
Capture of a functionally active methyl-CpG binding domain by an arthropod retrotransposon family
Abstract
The repressive capacity of cytosine DNA methylation is mediated by recruitment of silencing complexes by methyl-CpG binding domain (MBD) proteins. Despite MBD proteins being associated with silencing, we discovered that a family of arthropod Copia retrotransposons have incorporated a host-derived MBD. We functionally show how retrotransposon-encoded MBDs preferentially bind to CpG-dense methylated regions, which correspond to transposable element regions of the host genome, in the myriapod Strigamia maritima. Consistently, young MBD-encoding Copia retrotransposons (CopiaMBD) accumulate in regions with higher CpG densities than other LTR-retrotransposons also present in the genome. This would suggest that retrotransposons use MBDs to integrate into heterochromatic regions in Strigamia, avoiding potentially harmful insertions into host genes. In contrast, CopiaMBD insertions in the spider Stegodyphus dumicola genome disproportionately accumulate in methylated gene bodies compared with other spider LTR-retrotransposons. Given that transposons are not actively targeted by DNA methylation in the spider genome, this distribution bias would also support a role for MBDs in the integration process. Together, these data show that retrotransposons can co-opt host-derived epigenome readers, potentially harnessing the host epigenome landscape to advantageously tune the retrotransposition process.
Cytosine DNA methylation is a base modification associated with gene and transposable element repression in animals (Zemach and Zilberman 2010; Schübeler 2015; Deniz et al. 2019). Methylation is deposited by DNA methyltransferases on CpG dinucleotides (CpGs), but despite DNA methyltransferases being deeply conserved across animal genomes (Lyko 2018), there is extensive variability regarding the genome methylation levels and distribution between lineages. In vertebrates, there is widespread high methylation across the genome, mostly only absent from CpG island promoters and active regulatory regions (Schübeler 2015). In contrast, in invertebrates, methylation is “mosaic,” concentrated on active gene bodies and, in some instances, on transposable elements (Suzuki and Bird 2008). However, some invertebrate lineages have lost DNA methylation, such as Drosophila melanogaster and Caenorhabditis elegans, whereas others have lost methylation only on transposable elements, including most insects and crustaceans (Bewick et al. 2017; Gatzmann et al. 2018).
Critical to the function of cytosine DNA methylation are methylation “readers,” proteins capable of binding and interpreting the methylation state and subsequently altering the transcriptional output or chromatin environment (Law and Jacobsen 2010; Zhu et al. 2016). The major family of methylation readers are the methyl-CpG binding domain (MBD) proteins (Bogdanović and Veenstra 2009; Du et al. 2015). The 70-amino-acid-long MBD is responsible for binding to methylated CpGs, whereas most MBD family members have additional protein domains that can recruit silencing complexes (Du et al. 2015). Not all MBD family members are able to bind methylated cytosines, despite encoding an MBD; for instance, SETDB and BAZ2 chromatin remodelers or the mammalian MBD3 ortholog prefer unmethylated cytosines (Hendrich and Tweedie 2003). With the exception of MBD4, which is known for its role in DNA repair after methylated cytosine deamination, all other MBD family members are highly associated with gene repression and heterochromatin formation (Bogdanović and Veenstra 2009; Du et al. 2015). Thus, cytosine methylation and heterochromatin formation by MBD proteins are one of the main defense mechanisms that the host genome possesses to silence and control transposable elements (Levin and Moran 2011; Deniz et al. 2019).
In turn, transposable elements are engaged in a continual arms race with their hosts. To proliferate, transposons must develop strategies to escape from silencing mechanisms and targeting by the host. Among transposable elements, there are two main types depending on their replication strategy: DNA transposons, which are excised and copied in the genome as DNA, and retrotransposons, which have an intermediate RNA step before retrotranscription to DNA and integration (Wicker et al. 2007; Bourque et al. 2018). Retrotransposons are furtherly divided in two main types: long terminal repeat (LTR) retrotransposons, which possess repetitive sequences flanking the retrotransposon, and long interspersed nuclear elements (LINEs), which lack LTRs. Autonomous LTR-retrotransposons require at least a reverse transcriptase (RT) and an integrase (INT) to replicate by themselves; however, additional protein domains might have an influence in the retrotransposition process. Here, we report how a family of retrotransposons has benefited from integrating an MBD into their coding sequence, challenging our views on MBD function and retrotransposon evolution.
Results
While profiling the evolution of genes involved in DNA methylation in animal genomes, we serendipitously discovered that the centipede Strigamia maritima (Chipman et al. 2014) encoded hundreds of MBD containing proteins (Fig. 1A), in contrast to most animal genomes that encode between one and 10 MBD family members. Some of the Strigamia MBD-containing gene models also presented typical retrotransposon domains, such as INTs and RTs, specifically the RVT_2 domain characteristic of Copia retrotransposons (Wicker et al. 2007). By performing a de novo annotation of repetitive elements in the Strigamia genome, we confirmed that 98% of the MBD gene models were not host genes belonging to conserved gene families but were in fact in open reading frames (ORFs) belonging to retrotransposons. Some of the copies displayed well-conserved LTRs typical of Copia retrotransposons; thus, we called this new type of retrotransposon CopiaMBDs (Fig. 1B).
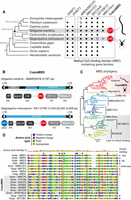
A family of arthropod Copia retrotransposons have incorporated an MBD into their coding sequence. (A) Cladogram showing a subset of animal species, taxonomic affiliation, and the presence/absence patterns of major DNA methylation enzymes (DNMT1 and DNMT3) and MBD gene family members encoded in their genomes. In red indicates the total number of MBDs belonging to Copia retrotransposons encoded in centipede and spider genomes. (B) Case examples of CopiaMBD structure from Strigamia and Stegodyphus genomes, as well as domain architecture of the Pol ORF. The protein domains as defined by Pfam (MBD PF01429, gag_pre PF13976, rve PF00665, RVT_2 PF07727, RNase_H PF00075). The domains as also annotated according to retroviral nomenclature conventions: (GAG) group-specific antigen; (PR) protease; (INT) integrase; (RT) reverse transcriptase; (RH) RNase H. (C) Maximum likelihood phylogeny of the MBD metazoan gene families. Nodal supports represent nonparametric bootstrap as computed by IQ-TREE. Shaded in red are the sequences belonging to Copia retrotransposons, shaded in gray are the MBD families known for not having methyl-binding activity despite encoding an MBD. Red branches indicate CopiaMBD Strigamia sequences, and orange branches indicate CopiaMBD Steodyphus sequences. (D) MBD multisequence alignment. Black triangles highlight amino acids known to influence the DNA binding ability of the MBD (Hendrich and Tweedie 2003). Shaded in red is the phenylalanine of the mammalian MBD3 responsible for its lack of methylcytosine binding activity. Amino acid color code is as per polarity described in the legend. (Hsap) Homo sapiens; (Xlae) Xenopus laevis; (Smar) Strigamia maritima; (Smim) Stegodyphus mimosarum. Silhouettes were obtained from http://phylopic.org/.
To test whether this retrotransposon family was specific to the centipede Strigamia, we used the MBD sequence to scan for its presence in other animal genomes. The only genomes in which we identified similarity hits were in the spiders Stegodyphus mimosarum (Sanggaard et al. 2014) and Stegodyphus dumicola (Liu et al. 2019), whereas it was not detected in other arachnid, pancrustacean, or myriapod genomes (Fig. 1A). We then asked whether CopiaMBDs in Strigamia and Stegodyphus evolved through recruiting MBDs independently or whether the MBD capture occurred once and was then vertically inherited. To test this, we built a phylogenetic tree of eukaryotic RTs belonging to Copia retrotransposons (Supplemental Fig. S1), confirming that all CopiaMBD are monophyletic and thus share a common ancestor that already encoded an MBD.
Given that MBDs had not been previously observed in retrotransposons, we next investigated the origins of the retrotransposon MBD by building a phylogenetic tree of MBD family proteins. This revealed that CopiaMBDs branched as a sister group to the MBD1/2/3 and MBD4/MECP2 clades, whereas BAZ2, SETDB, and a previously unreported MBD-Fbox family branched as an outgroup (Fig. 1C). Although the phylogeny did not allow us to specify the parental family of the retrotransposon MBDs, CopiaMBDs did show closer affiliation to MBD families known to be able to bind methylated cytosines. Congruently, the amino acids known to be responsible for DNA binding were conserved in most CopiaMBD copies, specifically the tyrosine residue known to be responsible for methylcytosine recognition (Fig. 1D; Hendrich and Tweedie 2003).
Transposons have been reported to jump across species, even between distantly related lineages (Schaack et al. 2010; El Baidouri et al. 2014; Peccoud et al. 2017). Given the patchy distribution of CopiaMBDs across arthropod genomes, we tested if there were any evidence for recent horizontal gene transfer between Strigamia and Stegodyphus. If CopiaMBDs had been acquired through horizontal transfer, they would be expected to show fewer changes across species than equivalent proteins that have been vertically inherited. However, we did not observe this when comparing the MBD branch lengths between conserved orthologs; in fact, CopiaMBDs showed more amino acid substitutions per site than any other MBD-encoding gene family (Supplemental Fig. S2A). When assessing the nucleotide synonymous substitution rates (Ks) between Strigamia and Stegodyphus CopiaMBDs, most copies presented saturated changes (Ks = 10), whereas those that were not saturated had similar substitution rates than those of conserved one-to-one orthologs (Supplemental Fig. S2B). Therefore, we found no support for a recent horizontal transfer for CopiaMBDs.
We next assessed whether the retrotransposon-encoded MBDs showed signs of purifying selection when comparing divergent CopiaMBD copies from the same species. MBDs showed an excess of synonymous substitutions versus nonsynonymous (Ka/Ks < 0.1), at the same extent as observed in the neighboring INT domain (Supplemental Fig. S3), which is critical for retrotransposon replication. Therefore, the MBD is actively conserved, suggesting retrotransposons benefit from its presence.
To explore the functional conservation of CopiaMBDs, we cloned the MBD of three Strigamia divergent copies (CopiaMBD 1, 2, 3), which only showed 46% identical amino acids between each other, and performed DNA affinity purification sequencing (DAP-seq) (Bartlett et al. 2017). For DAP-seq, native Strigamia genomic DNA was fragmented and adaptor ligated, incubated with in vitro–expressed CopiaMBDs fused to a HaloTag, and purified using magnetic separation. The same strategy was followed in parallel with PCR-amplified Strigamia genomic DNA libraries, which have lost the native DNA methylation configuration through amplification with unmethylated nucleotides (ampDAP-seq) (Bartlett et al. 2017). We then sequenced and mapped the purified DNA back to the Strigamia genome and found that all CopiaMBD DAP-seq profiles clustered together, whereas ampDAP-seq samples clustered aside, showing high correlation with the ampDAP-seq background empty HaloTag library (Spearman's correlation ≥ 0.87) (Supplemental Fig. S4A). When comparing the enrichment signal of CopiaMBDs to the background, only DAP-seq samples showed an enrichment, albeit somewhat lower for CopiaMBD 1 (Supplemental Fig. S4B). This indicates that although ampDAP-seq samples were almost indistinguishable from the ampDAP-seq background, DAP-seq samples showed strong sequence preferences.
We then identified thousands of CopiaMBD DAP-seq peaks enriched over background (7461–15,714), whereas ampDAP-seq libraries retrieved only a few hundred peaks (133–358) (Supplemental Fig. S5A). A large majority of peaks were overlapping between the CopiaMBD DAP-seq libraries, with a substantial fraction (24%–59%) of reads located in peaks, indicating a high signal-to-background ratio (Supplemental Fig. S5B). On the contrary, ampDAP-seq peaks showed very few overlapping peaks between samples, and <0.6% of the reads were located in peaks, underscoring lack of specificity (Supplemental Fig. S5B). Taken together, these data show that CopiaMBDs have a high affinity for binding natively methylated DNA compared with its unmethylated counterpart.
To confirm methyl-CpG binding affinity, we profiled the native methylome by whole-genome bisulfite sequencing (WGBS) of the matched Strigamia genomic DNA used for DAP-seq. This revealed that CopiaMBD DAP-seq peaks were strongly enriched in highly methylated regions, as well as showing a high density of CpGs (Fig. 2A). Additionally, CopiaMBD peaks showed motifs enriched in CpG sites (Supplemental Fig. S5C), which together indicates that retrotransposon-encoded MBDs show the typical binding affinity of canonical MBD family proteins (Baubec et al. 2013; Rube et al. 2016).
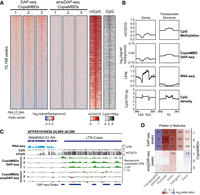
Retrotransposon-encoded MBDs preferentially bind methylated CpG-dense regions overlapping transposable elements. (A) Heatmap showing enrichment levels for three phylogenetically distinct CopiaMBDs based on Strigamia DAP-seq (native genome methylation) and ampDAP-seq (amplified genome depleted of native methylation) libraries, CpG methylation, and CpG density on a union of all three CopiaMBD DAP-seq peaks set. (B) Profiles of CpG methylation, CopiaMBD 3 DAP-seq enrichment, RNA-seq, and CpG density on Strigamia protein-coding genes and transposable elements. (C) Genome browser display showing enrichment tracks for DAP-seq, ampDAP-seq, and cytosine methylation. RNA-seq shown as counts per million (CPM), whole-genome bisulfite sequencing (WGBS) shown as the mC/C ratio at CpG sites, DAP-seq, and ampDAP-seq data shown as background subtracted CPM tracks (CopiaMBD signal minus empty pIX-HALO plasmid). (D) Heatmap showing the enrichment of CopiaMBD peaks on various genomic features. Colors represent log2(odds ratio), whereas untransformed odds ratios are shown for the significantly enriched tests (Fisher's exact test P < 0.01).
To further investigate the ability of CopiaMBDs to preferentially bind to methylated DNA, we took an orthogonal approach to peak calling. By using the WGBS data, we selected genomic regions with high CpG densities (>5 CpG/100 bp) that showed either high methylation (>0.8 mCG/CG) or no methylation (<0.2 mCG/CG), ensuring that most DNA molecules in the DAP-seq unamplified libraries belonging to those regions were either methylated or unmethylated. We then compared the coverage on those regions for CopiaMBD-incubated DAP-seq samples and for the empty-HaloTag background control. This confirmed that CopiaMBDs show enriched coverage on the methylated regions and depleted coverage for unmethylated regions (Supplemental Fig. S6A), confirming CopiaMBD preference for methylated DNA in a pool of molecules with equivalent CpG densities. Furthermore, we tested whether CopiaMBDs had a preference for either non-CG methylation or hemi-methylated sites in the Strigamia genome, finding no support for either context (Supplemental Fig. S7).
Although it is well established that genomic methylation is generally stable across developmental stages and tissues in invertebrates (Suzuki et al. 2013; Dixon et al. 2016; Libbrecht et al. 2016; Gatzmann et al. 2018), we wanted to test if cell-type heterogeneity could be confounding the alleged CopiaMBD preference for methylated DNA. By subsetting CpGs for those with reliable coverage (>10×), we observed opposite distributions of methylation levels on all genome CpGs compared with CpGs found on CopiaMBD DAP-seq peaks (Supplemental Fig. S6B). Whereas the majority of CpGs in the genome are unmethylated (0 mCG/CG), most CpGs on CopiaMBD DAP-seq peaks are fully methylated (1.0 mCG/CG). This confirms CopiaMBD's strong enrichment not only for methylated regions but also for sites for which molecules are 100% methylated. Testing the aggregated methylation levels for CopiaMBD DAP-seq peaks revealed a similar enrichment for high methylation levels (≥0.9 mCG/CG) (Supplemental Fig. S6C,D), thus confirming that CopiaMBD peaks are heavily methylated and not confounded by cell-type heterogeneity.
The Strigamia genome is sparsely methylated, as most invertebrate genomes are (Feng et al. 2010; Zemach et al. 2010; Schübeler 2015), showing cytosine DNA methylation concentrated on expressed gene bodies and silent transposable elements (Fig. 2B; Supplemental Fig. S8). However, the highest CpG densities are concentrated on unmethylated promoters and methylated transposable elements, suggesting that CopiaMBDs are more likely to bind transposable elements. Indeed, we confirmed this prediction, as CopiaMBD DAP-seq peaks were highly enriched on transposable elements (five to six odds ratio) while being significantly depleted on any other genomic features, including exons (Fig. 2C,D). Among the transposable elements overlapping CopiaMBD DAP-seq peaks, most LTR-retrotransposon classes were statistically enriched (q-value < 0.05) (Supplemental Fig. S9), including CopiaMBDs as well as Gypsy and non-MBD-encoding Copia elements. In contrast, most DNA transposons were depleted. In summary, this indicates that the MBD is likely guiding the insertion site of the new copies of the retrotransposon to highly methylated CpG-rich regions, which coincide with heterochromatic regions enriched in LTR-retrotransposons of the host genome.
To further investigate whether the MBD has a role in directing the insertion of CopiaMBDs, we characterized the insertion sites of young LTR-retrotransposon copies in the Strigamia and Stegodyphus genomes (LTR identity > 90%). By selecting the 150 flanking nucleotides 5′ and 3′ of the LTR-retrotransposons, we observed that CopiaMBDs were in regions with higher CpG densities than those of other LTR-retrotransposons, including Copias lacking an MBD, Gypsy, or Pao retrotransposon (one-sided Wilcoxon rank-sum test P value < 0.05) (Fig. 3A). Despite the insertion of the retrotransposon potentially causing the modification of the neighboring region's methylation status, we quantified the methylation levels of the 1000-bp flanking nucleotides of LTR-retrotransposons in Strigamia and Stegodyphus. CopiaMBD neighboring regions showed consistently higher methylation levels than those of other LTR-retrotransposons (Fig. 3B), thus suggesting that they might have inserted in previously methylated areas or that they are more likely to attract methylation after insertion. Of note, it is well established that genomic transposable element postintegration distribution is heavily influenced by selection (Sultana et al. 2019), given that deleterious insertions are less likely to be fixed in the population and therefore will not be detected. This is particularly evident when comparing distributions of Alu and LINE elements in mammalian genomes. Despite Alu and LINEs sharing the same integration machinery, they are very different in size and sequence composition, and are enriched in distinct genomic regions by a combination of selective pressures (Pavlíček et al. 2001). However, both Copia and Gypsy LTR-retrotransposons in Stegodyphus and Strigamia genomes show very similar distributions (Fig. 3); thus, it is likely that these distributions represent the expected distribution of LTR-retrotransposons after postintegration selection. Therefore, CopiaMBD distribution deviations from those of other LTR-retrotransposons seem to suggest an initial bias in integration preferences. Therefore, the combined evidence from flanking CpG density and methylation levels is congruent with a possible role of the MBD in directing the insertion localization of CopiaMBDs.
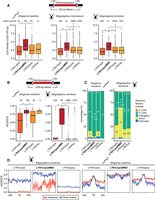
LTR-retrotransposon insertion distribution in the Stegodyphus and Strigamia genomes. (A) Distribution of CpG densities in the 150-bp flanking regions surrounding LTR-retrotransposons classified in four major classes. Asterisks represent Wilcoxon one-sided rank-sum test P < 0.01; the dashed gray line shows the genomic CpG density for each species. (B) Distribution of methylation levels in the 1000-bp flanking regions surrounding LTR-retrotransposons. (C) LTR-retrotransposon insertion intersections with genomic features based on an RNA-seq annotation (StringTie). Asterisks represent Fisher's exact two-sided test P < 0.01 comparing intergenic versus nonintergenic overlap proportions between LTR/CopiaMBD and LTR/Copia and LTR/Gypsy, respectively. (D) Average methylation levels on LTR-retrotransposon insertions divided by location within intergenic regions or gene bodies (nonintergenic categories in panel C). Thick line depicts mean methylation; shade, SE.
Finally, we could observe that CopiaMBD elements were statistically enriched in intergenic regions compared with Copia lacking an MBD and Gypsy retrotransposons in Strigamia (two-sided Fisher's exact test < 0.01) (Fig. 3C). This was congruent with the expectation based on the CopiaMBD binding patterns, depleted from gene bodies. In contrast, the LTR-retrotransposon distribution was the inverse in Stegodyphus. In Stegodyphus, CopiaMBDs were enriched in introns and depleted from intergenic regions compared with other LTR-retrotransposon copies (Fig. 3C). In fact, DNA methylation has been reported not to target transposable elements in the Stegodyphus genome (Liu et al. 2019). We further tested this by dividing LTR-retrotransposon elements into two categories: elements found within intergenic regions and elements found in gene bodies (UTRs, promoters, introns) (Fig. 3D). LTR-retrotransposons in gene bodies showed higher methylation levels than those in intergenic regions in Stegodyphus, corroborating that retrotransposons are rarely marked by methylation outside gene bodies in this species. Conversely, Strigamia LTR-retrotransposons are marked by methylation, irrespective of their genomic position. This indicates that the accumulation of CopiaMBDs toward distinct genomic elements might shift depending on the host epigenome patterns.
Discussion
Here, we show how a group of arthropod retrotransposons have acquired a functionally conserved MBD. As the MBD is located adjacent to the INT domain in the same ORF (no stop codons between both protein domains), we hypothesize that the MBD has a role in restricting the integration site of the newly retrotranscribed DNA. Despite the mature peptides encoding the INT and the MBD having the potential to be cleaved and separated by the CopiaMBD aspartic peptidase, this does not preclude the role of MBD in establishing integration preferences, as shown by the neighboring region characteristics of CopiaMBDs relative to other LTR-retrotransposons. Given the binding affinities of the MBD and the known poor sequence specificity of INTs (Sultana et al. 2017), it is possible that the new insertions will be guided by the MBD. In the Strigamia genome, such MBD-preferred regions would correspond to highly methylated transposable elements. This preference for repetitive regions would avoid potentially harmful effects to the host by not disrupting genes, at least in the Strigamia genome. This strategy would be analogous to chromoviruses, a type of Gypsy retrotransposon known to use chromodomains to direct the integration of new copies to heterochromatic regions (Gao et al. 2008). Similarly, many retrotransposons encode a PHD finger domain (Kapitonov and Jurka 2003; Pérez-Alegre et al. 2005), which is also capable of recognizing specific histone tail modifications (Sanchez and Zhou 2011). Therefore, encoding a protein domain such as MBD that influences integration would fit with the current knowledge about retrotransposons.
However, the MBD could, in principle, be unrelated to the retrotransposon integration process. Indeed, postintegration selection could influence the distinct distribution patterns observed for CopiaMBD elements. However, for selection to have a preponderant role in determining CopiaMBD distributions, there should be a selective advantage for CopiaMBD elements to be found on CpG-rich regions; a selective advantage that would not apply to other LTR-retrotransposons. Also, postintegration selection should reflect different evolutionary pressures in Strigamia and Stegodyphus, because the distribution of CopiaMBDs in each species shifts in opposite directions—depleted versus enriched on gene bodies, respectively—compared with that of other LTR-retrotransposons. For instance, CopiaMBD elements found in Stegodyphus intergenic regions would be more likely to be lost than other LTR-retrotransposons in the same regions, whereas CopiaMBDs in gene bodies would not be selected against. Furthermore, in order to explain why CopiaMBDs are found in regions with consistently higher methylation levels than other LTR-retrotransposons of comparable ages, DNMTs would need to have a mechanism to actively target CopiaMBDs after integration and spread DNA methylation to nearby regions in both Strigamia and Stegodyphus. Such a DNMT-targeting mechanism would not actively target other LTR-retrotransposons for methylation. Also, accumulation of CopiaMBDs in hypermethylated regions could be merely coincidental. In such scenarios, the MBD could have a role in other phases of the life cycle of the retrotransposon, unrelated to integration. Perhaps, this role of the MBDs could be related to interfering with host-related processes, for instance, silencing of endogenous genes. The role of well-characterized MBD-containing proteins in silencing is dependent on adjacent protein-interaction domains responsible for recruiting silencing complexes (Bogdanović and Veenstra 2009; Du et al. 2015). However, such protein-interaction domains are not detectable in CopiaMBDs. MBDs from CopiaMBDs could be competing for binding with host MBD-containing proteins, thus inhibiting the formation of heterochromatin, which could be beneficial toward integration. However, this would presumably be a very nonspecific process and would require high levels of CopiaMBD protein. It could also be argued that the MBDs have a beneficial role for the host, causing CopiaMBDs to be indirectly selected as a result. However, such beneficial roles are not fully consistent with the limited distribution of CopiaMBDs across arthropod genomes. In sum, the current data are more consistent with the MBDs having a role in the integration of CopiaMBDs, but other scenarios cannot be rejected. More data from additional arthropod lineages could perhaps offer supporting evidence for either hypothesis, as well as experimental approaches in model systems with comparable methylation patterns.
Regarding the limited distribution of CopiaMBDs across arthropods, we can rule out a recent horizontal transfer event between Stegodyphus and Strigamia. However, it is difficult to discriminate whether the presence of CopiaMBDs in only two distantly related species among the currently sequenced arthropod genomes is owing to vertical inheritance from the last common ancestor of arthropods or an ancient horizontal transfer event followed by rapid sequence divergence in the spider and the centipede lineages. Irrespective of the origins of CopiaMBDs, if the MBD influences integration, harboring an MBD might be evolutionary unstable for retrotransposons. Changes in the CpG density of transposable elements relative to gene bodies or a global reduction of methylation levels on transposable elements could explain why MBD-encoding transposons have been lost repeatedly or why they have not successfully colonized more genomes through horizontal transfer. For instance, most insect, crustacean, and spider genomes are very sparsely methylated (Feng et al. 2010; Zemach et al. 2010; Bewick et al. 2017; Gatzmann et al. 2018; Liu et al. 2019), and most methylation is restricted to transcribed gene bodies but absent from most transposable elements. In such a context, CopiaMBDs would preferentially insert in gene bodies. Consistently, we observed an accumulation of young CopiaMBD elements in introns of expressed genes in the spider Stegodyphus. This has the potential to be deleterious, as CopiaMBD could disrupt important genes upon insertion. In the spider genome, these detrimental effects could explain why CopiaMBDs are less abundant than other types of LTR-retrotransposons, including Copias lacking an MBD. However, given that Stegodyphus has a relatively very large genome (∼2.5 Gbs) with long introns, these insertions are less likely to be detrimental than in species with smaller genomes and compact introns, which accounts for most insect species sequenced to date. Therefore, despite encoding an MBD likely being beneficial for extant CopiaMBDs, this strategy is largely dependent on the host epigenomic environment.
Complementarily, the methylation landscape of the Strigamia genome is the first example of an arthropod genome with high methylation in both gene bodies and transposable elements. Given that the Strigamia genome shows many ancestral characteristics that contrast with insects and crustaceans (Chipman et al. 2014) and lacks whole-genome duplications such as those of chelicerates (Schwager et al. 2017), it is likely that the methylation landscape is more representative of the last arthropod common ancestor. Thus, the Strigamia methylome is an important resource toward understanding the gradual loss of DNA methylation in the arthropod phylum, especially in insects (Bewick et al. 2017; Provataris et al. 2018).
Retrotransposon-encoded chromodomains and PHD are widespread in eukaryotes, and thus, the evolutionary origin for the domain is very difficult to reconstruct. Instead, CopiaMBDs are restricted to arthropods, which strongly indicates that the MBD was co-opted from a host protein. Furthermore, CopiaMBD retrotransposons would be exceptional among retrotransposons if they use cytosine methylation to modify their integration site preference through an MBD. This also complements our recent report on how several retrotransposons have acquired cytosine methyltransferases from their hosts (de Mendoza et al. 2018), which is another clear example of how DNA methylation has been hijacked by transposons despite its widespread role in transposon silencing. Plenty of recent literature has focused on how transposable elements have been domesticated to fulfill advantageous functions for the host (Jangam et al. 2017; Bourque et al. 2018); however, this relationship is bidirectional, as cases such as CopiaMBDs show how transposons can also co-opt host proteins to their advantage. Despite it being well known that transposons can capture host genomic DNA (Cerbin and Jiang 2018), cases of incorporation of functional domains involved in epigenome regulation from the host are just starting to emerge (Iyer et al. 2014). Together, these cases reveal unexpected intricacies in the arms race between transposons and their hosts and how epigenome regulation is at the center of this battleground.
Methods
Sequence searches, alignment, and phylogenetic inference
The Pfam MBD hidden Markov model was scanned using HMMER3 (Eddy 2011) in a list of 58 proteomes spanning the whole diversity of animal phyla (Supplemental Table S1). This revealed how gene models from Strigamia and Stegodyphus were in fact unmasked retrotransposons. By taking the best hits for both species (choosing the longer ORFs encoding most retrotransposon-associated domains), subsequent CopiaMBD searches were performed with TBLASTN against nucleotide genome sequences, HMMER3 searches against AUGUSTUS (Stanke et al. 2008) intronless de novo protein annotations of ecdysozoan genomes, and phmmer against Reference proteomes. Domain architectures for all the hits were defined using Pfam database (Punta et al. 2012), NCBI Conserved Domains Database (Marchler-Bauer et al. 2015), and GyDB (Llorens et al. 2011).
To gather the sequences for the RT phylogeny (Supplemental Fig. S1), we used a combination of reference Copia RTs from Repbase (Bao et al. 2015) and the best hits encoding a RT (Pfam RVT_2) from CopiaMBD searches against metazoan genomes. RT sequences from Gypsy, Pao, DIRS, and Ngaro classes were included as outgroups. To avoid redundancy, CD-HIT (Fu et al. 2012) was used to cluster sequences with greater than 0.9 identity. To gather sequences for the MBD phylogeny (Fig. 1C), we used the Pfam MBD and scanned the proteomes for the 10 species listed in Figure 1A. CopiaMBDs were selected to represent the major clades obtained from the RT phylogeny (Supplemental Fig. S1).
Multisequence protein alignments were constructed using MAFFT (L-INS-I mode) (Katoh and Standley 2013), trimmed with trimAl (-gappyout mode) (Capella-Gutiérrez et al. 2009), and fed to IQ-TREE (default evolutionary model testing) (Nguyen et al. 2015) to obtain maximum likelihood phylogenies with 100 nonparametric bootstrap replicates to compute nodal supports. Alignments and phylogenies are available as Supplemental Tables S2 through S4.
Repeat annotation
RepeatModeler (Smit and Hubley 2008) was used to obtain a de novo transposable element annotation for Strigamia and Stegodyphus genomes. The resulting models did not capture the full diversity of CopiaMBDs for Strigamia, likely because of genome assembly problems such as gaps filled with Ns or collapsed copies into single sequences diminishing the repetitiveness of the sequences. Therefore, we used manually annotated full-length CopiaMBDs, showing ORFs encoding RT, MBD, and INT domain and conserved LTRs, to complement the RepeatModeler consensus FASTA file. These files were then used to annotate the genomes of both species using RepeatMasker (Smit et al. 2013–2015).
Complementarily, we used LTRharvest (Ellinghaus et al. 2008) to identify full-length LTR-retrotransposons, requiring at least 90% identity between both repeats and limiting the inter-LTR distance to 2000–9000 bp to avoid spurious hits. Those LTR-retrotransposons were classified according to their overlap with the RepeatMasker annotation, requiring an overlap with just one class of LTR-retrotransposon and encoding of ORFs with RT and INT domains. The presence of the MBD was also required to annotate CopiaMBDs. The ORFs within the LTRs were predicted using TransDecoder (Haas et al. 2013), and the domains were identified with HMMER3. The resulting genome coordinates and annotation are available in Supplemental Tables S5 through S7.
LTR-retrotransposon flanking regions were obtained using BEDTools (Quinlan and Hall 2010). To calculate CpG density of the LTR-retrotransposon flanking regions, we required at least 60% of the sequence (200 bp) not to be ambiguous nucleotides in the reference (Ns). The Ns were deducted from the total length of the region to obtain CpG densities.
Sequence conservation and divergence estimation
Branch length distances for MBDs were estimated using the “ape” package for R (R Core Team 2018; Paradis and Schliep 2019). The distance was calculated between the phylogeny tips in the case of the conserved gene families, whereas the distance between CopiaMBDs was calculated between the base of the monophyletic group of Stegodyphus sequences and the monophyletic group of Strigamia sequences. The inter-nodal distance would accurately reflect the distance before the family expanded in each lineage. Another maximum likelihood tree was computed, including the orthologs of the house spider Parasteatoda tepidariorum orthologs (Schwager et al. 2017), to rule out possible misannotations or alignment artifacts affecting branch length distance measurements and also to measure SETDB1/2 and MBD-Fbox distances between Strigamia and a spider species, as those families are absent in the Stegodyphus genome.
To analyze domain conservation between intra-species CopiaMBD subfamilies, codon alignments were obtained with PAL2NAL (Suyama et al. 2006), and Ka/Ks values were computed using the “seqinr” package for R (Charif and Lobry 2007). Sequences from Stegodyphus were not included given that their divergence time is too recent to calculate substitution parameters between copies.
To compare inter-species CopiaMBD subfamilies, we selected the longest ORFs encoding at least the MBD, INT, and RT domains for Stegodyphus and Strigamia and performed pairwise amino acid alignments using MAFFT between all copies (excluding intra-species comparisons) to maximize alignment length. Furthermore, the MBD, INT, and RT domains were extracted using HMMER3 for each copy, only selecting sequences that encoded at least 90% of the domain model length, and aligned separately. In parallel, we used OrthoFinder2 (Emms and Kelly 2018) to obtain orthologs between the seven species represented in Supplemental Figure S2B. We selected the BUSCO set of arthropod conserved genes (Simão et al. 2015) in Stegodyphus and used that subset to select one to one orthologs between Stegodyphus BUSCO genes and the rest of species. Pairwise one-to-one orthologs were aligned using MAFFT. All alignments were back-translated into nucleotides and trimmed using trimAl, and Ks values were obtained using “seqinr” in R.
Whole-genome bisulfite sequencing
WGBS by MethylC-seq was performed as described previously(Urich et al. 2015). Briefly, genomic DNA was extracted from frozen whole adult Strigamia centipedes. Three hundred nanograms of genomic DNA plus 0.1% (w/w) of unmethylated lambda genome DNA was then sheared to 200-bp fragments with a Covaris sonicator. The sheared DNA was purified and end-repaired, and methylated Illumina adaptors (BIOO Scientific) were ligated using the Lucigen AmpFREE low DNA library kit. The resulting library was bisulfite converted using EZ DNA Methylation-Gold kit (Zymo Research) and amplified with KAPA HiFi HotStart uracil + DNA polymerase (Kapa Biosystems). The library was sequenced with an Illumina HiSeq 1500. Bisulfite converted reads were trimmed using fastp (Chen et al. 2018) and then mapped using BS-Seeker2 (Guo et al. 2013) using Bowtie 2 (Langmead and Salzberg 2012) as the aligner (end-to-end), and PCR duplicates were removed with Sambamba (Tarasov et al. 2015). The bisulfite nonconversion rate was calculated using the total number of C calls divided by coverage on C positions on the lambda genome (0.32%). WGBS for S. dumicola was provided by the investigators of a recent publication (Liu et al. 2019) and can be found at NCBI Sequence Read Archive (SRA) accessions SRR8417342 and SRR8417350. The reference genomes for Strigamia and S. mimosarum were downloaded from Ensembl Metazoa, whereas S. dumicola is available from Liu et al. (2019).
DAP-seq and ampDAP-seq
Two micrograms of genomic DNA from the same extraction used for WGBS (adult Strigamia) was sonicated to 200 bp using a Covaris sonicator. The resulting fragments were purified, end-repaired, and ligated to Y-shaped adaptors as previously described (Bartlett et al. 2017). This unamplified library was used for the DAP-seq experiments. AmpDAP-seq libraries were generated using 15 ng of unamplified adaptor ligated DNA and amplified by PCR for 11 cycles.
Three MBDs from distinct clades of Strigamia CopiaMBDs were selected based on sequence conservation as well as spanning maximal diversity of sequences among CopiaMBDs. The MBDs plus 50 padding amino acids were cloned into pIX-HALO plasmids fused to an N-terminal HaloTag. The sequences of the inserts and the plasmids are available in Supplemental Table S8. Furthermore, we added a negative control using the empty plX-HALO plasmid only encoding the HaloTag. These plasmids were in vitro transcribed using the TNT SP6 coupled wheat germ extract system (Promega). The subsequent steps were performed following the standard DAP-seq protocol (Bartlett et al. 2017). For each pIX-HALO plasmid, 40 ng of Strigamia DAP-seq and ampDAP-seq libraries with unique Illumina multiplexing indexes was used in the affinity pull down. The pooled libraries were sequenced with an Illumina NextSeq instrument. The resulting reads were trimmed using fastp (Chen et al. 2018) and mapped to the Strigamia genome using Bowtie (Langmead et al. 2009) with “-m 1 -v 1 -q -S” parameters, and uniquely mapped reads were used to call peaks using MACS2 (Feng et al. 2012), requiring down-sampling and a q-value < 0.05. DAP-seq and ampDAP-seq libraries for the empty pIX-HALO plasmid were used as background for peak calling. Motif enrichment on motifs was obtained using HOMER (Heinz et al. 2010).
We used Bowtie 1 instead of Bowtie 2 for mapping DAP-seq and ampDAP-seq data because Bowtie 1 allows for a more stringent mapping. For genome assemblies such as that of Strigamia, the quality of the assembly or the heterozygosity cannot be taken for granted; thus, stringency is preferred. Furthermore, the protocol for DAP-seq recommends 2–4 million reads per sample for a genome the size of Arabidopsis thaliana (∼135 Mb) (Bartlett et al. 2017), and we sequenced an average of approximately 90 million paired-end reads per sample for a genome assembly of ∼173 Mb; thus, we estimated that we could afford to be stringent in the mapping step. However, to confirm the effects of this decision, we remapped the data using Bowtie 2 and found the same patterns as in Figure 2A.
Sequencing data were visualized using the IGV genome browser (Thorvaldsdóttir et al. 2013) and deepTools2 (Ramírez et al. 2014). Overlaps and coverage were obtained using BEDTools (Quinlan and Hall 2010). Statistical tests were computed using base R (R Core Team 2018).
RNA-seq processing
Strigamia RNA-seq was downloaded from NCBI SRA (https://www.ncbi.nlm.nih.gov/sra) from the accession numbers SRR1267274, SRR1267275, and SRR1267276. This RNA-seq samples corresponded to embryonic and adult Strigamia stages, as described previously (Chipman et al. 2014). S. dumicola RNA-seq corresponds to SRR8416255–SRR8416299, also described in a prior publication (Liu et al. 2019). The RNA-seq was mapped to the reference genome using HISAT2 (Kim et al. 2015), allowing one mismatch in the seed (-N 1) and a maximum intron length of 40 kb (--max-intronlen 40,000), and for computing gene expression, the reads with secondary alignments were excluded filtering the “ZS:” flag in the resulting BAM file. The expression levels were calculated using StringTie (Pertea et al. 2015) and the reference annotation from Ensembl Metazoa. To obtain an RNA-seq–based annotation for Strigamia and S. dumicola, we merged the RNA-seq alignments from HISAT2 (BAM files obtained using the “--dta” option) and computed a reference annotation using StringTie default parameters. The resulting transcript models were translated and annotated using TransDecoder, filtering out models that did not encode peptides longer than 50 amino acids and those that encoded transposon domains.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE118012.
Acknowledgments
We thank Michael Akam and Ken Siggens for providing Strigamia genomic DNA, Jon Cahn for assistance with the DAP-seq protocol, Ozren Bogdanović for critical reading of the manuscript, and Sam Buckberry for input on the bioinformatic analysis and comments on the manuscript. We also thank Jesper Smærup Bechsgaard and Trine Bilde for sharing S. dumicola data. This work was supported by the Australian Research Council (ARC) Centre of Excellence program in Plant Energy Biology (CE140100008). R.L. was supported by a Sylvia and Charles Viertel Senior Medical Research Fellowship, ARC Future Fellowship (FT120100862), and Howard Hughes Medical Institute International Research Scholarship. A.d.M. was funded by an EMBO long-term fellowship (ALTF 144-2014).
Author contributions: A.d.M. and R.L. designed the study. A.d.M. performed the experiments and did the analysis. J.P. performed the sequencing. R.L. supervised the study. A.d.M. and R.L. wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.243774.118.
- Received September 5, 2018.
- Accepted June 20, 2019.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








