments involving 9990 genes (Supplemental Table S1). To generate a “gene-wise” analysis as a comparison group, we applied the same mapping procedure to the single protein sequences of 18,805 RefSeq genes. To calibrate the optimal sliding window size, we conducted multiple rounds of burden analyses with varying window sizes (see Methods). We observed that at greater sliding window sizes, more PERs were detected; however, the ratio of aligned amino acids positions with patient variants versus without decreased (Supplemental Fig. S1). To ensure specificity, we decided to limit PERs to contain a minimum of 50% of amino acids with at least one disease association in any gene-family member. As a result, the analysis was calibrated to a sliding window of nine amino acids (Supplemental Fig. S1).
PERs detected in the family-wise and gene-wise approaches
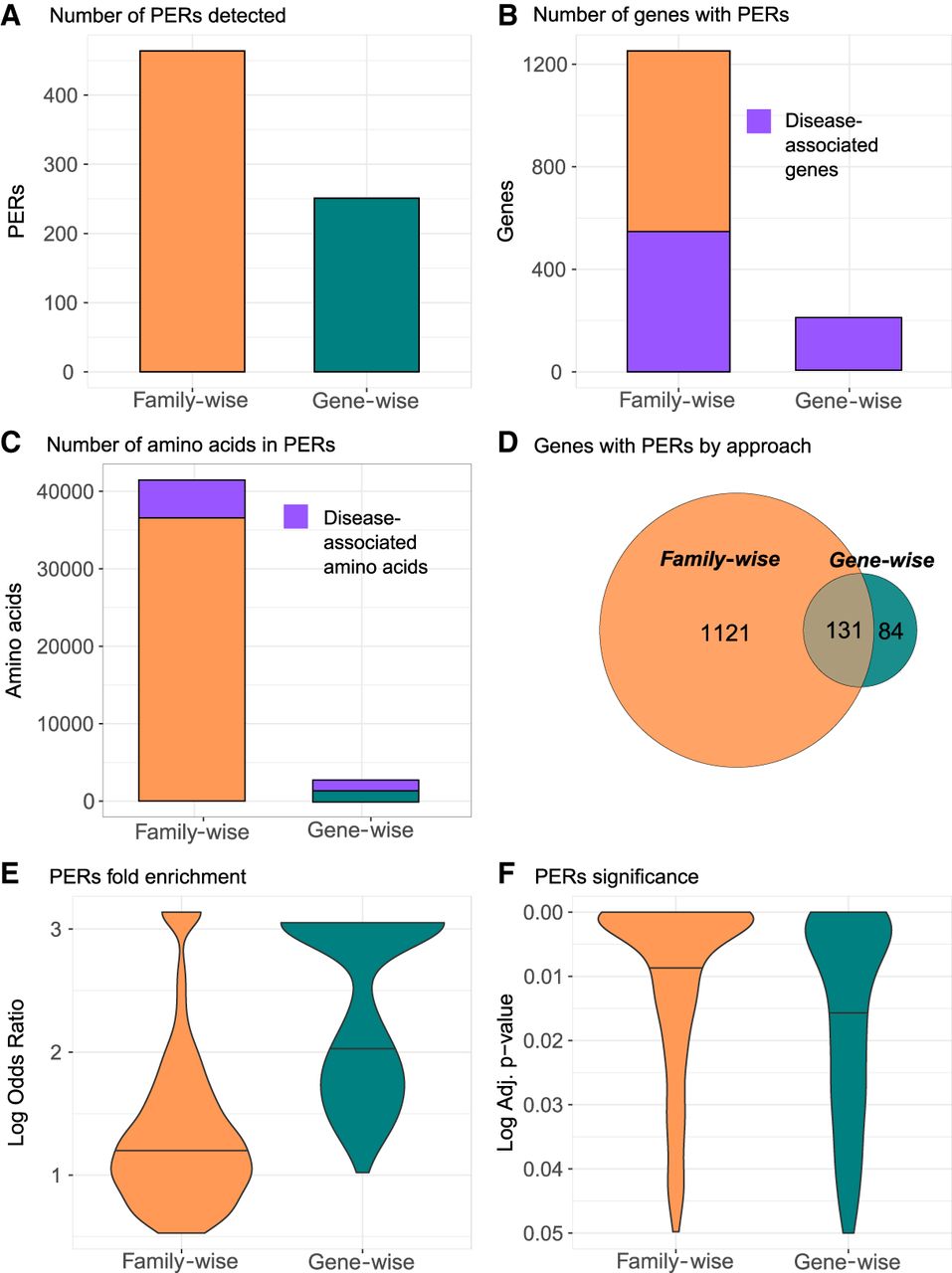
We identified 465 and 251 PERs in the family-wise and gene-wise analysis, encompassing 41,463 and 2639 amino acids, respectively (Fig. 2A). Collectively, a total of 42,713 amino acids from 1338 genes fall within PERs boundaries, which can be traced back to 128,139 nucleotides in the reference genome. For family-wise and gene-wise analysis, the complete list of genes and amino acids affected by PERs as well as the corresponding genomic coordinates in BED format are shown in Supplemental Table S2. We observe a 5.8-fold enrichment of genes with at least one PER in the family-wise analysis (n = 1252) than in the gene-wise analysis (n = 215). All genes in the gene-wise approach have been previously associated to disease; however, the family-wise approach was able to detect PERs in 700 genes not yet associated with a human phenotype (Fig. 2B). Similarly, among the amino acid positions within family-wise PERs, 88.4% (n = 36,660) have no prior disease association in comparison to 55.7% (n = 1471) observed in the gene-wise PERs (Fig. 2C). Given that the family-wise PERs are composed of several genes, the aligned amino acids covered by PERs are transferrable to all the gene-family members” protein sequences. In general, the family-wise approach identified more PERs, amino acids sites, and patient variants than the gene-wise approach. Overall, 83.9% of genes with at least one PER were identified exclusively through the family-wise approach (n = 1221), whereas 6.3% of the total genes (n = 84) were found exclusively in the gene-wise analysis. A total of 131 genes (9.8%) had PERs in both approaches (Fig. 2D). It is important to note that 66 out of the 84 (78.5%) genes with PERs exclusively found in the gene-wise analysis do not belong to a gene family. Considering the 131 genes with PERs detected in both methods, we found that out of the 155 PERs found in the gene-wise analysis, 143 (92.2%) were also captured by the family-wise analysis. The family-wise overlapping PERs were, on average, five amino acids larger than PERs found with the gene-wise approach (Supplemental Table S2). The average patient variant fold enrichment observed for PERs identified in the family-wise approach was lower than that observed for PERs identified by the gene-wise analysis (Fig. 2E). However, the corresponding association was more significant in the family-wise analysis compared with the gene-wise analysis (Fig. 2F). Taken together, PERs show an average size of 33 amino acids covering 5.48% of the affected protein sequence (Supplemental Table S2). The smallest PER detected was found in the SCNN1D gene, with a size of three amino acids (0.37% of protein sequence), whereas the largest was found in COL11A1 gene, with 350 amino acids (28.91% of protein sequence). Annotation of the total set of amino acids covered in PERs (n = 42,713) showed that 33,256 (77.8%) overlapped with known Pfam domains. The most frequent domain affected by PERs was the ion transport protein domain (PF00520), with 4174 amino acids overlapped by PERs, followed by collagen triple helix repeat domain (PF01391), with 2888 amino acids involved, and the intermediate filament protein domain (PF01391), with 2574 amino acids affected (Supplemental Fig. S2).
PERs detected with the family-wise and gene-wise burden analyses. Summary statistics for family-wise (orange) and gene-wise (green) approaches are shown for number of PERs detected (A), number of genes with PERs (B), and number of amino acids involved in in PERs (C). For B and C, the number of genes and amino acids associated to disease is shown in purple. (D) To reflect gene with PERs distribution by approach, a Venn diagram is shown. (E,F) Overall enrichment (log odds ratio) and significance (adjusted P-value) distribution of all PERs detected in each approach are shown in E and F, respectively.
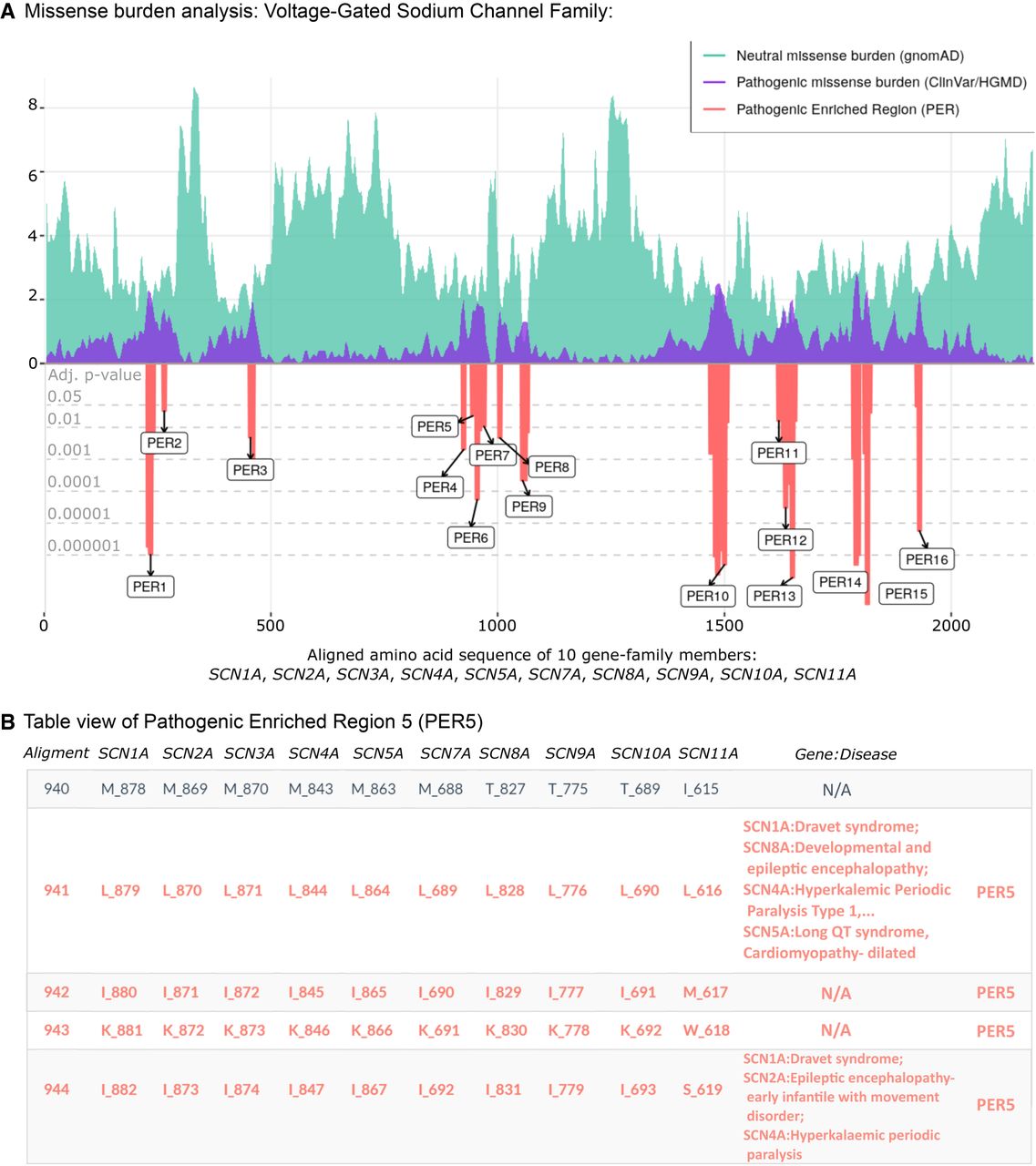
Illustrative example: the voltage-gated sodium channel gene family
We show the missense burden analysis results of the voltage-gated sodium channel gene family (family ID: 2614) composed of 10 paralogous genes: SCN1A, SCN2A, SCN3A, SCN4A, SCN5A, SCN7A, SCN8A, SCN9A, SCN10A, and SCN11A (Fig. 3). The alignment of the 10 protein sequences consists of 2188 amino acids, in which the patient and population missense variants were subsequently mapped. Clinical phenotypes from patient variants found in any gene-family member were aggregated into the corresponding aligned amino acid position. The missense burden analysis identified 16 PERs (Fig. 3A). Overall, regions with a drop in the distribution of population variants are increased for patient variants and vice versa. PER10 represented the longest patient variant enriched region, with 44 consecutive aligned amino acid sites from positions 1466 to 1509. As an example, we show the clinical phenotypes of patients carrying variants in PER5, located between aligned positions 941 to 949 (Fig. 3B). The patient variant enrichment within PER5 was based on missense variants in SCN1A (n = 4), SCN5A (n = 4), SCN4A (n = 3), SCN8A (n = 2) and SCN2A (n = 2), SCN1A (n = 1), and SCN9A (n = 1), representing patients with long QT syndrome, Brugada syndrome, Dravet syndrome, and a broad range of infantile epilepsies and epileptic encephalopathies. In contrast to the family-wise burden analysis, the gene-wise burden analysis was not able to identify PERs in any of the 10 voltage-gated sodium channel genes (Supplemental Fig. S3), indicating greater statistical power of family-wise burden analysis in this gene family.
PER viewer tool example. The voltage-gated sodium channel family. (A) Missense burden analysis of the voltage-gated sodium channel protein family (family ID: 2614.subset.3) composed by SCN1A, SCN2A, SCN3A, SCN4A, SCN5A, SCN7A, SCN8A, SCN9A, SCN10A, and SCN11A. Population and patient missense burden are shown in green and purple, respectively. Significant pathogenic enriched regions (PERs) identified are shown in the red negative area and are proportional to their adjusted P-values (gray horizontal lines). (B) Table view of pathogenic enriched region 5 (PER5; positions 941–949). Gene columns denote individual canonical sequence alongside corresponding amino acid position. Column “Gene:Disease” displays analogous diseases observed in the patient data set. N/A sites show aligned amino acids positions with no disease reported.
PER viewer
We developed an R-based online tool to make the full set of results accessible. The “PER viewer” is available at http://per.broadinstitute.org. The main features of PER viewer are shown in Supplemental Figure S4. The user can query any gene and search for its corresponding missense burden analysis results. If the gene belongs to a gene family, the results will be shown family-wise with the option to evaluate genes independently. For genes that do not belong to a gene family, the single gene burden analysis will be shown. Burden analyses and table browsing are displayed in the same format shown in Figure 3. The user can explore the burden observed in the population and patient data sets along the alignment or gene sequence at the amino acid level. Alignments, burden analyses, summary statistics, and the identification of PERs are available for download at PER viewer.
PERs on independent cohorts
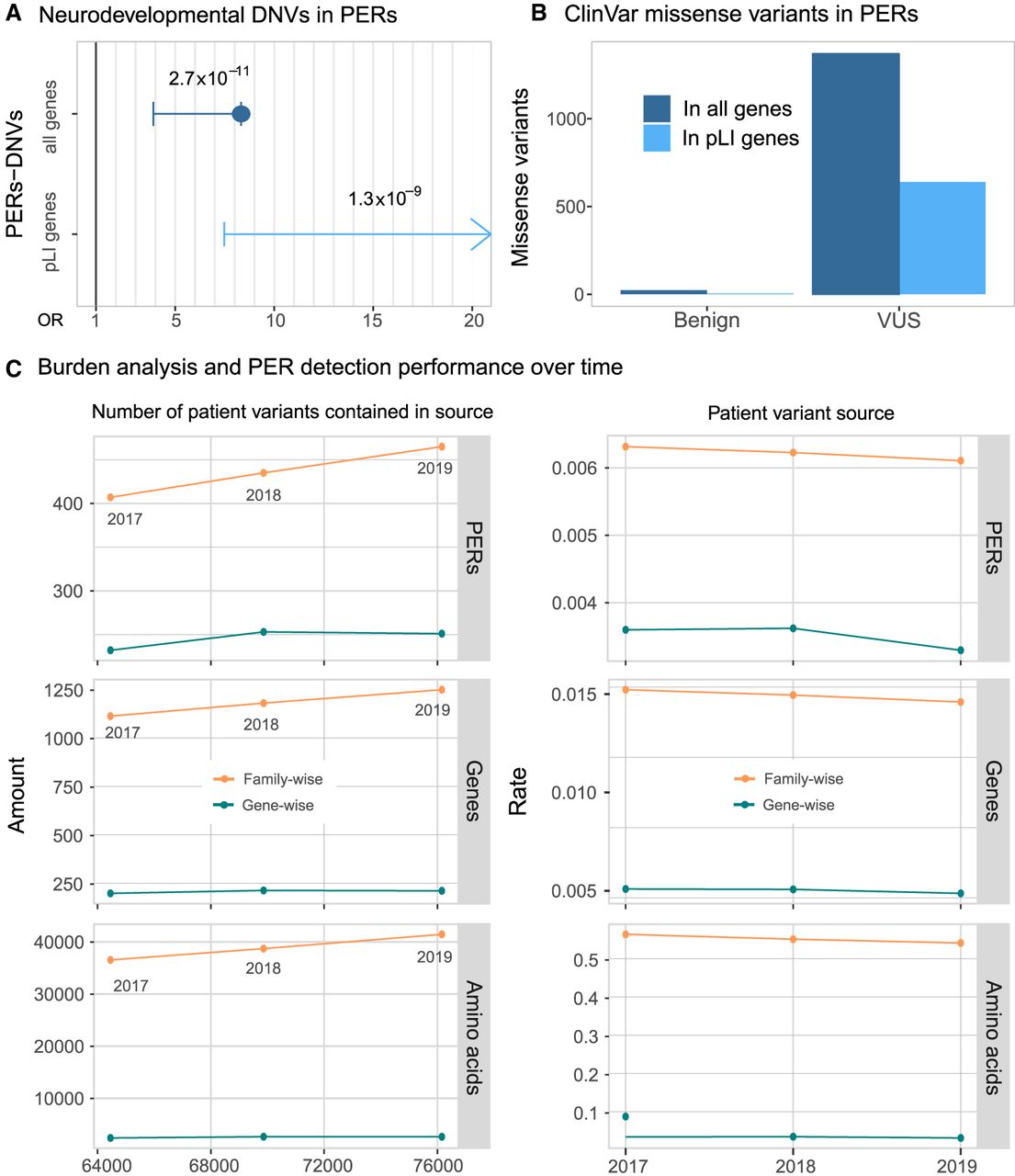
To test the utility of PER annotation in an independent data set, we evaluated the distribution of de novo missense variants (DNVs) within and outside of the identified PERs from a large neurodevelopmental (NDD) case-control cohort (Heyne et al. 2018). The data set included 6753 patients with 4404 missense DNVs identified and 1911 unaffected siblings with 768 missense DNVs identified (Fig. 4A). Patient missense DNVs (n = 228) were 8.33-fold enriched within PERs compared with control missense DNVs (OR = 8.33, 95% C.I. = 3.90-Inf, P-value = 2.72 × 10−11). The fold enrichment of patient variants in PERs was even greater when we restricted the analysis to constrained genes (pLI > 0.9) (Lek et al. 2016). For this group of haploinsufficient genes, no patient DNV enrichment was observed (OR = Inf, 95% C.I. = 7.48-Inf, P-value = 1.34 × 10−9). It is not expected that all patient DNVs are pathogenic. In an additional analysis, we evaluated the distribution of benign and unknown significance (VUS) missense variants reported in the complete ClinVar release (October 2019). We found 23 benign variants and 1370 VUS missense variants within PERs (Fig. 4B). We note that 16 (70%) of the 23 benign variants came from single submitters, and none of them were evaluated with the established guidelines criteria for variant interpretation (Richards et al. 2015). We compared the number of ClinVar pathogenic and benign variants inside and outside PERs and observed a 106.15-fold enrichment for pathogenic variants (OR = 106.15, 95% C.I. = 70.66-Inf, P-value <2.2 × 10−16) inside PERs boundaries. Finally, to explore if the number of PERs is increasing over time, we conducted burden analyses using patient missense variants (ClinVar/HGMD) from three different time points against the same set of population variants: (1) missense variants reported until December 2017 (patient variants = 64,458), (2) until December 2018 (patient variants = 69,863), and (3) until October 2019 (current; patient variants = 76,153). We observe a consistent increase in the number of PERs, genes, and amino acids involved (Fig. 4C). Family-wise PER detection found 407 (genes = 1116; amino acids = 36,552) and 435 (genes = 1183; amino acids = 38,731) PERs with patient variants reported until 2017 and until 2018, respectively. In comparison, 465 PERs (genes = 1252; amino acids = 41,464) were detected with the current release (October 2019). The PERs detected with the current 2019 set of patient variants are able to capture 6.89% and 13.43% more amino acids than with the 2018 and 2017 patient variant sources, respectively. We note that the increase of power is driven mostly by the family-wise analysis because PERs detected with the gene-wise analysis showed more stable numbers (2017 = 232 PERs; 2018 = 253 PERs; 2019 = 251 PERs) (Fig. 4C, left). Regardless of the method, the overall significance of PERs also increases slightly over time (average −log P-value: 2017 = 3.65; 2018 = 3.68; 2019 = 3.75). However, we note that the annual rate of new PERs decreased over time (Fig. 4C, right).
Disease-causing variants are enriched in PERs. (A) Neurodevelopmental disorder DNVs inside PERs. Case and control comparison of DNVs inside PERs retrieved from Heyne et al. (2018) is shown for all genes (blue; OR = 8.33, 95% C.I. = 3.90-Inf, P-value = 2.72 × 10−11) and genes with high probability of being loss-of-function intolerant (light blue; OR = Inf, 95%C.I. = 7.48-Inf, P-value = 1.34 × 10−9). Fold enrichment observed in cases was calculated with a one-sided Fisher's exact test. Resulting odds ratio (OR) with 95% confidence and corresponding P-values are shown in the horizontal axis. (B) ClinVar missense variants (from October 2019 release) inside PERs with benign and unknown (VUS) clinical significance. The number of variants observed is shown considering all genes (blue) and pLI > 0.9 genes only (light blue). (C) Burden analysis performance over time. PER detection was performed with patient variants reported until 2017 and 2018 and compared with the current 2019 data set analysis. (Left) Overall amount of PERs, amino acid, and genes detected as a function of the number of input patient variants. (Right) Rate of PERs, genes, and amino acids detected per patient variant contained in 2017, 2018, and 2019 sources.
Discussion
The present work compares the exome-wide distribution of missense variants from the general population with patient variants across single-protein sequences and gene-family protein sequence alignments. The family-wise approach was more sensitive and powerful than the basic gene approach in PER detection (Fig. 2). Missense variants in amino acid positions within PERs are more likely to be classified as pathogenic rather than benign. These regions, enriched for patient variants and depleted for population variants, likely encompass functionally essential protein features. We show that 77.8% of amino acids captured by PERs overlapped with conserved functional domains. The remaining 22.2% of sites can still provide additional biological insights, suggesting novel functional regions that might not be directly captured by traditional annotation (McLaren et al. 2016). The generated exome-wide map of PERs can be used as an additional criterion for variant interpretation. Specifically, PER annotation and evaluation could be included in the “PM1” category of the American College of Medical Genetics and Genomics (ACMG) guidelines. PM1 is defined as “variants located in a mutational hot spot and/or critical and well-established functional domain without benign variation” (Richards et al. 2015). Furthermore, the statistical framework designed to detect PERs provides fold enrichments and 95% confidence intervals that can be integrated into Bayesian tools based on ACMG guidelines (Tavtigian et al. 2018). It has been estimated that an observed fold enrichment above 18.7 can be considered as a strong criterion for variant interpretation. Thus, 26.01% of all PER sites could be further incorporated as a strong criterion for variant interpretation. In this regard, for each PER, genomic coordinates, the corresponding enrichment values, and significance are included in Supplemental Table S2.
Identification of functional essential domains and sites across single protein sequences represents a challenge for rare Mendelian disorders. The number of patient variants annotated for most genes is still small and limits variant interpretation and prediction score development. However, the number of variants and quality of interpretation has been increasing exponentially during the past years (Harrison et al. 2017). Our analysis with previous, smaller releases of ClinVar and HGMD with fewer patient variants (Fig. 4C) suggests that more PERs remain to be identified with future larger variant data sets.
Our approach aggregates variants across analogous sites within gene families to a single unit, hypothesizing that functionally essential sites across related proteins are conserved. We observed that the distribution pattern of patient and population variants across protein sequences was similar across gene-family members, which yielded in a larger number of PERs and genes with PERs in the family approach compared with the single-gene approach (Fig. 2). Similar sequence grouping approaches have been conducted over homologous protein domains (Wiel et al. 2017), defined as functional subunits that can be present in a broad spectrum of unrelated proteins (Finn et al. 2016). In this regard, a recent study conducted a similar approach to detect domains or exons enriched with pathogenic variants based on ClinVar and gnomAD variants. They reported 259 genes in which there is a significant relationship between intolerance scores and the location of pathogenic missense mutations (Hayeck et al. 2019). We note that 40.9% (n = 108) of these genes have PERs. Collectively, these studies are not mutually exclusive but rather complementary to our results and provide additional tools and regions that should also be considered for variant interpretation. In contrast to domain-wise approaches, our missense burden analyses were performed on functionally redundant genes. Paralogous genes have accumulated a significant amount of disease variants because they can be masked by paralog functional redundancy (Chen et al. 2013b; Barshir et al. 2018). Paralog families can leverage additional insights in the context of sequence grouping approaches.
In comparison with other variant interpretation tools such as MTR (Traynelis et al. 2017), VEST 3.0 (Carter et al. 2013), or CADD (Kircher et al. 2014), PER viewer does not provide a score for all possible substitutions but instead provides a set of amino acids regions in which pathogenic variants accumulate significantly. PERs are able to capture aligned amino acids sites, regardless of disease association, which allows variant interpretation even if no missense variant has been previously reported (e.g., lysine index position 943) (Fig. 2B). The family-wise variant annotation allows us to manually inspect variants across the alignment index position, which can be useful for biological and clinical interpretation. For example, in the voltage-gated sodium channel gene-family example, we observe at index position 941 the fully paralog conserved leucine (PER5) (Fig. 2B) with pathogenic variants in the genes SCN1A, SCN8A, SNC4A, and SCN5A. Here, future variants found inside the genes SCN2A or SCN11A at the same alignment index position (leucine 870 or 690, respectively) are more likely to be pathogenic, reflecting the practical use of our family-wise approach. In fact, paralogous annotation and variant interpretation transfer has been explored before and could be considered as common practice in the field of electrophysiology (Ware et al. 2012; Walsh et al. 2014).
Our approach has several limitations. First, the missense burden analysis and statistical identification of PERs is highly dependent upon the number and quality of variants used as references for the population and patient data sets. We cannot rule out that missense variants outside PER boundaries are pathogenic; rather, we are prioritizing variants within these regions. Similarly, the ascertainment of variants for specific genes can be skewed, for example, different sequencing coverage of patient and population variants. As we showed with the burden analyses performed with older releases of patient variants, it is likely that more and stronger PERs will be identified as the population and patient databases continue to grow in size and quality. Second, paralogs belonging to the same family may evolve different functions through the development of specific domains (Pires-daSilva and Sommer 2003; Dos Santos and Siltberg-Liberles 2016). Upon alignment, gene-specific domains not present in other family members will not show conservation; they are therefore less likely to reach significance in the family-wise burden analysis. Nevertheless, if gene-specific domains are in fact enriched for pathogenic variants, the gene-wise approach could still identify PERs in such regions. Third, functional redundancy among paralogs does not guarantee the same degree of tolerance or intolerance to variation. Burden analyses, including genes tolerant to variation, will introduce noise and may mask specific signals. Similarly, genes with no pathogenic variants decrease the chances of reaching significance in regions with pathogenic variants in other family members. Fourth, our approach is able to identify protein regions constrained for variants in the general population and likely disease causing when mutated. Protein regions that can confer risk to disease through low penetrance variants or late onset of disease after typical reproductive age are unlikely to be identified in PERs owing to little constraint in the general population (Bodmer and Bonilla 2008). Finally, our analysis and the PERs detected are limited to canonical transcripts. Testing all combinations of transcripts alignments in the burden analysis would have made it very difficult to reach significance after multiple testing.
The ACMG guidelines (Richards et al. 2015) have made considerable efforts to provide guidelines and standardize criteria for pathogenicity assignment. Nevertheless, ∼49.49% of missense variants in ClinVar (October 2019) either have conflicting reports of pathogenicity, have no interpretation at all, or are annotated as VUSs (Landrum et al. 2016). With increasing data, machine learning approaches are likely to outperform older variant prediction algorithms such as PolyPhen and SIFT (Itan and Casanova 2015). However, they lack the ability to understand why a given prediction score is high or low, limiting translation into therapeutics and biology. With the PER viewer, we are able to collect the phenotypes observed from a given region in an online tool that can simultaneously serve as an intuitive variant interpretation tool. Our framework is not restricted to the aforementioned resources and can be implemented with alternatives inputs. For example, missense burden analysis using the Catalogue of Somatic Mutations in Cancer (COSMIC) (Forbes et al. 2017) may be of use in the detection of cancer-specific PERs.
PERs will empower gene discovery studies by facilitating the identification of specific regions within these candidate disease genes. This will have an immediate impact on the prioritization of candidate variants for researchers and molecular diagnostic laboratories evaluating variants within PERs.
Methods
Population missense variants
Protein-coding variants from the general population were retrieved from gnomAD public release 2.0.2 (Lek et al. 2016). Exonic variants were downloaded in the variant call format (VCFs) following gnomAD guidelines (http://gnomad.broadinstitute.org/downloads). Missense variants were extracted using VCFtools (Danecek et al. 2011) based on the consequence “CSQ” field. The CSQ field is preannotated by gnomAD with the Variant Effect Predictor (VEP) software (Ensembl v92) and provides information on 68 features, including gene/transcript, cross-database identifiers, as well as the desired molecular consequence. All annotations refer to the human reference genome version GRCh37.p13/hg19. Entries passing gnomAD standard quality controls (filter = “PASS” flag) and annotated to a canonical gene transcript (CSQ canonical = “YES” flag) were extracted. The canonical transcript is defined as the longest CCDS translation with no stop codons according to Ensembl (Hunt et al. 2018). Missense variants calls were merged into one single file, matching amino acid position and annotation. The final “population” data set contains all missense variants within canonical transcripts found in the general population.
Patient missense variants
Disease-associated missense variants were retrieved from two sources: the ClinVar database (ClinVar; release October 2019) (Landrum et al. 2016) and HGMD professional release 2019.2 (Stenson et al. 2003). ClinVar variants were downloaded directly from the ftp site (ftp://ftp.ncbi.nlm.nih.gov/pub/clinvar/) in a table format. Molecular consequence was inferred through the analysis of the Human Genome Variation Society (HGVS) sequence variant nomenclature field (den Dunnen et al. 2016). Specifically, when the variant was reported to cause an amino acid change different to the reference, it was subsequently annotated as a missense variant (e.g., p.Gly1046Arg). To increase stringency, ClinVar missense variants exclusively classified as “pathogenic” and/or “likely pathogenic” were considered. Missense variants with conflicting or ambiguous clinical significance (e.g., “pathogenic, other”) were excluded from the study. The HGMD data were directly filtered for “missense variants,” “high-confidence” calls (hgmd_confidence = “HIGH” flag), and “disease-causing” state (hgmd_variantType = “DM” flag). All annotations refer to the human reference genome version GRCh37.p13/hg19, and variants belonging to noncanonical transcripts were removed. Our approach is based in the study of missense variants mapped within canonical protein sequences with a consensus coding sequence (CCDS) (Supplemental Table S1), which are stable across different human genome assemblies (Pruitt et al. 2009). Thus, using GRCh38 annotations would not significantly affect our conclusions. Because ClinVar and HGMD are not mutually exclusive, we took the union of both resources and removed duplicated entries by comparing HGVS annotations. The final “patient” data set contains patient-derived missense variants and their corresponding disease annotation.
Gene-family definition
Gene families were retrieved following the method previously described (Lal et al. 2017). Briefly, we downloaded the human paralog definitions from the Ensembl BioMart system (Kinsella et al. 2011). Noncoding genes and genes without a HUGO Gene Nomenclature Committee (HGNC) (Yates et al. 2017) symbol were excluded. Similarly, gene families with fewer than two HGNC genes were filtered out. For all analyses, we used one transcript per gene, keeping only the canonical version according to Ensembl. To construct a family-wise FASTA file, respective CCDSs were downloaded for all canonical transcripts from the UCSC Table Browser (Karolchik et al. 2004). Family protein sequence alignment was conducted with MUSCLE (Edgar 2004). Younger evolutionary paralogs show higher functional redundancy (Chen et al. 2013a). To avoid alignments of strongly diverging sequences and to enrich for overall similarity, we filtered out families with <80% similarity in their overall protein sequence (Dufayard et al. 2005). In total, we used 2871 gene families comprising 9990 genes. Paralog gene-family structure and canonical protein sequences are shown in Supplemental Table S1. Population and patient data sets containing all missense variants analyzed in the present study (“input.gnomad” and “input.clinvar-hgmd,” respectively) are available in the Supplemental Code and at our GitHub repository (https://github.com/dlal-group/PERs/). Because access to HGMD professional release 2019.2 is restricted, the genomic coordinates and phenotypes of HGMD missense variants are not included in the patient variant input file (“input.clinvar-hgmd”). Here, HGMD missense variants contain only the observed protein exchange (e.g., Arg109Phe), which allows one to entirely reproduce the burden analyses and PER detection reported in this study.
Missense variant mapping
The population and patient missense variants were independently mapped to corresponding amino acids in all gene-family protein sequence alignments. Population and patient missense variant mapping was conducted using a binary annotation: “0” for amino acids with no missense variant reported and “1” for residues with at least one missense variant reported. We expected that constrained regions across the gene-family alignment will be enriched with amino acids marked as “0,” whereas disease-sensitive regions will cluster amino acids marked with “1.” We found that gene-family alignment regions with more gaps are less conserved than aligned amino acids and are more likely to not be functionally essential. Thus, in the population variant mapping, gaps introduced in any gene-family member were also assigned a “1” state as if they were mutated to penalize less-conserved sites. For the patient variant mapping, the gaps were kept as “0.” Because every missense variant contained in the patient subset was associated with at least one phenotype in one gene, multiple genes and diseases were aggregated in aligned residues upon alignment. This information was collected in an additional “Gene:Disease” field for further follow up.
Missense burden analysis—family-wise
We performed statistical comparisons between population and patient variants mapped to protein family alignments. Specifically, we applied sliding windows of nine amino acids across index positions of the paralog alignments with a 50% overlap to increase sensitivity (Fig. 1A). We summed the number of “0” and “1” sites inside and outside the window across the whole alignment index. A one-sided Fisher's exact test with 95% confidence was performed over each sliding window, comparing general population and patient counts inside the window against the corresponding counts outside of it. For example, a burden analysis based on a sliding window of size 5 will first test the counts of index positions 1 to 5 against the counts found from position 6 to the end of the alignment (Fig. 1). Bonferroni multiple testing adjustment was applied, accounting for the total number of sliding windows tested for each gene-family alignment. Sliding windows with adjusted P-values below 0.05 were considered significant and subsequently called PERs. If two or more consecutive sliding windows were found significant, the final PER reported will reflect the fusion of all consecutive significant windows boundaries. To identify the optimal sliding window size, the analysis was executed with multiple sliding window sizes—from three up to 31 amino acids—to evaluate the window size sensitivity and specificity (Supplemental Fig. S1). Sensitivity was measured by the number of significant regions detected, amino acids involved, and gene families affected. Specificity of the analysis was measured by the ratio between the number of amino acids sites inside PERs with no disease associations and the number of amino acids inside PERs with disease associations (i.e., in at least one family gene member). Missense variant mapping and sliding window counts were performed with an in-house Perl script. Fisher's exact tests, Bonferroni adjustment, and plots were performed with the R statistical software (R Core Team 2011).
Missense burden analysis—gene-wise
The missense variant mapping and burden analysis protocols were further applied to all RefSeq genes independently to evaluate gene-wise enrichment. For all 18,805 canonical transcripts, their respective CCDS was downloaded from the UCSC Table Browser (Karolchik et al. 2004). The missense variant mapping and burden analyses were conducted using the same Perl scripts, treating each gene as a one-member “family.” Perl (Part-1-missense-aligner.pl) and R (Part-2-burden-analysis.R) scripts used to carry out both family-wise and gene-wise missense burden analyses are available in the Supplemental Code and at our GitHub repository (https://github.com/edoper/PERs). Additionally, we provide a tutorial, test data, and expected output that allow users to carry out PER detection on any given alignment or gene file. PER Pfam domain annotation was performed with VEP software (McLaren et al. 2016) using the genomic coordinates of PERs for both gene-wise and family-wise analysis approaches.
Development of PER viewer
Population and patient missense burden calculations as well as the identification of significant regions within genes and gene families were made publicly available through the PER viewer (http://per.broadinstitute.org). PER viewer was developed with the Shiny framework of R studio, which transforms regular R code into HTML that can be displayed by any web browser. Precalculated burden analyses for all genes and gene families (Supplemental Table S1) were deployed in a Google virtual machine (VM) using the googleComputeEngineR package (https://cloudyr.github.io/googleComputeEngineR/). All graphs shown in the present work and by the online tool are based on the ggplot2 R library (Wickham 2009).
Software availability
The complete set of burden analyses for all gene families and genes is freely available on the PER viewer (http://per.broadinstitute.org). The website was implemented with R shiny framework, and all major browsers are supported. The Supplemental Code and our GitHub repository (https://github.com/dlal-group/PERs/) contain the source code and missense variants able to perform the missense burden analysis (Perl script: Part-1-missense-aligner) and PER detection (R script: Part-2-burden-analysis). Here, we included a detailed tutorial with test data and expected output that will allow the user to replicate entirely our burden analysis, patient versus population burden plots, and PER detection. The software is supported on Linux and freely available to noncommercial users under a MIT license.
Acknowledgments
We thank the Genome Aggregation Database (gnomAD) and the groups that provided exome- and genome-wide data to this resource. A full list of contributing groups can be found at http://gnomad.broadinstitute.org/about. We also thank the researchers and clinicians involved in the generation of clinical data contained in ClinVar and HGMD, as well as the respective patients and their families. E.P.-P. was supported by Dravet Syndrome Foundation research grant to D.L.
Author contributions: E.P.-P. and D.L. conceived and designed the study; P.M., A.P., A.O.-L., P.N., and M.D. supervised the study; E.P.-P., S.I., L.-M.N., J.A.C., and H.O.H. gathered and analyzed the data; E.P.-P. and J.D. designed and developed the web interface; and E.P.-P., P.M., and D.L. drafted the manuscript with input from all authors.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.252601.119.
References
- ↵The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. 10.1038/nature15393
- ↵Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. 10.1038/nmeth0410-248
- ↵Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. 2011. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet 12: 745–755. 10.1038/nrg3031
- ↵Barshir R, Hekselman I, Shemesh N, Sharon M, Novack L, Yeger-Lotem E. 2018. Role of duplicate genes in determining the tissue-selectivity of hereditary diseases. PLoS Genet 14: e1007327. 10.1371/journal.pgen.1007327
- ↵Bartha I, di Iulio J, Venter JC, Telenti A. 2018. Human gene essentiality. Nat Rev Genet 19: 51–62. 10.1038/nrg.2017.75
- ↵Bodmer W, Bonilla C. 2008. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet 40: 695–701. 10.1038/ng.f.136
- ↵Carter H, Douville C, Stenson PD, Cooper DN, Karchin R. 2013. Identifying Mendelian disease genes with the Variant Effect Scoring Tool. BMC Genomics 14: S3. 10.1186/1471-2164-14-S3-S3
- ↵Chen S, Krinsky BH, Long M. 2013a. New genes as drivers of phenotypic evolution. Nat Rev Genet 14: 645–660. 10.1038/nrg3521
- ↵Chen W-H, Zhao X-M, van Noort V, Bork P. 2013b. Human monogenic disease genes have frequently functionally redundant paralogs. PLoS Comput Biol 9: e1003073. 10.1371/journal.pcbi.1003073
- ↵Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloğlu A, Özen S, Sanjad S, 2009. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci 106: 19096–19101. 10.1073/pnas.0910672106
- ↵Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. 2005. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 15: 901–913. 10.1101/gr.3577405
- ↵Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. 10.1093/bioinformatics/btr330
- ↵den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, Roux A-F, Smith T, Antonarakis SE, Taschner PEM. 2016. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat 37: 564–569. 10.1002/humu.22981
- ↵Dos Santos HG, Siltberg-Liberles J. 2016. Paralog-specific patterns of structural disorder and phosphorylation in the vertebrate SH3–SH2–tyrosine kinase protein family. Genome Biol Evol 8: 2806–2825. 10.1093/gbe/evw194
- ↵Dufayard J-F, Duret L, Penel S, Gouy M, Rechenmann F, Perrière G. 2005. Tree pattern matching in phylogenetic trees: automatic search for orthologs or paralogs in homologous gene sequence databases. Bioinformatics 21: 2596–2603. 10.1093/bioinformatics/bti325
- ↵Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797. 10.1093/nar/gkh340
- ↵Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, 2016. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44: D279–D285. 10.1093/nar/gkv1344
- ↵Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, 2017. COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res 45: D777–D783. 10.1093/nar/gkw1121
- ↵Ge X, Gong H, Dumas K, Litwin J, Phillips JJ, Waisfisz Q, Weiss MM, Hendriks Y, Stuurman KE, Nelson SF, 2016. Missense-depleted regions in population exomes implicate ras superfamily nucleotide-binding protein alteration in patients with brain malformation. NPJ Genom Med 1: 16036. 10.1038/npjgenmed.2016.36
- ↵Gilissen C, Hoischen A, Brunner HG, Veltman JA. 2012. Disease gene identification strategies for exome sequencing. Eur J Hum Genet 20: 490–497. 10.1038/ejhg.2011.258
- ↵Grantham R. 1974. Amino acid difference formula to help explain protein evolution. Science 185: 862–864. 10.1126/science.185.4154.862
- ↵Gussow AB, Petrovski S, Wang Q, Allen AS, Goldstein DB. 2016. The intolerance to functional genetic variation of protein domains predicts the localization of pathogenic mutations within genes. Genome Biol 17: 9. 10.1186/s13059-016-0869-4
- ↵Harrison SM, Dolinsky JS, Johnson AEK, Pesaran T, Azzariti DR, Bale S, Chao EC, Das S, Vincent L, Rehm HL. 2017. Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet Med 19: 1096–1104. 10.1038/gim.2017.14
- ↵Hayeck TJ, Stong N, Wolock CJ, Copeland B, Kamalakaran S, Goldstein DB, Allen AS. 2019. Improved pathogenic variant localization via a hierarchical model of sub-regional intolerance. Am J Hum Genet 104: 299–309. 10.1016/j.ajhg.2018.12.020
- ↵Heyne HO, Singh T, Stamberger H, Abou Jamra R, Caglayan H, Craiu D, De Jonghe P, Guerrini R, Helbig KL, Koeleman BPC, 2018. De novo variants in neurodevelopmental disorders with epilepsy. Nat Genet 50: 1048–1053. 10.1038/s41588-018-0143-7
- ↵Hunt SE, McLaren W, Gil L, Thormann A, Schuilenburg H, Sheppard D, Parton A, Armean IM, Trevanion SJ, Flicek P, 2018. Ensembl variation resources. Database (Oxford) 2018: bay119. 10.1093/database/bay119/5255129
- ↵Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E, Karyadi D, 2016. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 99: 877–885. 10.1016/j.ajhg.2016.08.016
- ↵Itan Y, Casanova J-L. 2015. Can the impact of human genetic variations be predicted? Proc Natl Acad Sci 112: 11426–11427. 10.1073/pnas.1515057112
- ↵Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. 2004. The UCSC Table Browser data retrieval tool. Nucleic Acids Res 32: D493–D496. 10.1093/nar/gkh103
- ↵Kinsella RJ, Kähäri A, Haider S, Zamora J, Proctor G, Spudich G, Almeida-King J, Staines D, Derwent P, Kerhornou A, 2011. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxford) 2011: bar030. 10.1093/database/bar030/465356
- ↵Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46: 310–315. 10.1038/ng.2892
- ↵Kumar P, Henikoff S, Ng PC. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4: 1073–1081. 10.1038/nprot.2009.86
- ↵Lal D, May P, Samocha KE, Kosmicki JA, Robinson EB, Møller RS, Krause R, Nüernberg P, Weckhuysen S, Jonghe PD, 2017. Gene family information facilitates variant interpretation and identification of disease-associated genes. bioRxiv 10.1101/159780
- ↵Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, 2016. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44: D862–D868. 10.1093/nar/gkv1222
- ↵Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature 536: 285–291. 10.1038/nature19057
- ↵Liu X, Wu C, Li C, Boerwinkle E. 2016. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat 37: 235–241. 10.1002/humu.22932
- ↵McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. 2016. The Ensembl Variant Effect Predictor. Genome Biol 17: 122. 10.1186/s13059-016-0974-4
- ↵Melloni GEM, de Pretis S, Riva L, Pelizzola M, Céol A, Costanza J, Müller H, Zammataro L. 2016. LowMACA: exploiting protein family analysis for the identification of rare driver mutations in cancer. BMC Bioinformatics 17: 80. 10.1186/s12859-016-0935-7
- ↵Miosge LA, Field MA, Sontani Y, Cho V, Johnson S, Palkova A, Balakishnan B, Liang R, Zhang Y, Lyon S, 2015. Comparison of predicted and actual consequences of missense mutations. Proc Natl Acad Sci 112: E5189–E5198. 10.1073/pnas.1511585112
- ↵Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. 2013. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 9: e1003709. 10.1371/journal.pgen.1003709
- ↵Pires-daSilva A, Sommer RJ. 2003. The evolution of signalling pathways in animal development. Nat Rev Genet 4: 39–49. 10.1038/nrg977
- ↵Pruitt KD, Harrow J, Harte RA, Wallin C, Diekhans M, Maglott DR, Searle S, Farrell CM, Loveland JE, Ruef BJ, 2009. The consensus coding sequence (CCDS) project: identifying a common protein-coding gene set for the human and mouse genomes. Genome Res 19: 1316–1323. 10.1101/gr.080531.108
- ↵R Core Team. 2011. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/.
- ↵Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–423. 10.1038/gim.2015.30
- ↵Samocha KE, Kosmicki JA, Karczewski KJ, O'Donnell-Luria AH, Pierce-Hoffman E, MacArthur DG, Neale BM, Daly MJ. 2017. Regional missense constraint improves variant deleteriousness prediction. bioRxiv 10.1101/148353
- ↵Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GLA, Edwards KJ, Day INM, Gaunt TR. 2013. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat 34: 57–65. 10.1002/humu.22225
- ↵Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NST, Abeysinghe S, Krawczak M, Cooper DN. 2003. Human Gene Mutation Database (HGMD®): 2003 update. Hum Mutat 21: 577–581. 10.1002/humu.10212
- ↵Tavtigian SV, Greenblatt MS, Harrison SM, Nussbaum RL, Prabhu SA, Boucher KM, Biesecker LG. 2018. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet Med 20: 1054–1060. 10.1038/gim.2017.210
- ↵Traynelis J, Silk M, Wang Q, Berkovic SF, Liu L, Ascher DB, Balding DJ, Petrovski S. 2017. Optimizing genomic medicine in epilepsy through a gene-customized approach to missense variant interpretation. Genome Res 27: 1715–1729. 10.1101/gr.226589.117
- ↵Walsh R, Peters NS, Cook SA, Ware JS. 2014. Paralogue annotation identifies novel pathogenic variants in patients with Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia. J Med Genet 51: 35–44. 10.1136/jmedgenet-2013-101917
- ↵Ware JS, Walsh R, Cunningham F, Birney E, Cook SA. 2012. Paralogous annotation of disease-causing variants in long QT syndrome genes. Hum Mutat 33: 1188–1191. 10.1002/humu.22114
- ↵Wickham H. 2009. ggplot2: elegant graphics for data analysis. Springer-Verlag, New York.
- ↵Wiel L, Venselaar H, Veltman JA, Vriend G, Gilissen C. 2017. Aggregation of population-based genetic variation over protein domain homologues and its potential use in genetic diagnostics. Hum Mutat 38: 1454–1463. 10.1002/humu.23313
- ↵Xue Y, Ankala A, Wilcox WR, Hegde MR. 2015. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med 17: 444–451. 10.1038/gim.2014.122
- ↵Yates B, Braschi B, Gray KA, Seal RL, Tweedie S, Bruford EA. 2017. Genenames.org: the HGNC and VGNC resources in 2017. Nucleic Acids Res 45: D619–D625. 10.1093/nar/gkw1033