
PepQuery enables fast, accurate, and convenient proteomic validation of novel genomic alterations
- Bo Wen1,2,
- Xiaojing Wang1,2 and
- Bing Zhang1,2
Abstract
Massively parallel or second-generation sequencing-based genomic studies continuously identify new genomic alterations that may lead to novel protein sequences, which are attractive candidates for disease biomarkers and therapeutic targets after proteomic validation. Integrative proteogenomic methods have been developed to use mass spectrometry (MS)-based proteomics data for such validation. These methods replace the reference sequence database in proteomic database searching with a customized protein database that incorporates sample- or disease-specific sequences derived from DNA or RNA sequencing, thus enabling the identification of novel protein sequences. Although useful, this spectrum-centric approach requires a full evaluation of all possible spectrum-peptide pairs, which is time-consuming, error-prone, and difficult to apply. Here, we present PepQuery, a peptide-centric approach that focuses on only novel DNA or protein sequences of interest. PepQuery allows quick and easy proteomic validation of genomic alterations without customized database construction. We demonstrated the sensitivity and specificity of the approach in validating completely novel proteins, novel splice junctions, and single amino acid variants using simulations and experimental data. Notably, enabling unrestricted modification searching in PepQuery reduced false positives by up to 95%. We implemented PepQuery as both web-based and stand-alone applications. The web version provides direct access to more than half a billion MS/MS spectra from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) and other cancer proteomic studies. The stand-alone version supports batch analysis and user-provided MS/MS data. PepQuery will increase the usage of proteogenomics beyond the proteomics community and will broaden the application of proteogenomics in personalized medicine.
Massively parallel or second-generation sequencing-based genomic studies, especially cancer genomic studies, continuously identify new genomic abnormalities such as single nucleotide variants (SNVs), insertions and deletions (INDELs), RNA edits, novel junctions, fusions, and novel transcription regions. Some abnormal DNA and RNA sequences encode novel, disease-relevant proteins, which are promising candidates of disease biomarkers, drug targets, and neoantigens. The first step in translating these genomic discoveries into clinical practice is to validate their expression at the protein level. The tandem mass spectrometry (MS/MS)-based shotgun proteomics provides an excellent opportunity for such validation.
Traditional shotgun proteomics data analysis relies on searching all MS/MS spectra against a reference protein sequence database (Nesvizhskii 2006), such as RefSeq, UniProt, or Ensembl, and thus is unable to identify any novel, disease-specific sequences (Fig. 1A). An emerging proteogenomic approach derives customized, sample-specific protein databases from DNA and RNA sequencing data (Li et al. 2011; Wang et al. 2012), making it possible to identify novel peptides that are present in a specific sample but not included in the reference databases (Fig. 1B).
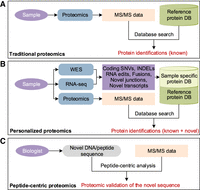
Proteomics data analysis strategies. (A) Traditional proteomics data analysis using a common reference protein database. (B) Proteogenomics approach using customized, sample-specific protein sequence databases. (C) Peptide-centric proteomics focuses on individual novel peptides of interest instead of interpreting all MS/MS spectra.
The customized database approach has been demonstrated in many proteogenomic studies (Zhang et al. 2014, 2016; Mertins et al. 2016), and there is an increasing need from the genomics community to reutilize published proteomic data sets to search for proteomic evidence of putative novel coding sequences predicted from genomics data. The putative novel coding sequences may come as a batch, but many times, the investigators are simply interested in an SNV, an INDEL event, a possible stop codon read-through, an intron retention, a novel junction, an upstream open reading frame, or a circular RNA, etc. An intuitive web-based query system providing direct access to the MS/MS data would be the most effective to serve this purpose.
Both traditional and customized database searching approaches are MS/MS spectrum-centric (Ting et al. 2015), and the common goal is to comprehensively interpret all MS/MS spectra through database searching (Fig. 1A,B). Therefore, validating even a single coding SNV requires customized database construction, a full evaluation of all peptide-spectrum pairs, and then extracting the peptide-spectrum matches (PSMs) involving the specific coding SNV. This process is time-consuming, with most of the time spent on database preparation and evaluating peptide-spectrum pairs irrelevant to the novel peptide sequences of interest. Therefore, it is not feasible for real-time web applications. A peptide-centric analysis, as suggested in a few previously published perspective articles (Noble 2015; Ting et al. 2015), could provide a novel solution to this problem.
Here, we report a peptide-centric method named PepQuery for validating putative novel protein coding sequences (Fig. 1C). PepQuery is conceptually similar to BLAST (Altschul et al. 1990). Whereas BLAST allows users to query a sequence database with a sequence of interest to look for sequence similarity, PepQuery allows users to query an MS/MS spectra database with a novel peptide or DNA sequence of interest to look for PSMs. Unlike the spectrum-centric analysis, PepQuery analyzes only the peptide-spectrum pairs involving the novel sequence, making it possible to support real-time, web-based analysis. Statistical evaluation of the PSM scores in peptide-centric analysis represents a new challenge, which we addressed with a new strategy. A spectrum matched to a novel peptide may correspond better to reference peptides with modifications, such as post-translational modifications (PTMs), chemical modifications, and artifacts of sample handling. Due to computational complexity, it is still difficult to consider all modifications in database searching, and this may result in false positive identifications in proteogenomic studies. The peptide-centric analysis in PepQuery provides an additional advantage of reducing false positives by comprehensive consideration of sequence modifications. We demonstrated the sensitivity and specificity of the method in identifying novel proteins, novel splice junctions, and single amino acid variants using simulations and experimental data. PepQuery is available as both web-based and stand-alone applications (http://www.pepquery.org).
Results
PepQuery workflow
PepQuery takes as input a novel peptide, protein, or DNA sequence, or novel genomic features in the VCF, BED, or GTF file format, and the workflow includes five major steps: (1) target peptide sequence preparation and initial filtering; (2) candidate spectra retrieval and PSM scoring; (3) competitive filtering based on reference sequences; (4) statistical evaluation; and (5) competitive filtering based on unrestricted modification searching (Fig. 2; Methods). For PSM scoring, we implemented the Hyperscore used in X!Tandem (Craig and Beavis 2004) and the multivariate hypergeometric distribution (MVH) score used in MyriMatch (Tabb et al. 2007). For statistical evaluation of the PSM scores, permutation P-values are calculated based on randomly shuffled peptide sequences (Methods). The spectra with statistically significant matches to the query peptide that cannot be better explained by unmodified or modified reference sequences are reported. Annotated PSMs can be visualized in the tool for manual evaluation.
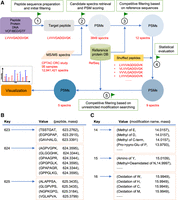
PepQuery workflow. (A) The PepQuery workflow involves five major steps: (1) target peptide sequence preparation and initial filtering; (2) candidate spectra retrieval and PSM scoring; (3) competitive filtering based on reference sequences; (4) statistical evaluation; and (5) competitive filtering based on unrestricted post-translational modification searching. The red text illustrates a real example in which a variant peptide LVVVGADGVGK is used to query the CPTAC colorectal cancer (CRC) data set with 95 samples and 12,941,421 spectra, using RefSeq as the reference protein database. Whereas existing methods require pairwise analysis between all 12,941,421 spectra and all RefSeq-derived peptide sequences plus the variant peptide sequence, PepQuery focuses only on the spectra that are relevant to the novel peptide, which reduces computational time and also allows more comprehensive analysis of these spectra. Illustration of peptide (B) and modification (C) indexing methods.
With the existing customized database approach, identifying a novel peptide sequence as shown in Figure 2 in the Clinical Proteomic Tumor Analysis Consortium (CPTAC) colorectal cancer data set (Zhang et al. 2014) will require constructing a customized database that includes both reference protein sequences (e.g., from RefSeq) and the novel sequence, and then evaluating all possible spectrum-peptide pairs between over 12 million MS/MS spectra and over one million unique peptide sequences. In contrast, PepQuery starts with the novel peptide sequence of interest, evaluates only peptide-spectrum pairs involving the novel peptide, and thus reduces the computational time by several orders of magnitude (Fig. 2). Moreover, the peptide-centric approach quickly narrows down to a small number of candidate spectra, which makes it possible to perform unrestricted modification searching in the last step to identify alternative interpretations of the candidate spectra in real-time applications.
Validating completely novel protein sequences
To evaluate the performance of PepQuery for validating completely novel protein sequences, we used a spiked-in MS/MS proteomic data set from the Proteome Informatics Research Group (iPRG) 2015 study (Choi et al. 2017). This data set was generated from yeast cells with six spiked-in nonyeast proteins. Here, we used the yeast proteome as the background, and the peptides derived from the six spiked-in nonyeast proteins were treated as the ground truth of “novel peptides.” The goal was to investigate whether PepQuery could accurately identify the “novel peptides” using individual “novel peptides,” the MS/MS data, and the yeast reference proteome as input. Because some of the peptides derived from the spike-in proteins may not be detectable in the MS/MS experiment, we first performed a standard database search using IPeak (Wen et al. 2015) to determine which peptides from the spiked-in proteins were detectable in the MS/MS experiment with 1% false discovery rate (FDR) at both PSM and protein levels. IPeak integrates search results from three search engines, MS-GF+, MyriMatch, and X!Tandem. Although this does not necessarily produce the exact set of detectable peptides from the spike-in proteins, using multiple search engines has been shown to achieve higher sensitivity and specificity compared to individual search engines (Wen et al. 2015). The IPeak analysis identified a total of 93 peptides from the six spiked-in proteins (Supplemental Table S1). The 93 peptides were used as gold-standard positives for novel protein sequences (Supplemental Fig. S1). Meanwhile, we randomly selected 10,000 tryptic peptides from Escherichia coli as gold-standard negatives. Searching the gold-standard peptide sequences against the iPRG data set using PepQuery, we obtained true positive rates (TPRs, a.k.a., sensitivity) of 94.62% and 97.85% (Fig. 3A; Supplemental Tables S2, S3) and false positive rates (FPRs, a.k.a., 1-specificity) of 0.05% and 0.05% (Fig. 3B; Supplemental Tables S4, S5) based on the Hyperscore and MVH score, respectively. The precisions of using Hyperscore and MVH after unrestricted filtering were 94.62% and 94.79%, respectively. These results demonstrate high sensitivity and specificity of PepQuery.
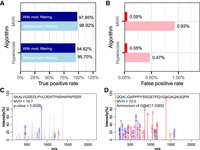
Evaluation of the performance of PepQuery for validating completely novel protein sequences. (A) Sensitivity and (B) specificity evaluation for novel peptide sequence validation. Mod. filtering means unrestricted modification searching-based filtering. (C) A spectrum matched to a gold-standard negative peptide without unrestricted modification searching-based filtering. Blue and red colors indicate matched peaks, whereas gray indicates unmatched peaks. (D) The same spectrum in C can be matched to a reference peptide with an ammonium salt modification on aspartic acid with very few unmatched peaks and a high score.
Notably, filtering based on unrestricted modification searching reduced the false positives by 89% (from 47 peptides to five peptides) and 95% (from 93 peptides to five peptides) for the two scoring algorithms, respectively (Fig. 3B), whereas the true positives were only reduced by 1% (Fig. 3A). To illustrate the merit of this approach in reducing false positives, a spectrum incorrectly matched to an E. coli peptide (false positive) without unrestricted modification searching-based filtering (MVH score = 19.7, P = 0.0028) (Fig. 3C) was matched by PepQuery to a yeast peptide with an ammonium salt modification on aspartic acid (D) (MVH score = 70.0) (Fig. 3D). When including ammonium salt modification on aspartic acid as a variable modification in a standard database searching of the iPRG 2015 data set using MyriMatch, we found that 306 (1.33%) out of the 22,678 identified peptides had the same ammonium salt modification, suggesting its origin from artifacts of sample handling. All spectra incorrectly matched to an E. coli peptide without unrestricted modification searching-based filtering and the alterative matches to a modified yeast peptide were annotated and presented in Supplemental Figure S2. These data suggest that competitive filtering with unrestricted modification searching provides a powerful means to reduce false positives in proteogenomic studies.
Validating novel splice junctions
To evaluate the ability of PepQuery to validate novel splice junctions, we applied PepQuery to a data set from a published study on novel splice-junction peptide identification (Sheynkman et al. 2013). The MS/MS data were generated from the Jurkat cells, and the study reported a total of 55 unique splice-junction peptides (Supplemental Table S6). The 55 peptides were searched against the MS/MS data using PepQuery and 36 (65%) and 39 (71%) were confirmed based on the Hyperscore and MVH score, respectively (Fig. 4A; Supplemental Tables S7, S8). Among the 16 peptides not confirmed by MVH (i.e., false negatives), nine were removed by competitive filtering based on reference sequences and five were removed by competitive filtering based on unrestricted modification searching. Thus, 87% of the MVH-based “false negatives” were likely to be false positive discoveries in the original study (Fig. 4C) and so were 84% of the Hyperscore-based “false negatives” (Fig. 4B). These data demonstrate that PepQuery effectively removes false identifications reported in published proteogenomic studies.
Evaluation of the ability of PepQuery to validate novel splice junctions. (A) Novel splice-junction peptide identification result. (B,C) Classification of the novel junction peptides reported by the original study but not by PepQuery (orange sections in the large circles). I: Peptides removed by competitive filtering based on reference sequences. II: Peptides removed by competitive filtering based on unrestricted modification searching. III: Remaining false negatives.
Validating single amino acid variants
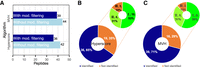
To evaluate the ability of PepQuery to validate single amino acid variants (SAAVs), we used simulation data derived from the iPRG 2015 study (Methods). For each of the 93 peptides from the six spiked-in nonyeast proteins, we generated two versions of variant peptides, each with a randomly introduced SAAV. One version served as a gold-standard negative. The other version was added to the reference database to replace the original reference peptide sequence. With regard to this new reference sequence, the original peptide became an SAAV peptide. Because the original peptide had matched spectra in the MS/MS data, it was used as a gold-standard positive sequence in our evaluation (Supplemental Fig. S3). On the basis of 100 sets of gold-standard positives, gold-standard negatives, and expanded reference databases, PepQuery achieved average TPRs of 93.53% and 96.27% (Fig. 5A) and average FPRs of 0.41% and 0.26% (Fig. 5B) using the Hyperscore and MVH score, respectively. Filtering based on unrestricted modification searching reduced the false positives by 75% and 86% for the two scoring algorithms, respectively (Fig. 5B), with only 1.56% and 1.96% reduction on the true positives, respectively (Fig. 5A). These results are comparable to those from the novel protein sequence study (Fig. 3A,B) and further highlight the sensitivity and specificity of PepQuery in SAAV validation and the benefits of competitive filtering with unrestricted modification searching.
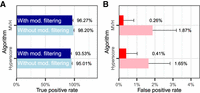
Evaluation of the ability of PepQuery to validate single amino acid variants (SAAVs). (A) Sensitivity and (B) specificity evaluation for validating single amino acid variants based on 100 simulation studies.
Applying PepQuery to large proteomic data sets
An important emerging application of PepQuery is to identify proteomic evidence for genomic findings using large, publicly available proteomic data sets. To demonstrate the utility of PepQuery in this scenario, we used the colorectal cancer data set produced by CPTAC (Zhang et al. 2014). The 4084 SAAV peptide-sample pairs reported by the original paper, which involves 799 fully tryptic SAAV peptides and 79 samples, were used as gold-standard positives for this study, whereas all other 59,037 pairs between the 799 SAAV peptides and the 79 samples were used as gold-standard negatives (Methods; Supplemental Table S9). Using PepQuery, each of the 799 SAAV peptides was searched against all 10,753,601 MS/MS spectra from the 79 samples. Annotated spectra for PSMs identified based on Hyperscore and MVH score can be found in Supplemental Figures S4 and S5, respectively. The Hyperscore and MVH score-based analyses showed TPRs of 78.04% and 90.96% (Fig. 6A) and FDRs of 2.01% and 2.40% (Fig. 6B), respectively (Supplemental Tables S10, S11). The TPRs are lower than those observed in the simulation study, whereas the FPRs are higher.
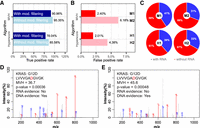
Evaluation of the performance of PepQuery for validating SAAVs in large data sets. (A) Sensitivity and (B) specificity evaluation for validating SAAVs in a large cancer data set. (C) A large proportion of the false positives in B are supported by RNA-seq evidence. (D,E) Two spectra supporting the KRAS G12D mutation in a sample, in which the mutation was supported by DNA but not RNA evidence.
We note that, unlike the simulation study, the “gold-standards” used in this analysis were derived from the original customized database searching results and are not perfect ground truth. In the original study, database searching used MyriMatch (MVH) but not Hyperscore, which explains the much lower TPR observed for the Hyperscore-based analysis. Furthermore, competitive filtering based on unrestricted modification searching reduced the true positives by 7.54% and 4.39% for the two algorithms, respectively (Fig. 6A). The original study considered only a few PTMs. Therefore, some of the gold-standard positives may be false because the spectra supporting the SAAV peptides corresponded better to modified reference sequences.
On the other hand, some of the false positives may be true. Among the SAAV peptide-sample pairs identified by PepQuery but not the original study (Fig. 6B), we were able to find RNA-seq evidence for 39% of the Hyperscore-based “false positives” (H1, 457 SAAV peptide-sample pairs) and 41% of the MVH score-based “false positives” (M1, 576 SAAV peptide-sample pairs) (Fig. 6C; Supplemental Tables S10, S11). These SAAV peptide-sample pairs were missed in the original report because RNA-seq data from these samples only had one or two reads covering corresponding SNVs, and a minimum of three-read depth was required for including an SNV in a sample-specific customized database (Zhang et al. 2014). We also found that unrestricted modification searching-based filtering increased the proportion of “false positives” that could be supported by RNA-seq evidence (Fig. 6C, H1 vs. H2 and M1 vs. M2).
RNA-seq also may fail to detect some SAAVs. For example, despite a lack of RNA-seq evidence, PepQuery identified two spectra supporting the KRAS G12D mutation in the sample TCGA-AA-A02O-01A (Fig. 6D,E). We manually checked the cBioPortal (Cerami et al. 2012), and the KRAS G12D mutation was indeed reported for this sample. Taken together, PepQuery identified hundreds of SAAV events that are supported by genomic data but are missing in the original customized database searching results, and unrestricted modification searching-based filtering helped remove false positives.
FDR estimation
To estimate FDR corresponding to the PepQuery P-value cutoff of 0.01 used in this study, we performed two additional analyses. The first analysis used a human-E. coli spike-in data set (317,502 MS/MS spectra) from a published study (Shen et al. 2017), in which E. coli protein lysate was spiked into a human cell digest. We searched the MS/MS data against the combined human-E. coli SWISS-PROT database using X!Tandem, MyriMatch, and MS-GF+ and identified 4540 unique E. coli peptides and 30,099 unique human peptides. The 4540 E. coli peptides were used as the target peptides. We further generated 4540 reverse peptides based on the target peptides. After removing reverse peptides that could be matched exactly to a human or E. coli protein sequence, we got 4512 decoy peptides. We searched the 9052 target and decoy peptide sequences against the human-E. coli spike-in data set using PepQuery and a P-value cutoff of 0.01. As shown in Table 1, the FDRs were 0.84% and 0.37% in the Hyperscore- and MVH score-based analyses, respectively.
FDR evaluation based on a human-E. coli spike-in data set
In the second analysis, we further evaluated the impact of sample size on PepQuery FDR using the CPTAC colon cancer data set (Zhang et al. 2014) with a total of 79 samples. We searched the MS/MS data against the RefSeq human protein sequences database using X!Tandem, MyriMatch, and MS-GF+ and identified 77,069 unique human peptides. We performed FDR estimation for randomly selected sets of one sample, 19 samples, 39 samples, 59 samples, and 79 samples. For each sample set, we randomly selected 2000 target peptides and generated 2000 decoy peptides. We searched all 4000 peptides against the MS/MS data from the same sample set using PepQuery and a P-value cutoff of 0.01. The analysis was repeated five times for each sample set. We observed increased FDRs with increasing sample size number (Table 2). Nevertheless, the FDRs were still <3% when all 79 samples were analyzed. These results suggest that a 1% PepQuery P-value cutoff coupled with unrestricted modification searching-based filtering provides well-controlled FDR for small to relatively large sample sizes.
Impact of sample size on FDR
Discussion
PepQuery is specifically designed for proteomic validation of novel genomic alterations. It is peptide-centric and is conceptually different from existing spectrum-centric approaches. The peptide-centric approach greatly reduces computational time and enables analysis of more than 1000 modifications in real-time applications. Indeed, searching one peptide sequence against the whole CPTAC colorectal cancer data set required <30 sec on a computer with one physical CPU and 16 cores and 64 GB of memory. To allow easy access to MS/MS data for proteogenomic analysis, we implemented PepQuery both as a standalone application and as a web application (http://www.pepquery.org) (Supplemental Fig. S6). The web application makes more than half a billion MS/MS spectra from more than 40 proteomic data sets available online for proteogenomic studies. New MS/MS data sets can be easily added to the web application for public access. The stand-alone application allows users to query their own proteomic data sets, and the analysis can be performed in the batch. For the first time, PepQuery makes MS/MS data directly available and useful to scientists outside the proteomics community.
The current version of PepQuery only supports searching one data set at a time. One obvious future improvement is to allow users to search multiple data sets or even all data sets at the same time. This will require overcoming several challenges. First, different data sets require different search parameters. Second, increased sample size would lead to a higher FDR, which may require new methods for FDR control. Third, the time required for searching all data sets may be too long for the web application, and a more efficient searching algorithm will be needed.
We demonstrated the sensitivity and specificity of the method in validating completely novel proteins, novel splice junctions, and SAAVs using simulations and both spiked-in and complex tumor data sets. The two scoring algorithms showed comparable results in all studies (Supplemental Fig. S7), highlighting the robustness of PepQuery analyses. Unlike the target-decoy approach used in spectrum-centric analysis that generates FDR estimation for a set of PSMs, PepQuery provides a direct statistical measurement for each PSM, and a 1% PepQuery P-value cutoff produced well-controlled FDRs in our FDR estimation study.
Competitive filtering based on unrestricted modification searching effectively reduces false positives, which is essential in proteogenomics. The current version of PepQuery considers one modification at a time in unrestricted modification searching. This may miss true matches with more than two modifications on the same peptide. We will include modification combinations in future development.
Both the stand-alone and web applications allow easy visualization of the spectra supporting variant peptides for manual assessment (Fig. 6D,E). For each PSM that passed the statistical evaluation, the PepQuery web application provides three annotated spectra in the report, including the novel peptide PSM, the best PSM from the reference database searching, and the best PSM from the unrestricted modification-based searching. This allows users to manually investigate the reliability of the novel peptide PSM identification and to manually compare the novel peptide match and modification peptide match (Supplemental Fig. S6). We anticipate that PepQuery will increase the usage of proteogenomics beyond the proteomics community and will broaden the application of proteogenomics in personalized medicine.
Methods
Overview of the PepQuery Workflow
PepQuery takes as input a peptide, protein, or DNA sequence, or a novel genomic feature in the VCF, BED, or GTF file format. In addition, a reference protein database and an MS/MS data set need to be specified. The PepQuery workflow includes five major steps: target peptide sequence preparation and initial filtering; candidate spectra retrieval and PSM scoring; competitive filtering based on reference sequences; statistical evaluation; and competitive filtering based on unrestricted modification searching (Fig. 2). Detailed information on each of these steps is provided below.
Target peptide sequence preparation and initial filtering
Peptide sequence input is used directly. Protein sequence input is digested in silico into peptides with the same enzyme used to generate the selected MS/MS data set. DNA sequence input is translated into protein sequences using a frame specified by the user and then digested. For VCF, BED, or GTF files, the software PGA (Wen et al. 2016) is used to translate the events in the file to protein sequences before digestion. In all cases, only peptides without exact match in the selected reference protein database are retained for further analysis.
Candidate spectra retrieval and PSM scoring
Each query peptide sequence is searched against the MS/MS data set specified, and candidate spectra are identified based on
the mass difference between a spectrum and the query peptide and a prespecified precursor mass tolerance. For the web application,
the precursor mass tolerance for each data set is determined based on the instrument setting for generating that data set.
For the stand-alone version, the precursor mass tolerance can be set by users. For each candidate spectra, two scoring algorithms
are used to calculate PSM scores. The Hyperscore calculation is similar to X!Tandem (Craig and Beavis 2004):
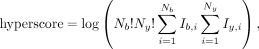 where Nb is the number of matched b-ions, Ny is the number of matched y-ions, Ib,i are the intensities of matched b-ions, and Iy,i are the intensities of matched y-ions. The multivariate hypergeometric distribution score calculation is similar to MyriMatch (Tabb et al. 2007):
where Nb is the number of matched b-ions, Ny is the number of matched y-ions, Ib,i are the intensities of matched b-ions, and Iy,i are the intensities of matched y-ions. The multivariate hypergeometric distribution score calculation is similar to MyriMatch (Tabb et al. 2007):
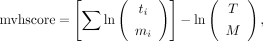 where ti is the number of peaks from a particular intensity class in the spectrum, mi is the number of peaks from a particular intensity class matched to the peptide derived peak list, T is the total number of locations in the spectrum, and M is the total number of peaks predicted from the peptide sequence.
where ti is the number of peaks from a particular intensity class in the spectrum, mi is the number of peaks from a particular intensity class matched to the peptide derived peak list, T is the total number of locations in the spectrum, and M is the total number of peaks predicted from the peptide sequence.
Competitive filtering based on reference sequences
Candidate spectra are searched against the specified reference protein database, and those with a better match to sequences in the reference database than to the target sequence are removed.
Statistical evaluation
For each remaining PSM, randomly shuffled sequences derived from the peptide in the PSM are used to evaluate the statistical
significance of the match. Specifically, 10,000 unique random peptides are generated by randomly shuffling of the original
peptide sequence. The resulted random peptide sequences have the same amino acid composition as the original sequence. When
the length of the original peptide sequence is too short to generate 10,000 unique random peptides, all possible random peptides
are generated. For each random peptide, the Hyperscore and the MVH score are calculated to quantify the match between the
random peptide and the spectrum in the PSM. Based on each of the scoring algorithms, a P-value is then calculated for the PSM:
 where Ns is the number of random peptides with a higher score than the original PSM scores, and N is the total number of random peptides generated. Only PSMs with a P-value ≤ 0.01 are retained for the unrestricted modification searching-based filtering.
where Ns is the number of random peptides with a higher score than the original PSM scores, and N is the total number of random peptides generated. Only PSMs with a P-value ≤ 0.01 are retained for the unrestricted modification searching-based filtering.
Competitive filtering based on unrestricted modification searching
All spectra involved in the remaining PSMs are searched against the selected protein reference database while considering all modifications from the Unimod database (http://www.unimod.org/) except for amino acid substitutions. Using the same scoring algorithm, if a spectrum has a better match to a modified peptide from the reference protein database than to the target peptide, the original identification is rejected. To speed up the searching, a peptide index and a modification index are generated. For a given protein reference database and user-specified fixed modifications and digestion parameters, a peptide index is generated for nonredundant peptides as shown in Figure 2B. This index is a hash map in which the integer values of the peptide masses are the key and the corresponding peptide sequences and masses are the values. The peptide indexing takes just a few seconds on a typical computer. The modification index is a hash map in which integer values of the modification masses are the key and the corresponding modification objects are the values as shown in Figure 2C.
Software implementation and support
PepQuery is available as a stand-alone application as well as a web application. The stand-alone version is written in Java and is platform-independent. The web version is developed using R Shiny (https://cran.r-project.org/web/packages/shiny/index.html). The multithreading technology is fully utilized in PepQuery to speed up the peptide identification. Both versions can be accessed through the PepQuery website (http://www.pepquery.org). Currently, using VCF, BED, and GTF files as input is only supported in the stand-alone version. For the stand-alone version, PSMs can be visualized using PDV (Li et al. 2018) at http://pdv.zhang-lab.org. Support is available via a Google users group (https://groups.google.com/forum/#!forum/pepquery) and a gitter channel (https://gitter.im/PepQuery/). This provides multiple venues for users to have their questions answered quickly and efficiently.
Mass spectrometry data sets and analysis
The spiked-in data set from the iPRG 2015 study (Choi et al. 2017) was generated from yeast cells with six spiked-in nonyeast proteins. The MS/MS data (JD_06232014_sample1-A.mgf) and the reference protein database were downloaded from ftp://iprg_study@ftp.peptideatlas.org/ (password ABRF329). The MS/MS data were searched using three search engines (MyriMatch v2.2.10165, X!Tandem v2017.2.1.2, and MS-GF+ v2017.01.13) against the mixed protein database (6622 yeast protein sequences + 6 spiked-in protein sequences) with decoy sequences through IPeak (Wen et al. 2015). Parameters for database searching were set as follows: Fixed modifications, Carbamidomethyl (C); variable modifications, Oxidation (M), acetylation on protein N-terminus and Deamidated (NQ); Precursor ion mass tolerance, 10 ppm; MS/MS mass tolerance, 0.02 Da; Enzyme specificity, trypsin; maximum missed cleavages, 2. The search results were integrated by IPeak with 1% PSM-level FDR. The same searching parameters were used for PepQuery. For the Jurkat proteome study, the MS/MS data and the reference database (Sheynkman et al. 2013) were downloaded from http://www.peptideatlas.org/PASS/PASS00215. The raw data were converted into MGF files using MSconvert (ProteoWizard, version 3.0.10462) (Chambers et al. 2012). The 55 junction peptides were retrieved from the supplemental file of the original paper. The parameters of PepQuery were set as follows: Fixed modifications, Carbamidomethyl (C); variable modifications, Oxidation (M); Precursor ion mass tolerance, 10 ppm; MS/MS mass tolerance, 0.05 Da; Enzyme specificity, trypsin; maximum missed cleavages, 2. The human-E. coli spike-in data set from a previous study (Shen et al. 2017) was downloaded from PRIDE (Jones et al. 2006) through the accession number PXD005590. The raw data were converted into MGF files using MSconvert (ProteoWizard, version 3.0.10462). The MS/MS data were searched using three search engines (MyriMatch v2.2.10165, X!Tandem v2017.2.1.2, and MS-GF+ v2017.01.13) against the mixed protein database (04/20/2018, 4443 E. coli protein sequences + 20,317 human protein sequences) with decoy sequences through IPeak. Parameters for database searching were set as follows: Fixed modifications, Carbamidomethyl (C); variable modifications, Oxidation (M); Precursor ion mass tolerance, 10 ppm; MS/MS mass tolerance, 0.05 Da; Enzyme specificity, trypsin; maximum missed cleavages, 2. The search results from each search engine were processed by Percolator (Kall et al. 2007) and then filtered with q value ≤ 0.001. The peptides identified by all the three search engines were retained for downstream analysis. The same searching parameters were used for PepQuery. The colorectal cancer data set was downloaded from the CPTAC data portal (https://cptac-data-portal.georgetown.edu/cptac/s/S022) (Zhang et al. 2014). Raw data were converted into MGF files using MSconvert (ProteoWizard, version 3.0.10462) (Chambers et al. 2012). Samples with replicates in the MS/MS or RNA-seq analysis were removed, and 799 fully tryptic SAAV peptides from the remaining 79 samples were retrieved from the supplemental file of the original paper. To be consistent with the original study, the parameters of PepQuery were set as follows: Fixed modifications, Carbamidomethyl (C); variable modifications, Oxidation (M); Precursor ion mass tolerance, 20 ppm; MS/MS mass tolerance, 0.5 Da; Enzyme specificity, trypsin; maximum missed cleavages, 2. The reference protein database was the same as the original study. All identification results of PepQuery were filtered by P-value ≤ 0.01. The MS/MS data were also searched using three search engines (MyriMatch v2.2.10165, X!Tandem v2017.2.1.2, and MS-GF+ v2017.01.13) against the RefSeq human database (33,820 human protein sequences) with decoy sequences through IPeak. Parameters for database searching were set as follows: Fixed modifications, Carbamidomethyl (C); variable modifications, Oxidation (M); Precursor ion mass tolerance, 20 ppm; MS/MS mass tolerance, 0.5 Da; Enzyme specificity, trypsin; maximum missed cleavages, 2. The search results from each search engine were processed by Percolator and then filtered with q value ≤ 0.001. The peptides identified by all the three search engines were retained for downstream analysis.
Performance evaluation for validating SAAVs
In order to evaluate the true positive rate and false positive rate of PepQuery for validating SAAVs, we used a simulation
data set and a real complex tumor data set. The simulation study was based on the spiked-in data set from the iPRG 2015 study
(Choi et al. 2017). This data set was generated from yeast cells with six spiked-in proteins. We analyzed the MS/MS data by IPeak (Wen et al. 2015) and identified a total of 93 peptides from the six spiked-in proteins with 1% PSM FDR. For each of the 93 peptides, we generated
two versions of variant peptides, each with a randomly introduced SAAV. One version served as a gold-standard negative. The
other version was added to the reference database to replace the original reference peptide sequence. With regard to this
new reference sequence, the original sequence became a gold-standard positive SAAV sequence. The gold-standard peptides were
taken as input to PepQuery and searched against the MS/MS data, using the new reference protein database. The FPR and TPR
were calculated as

 where T is the number of identified gold-standard positives, F is the number of identified gold-standard negatives, TP and FP are the total numbers of the gold-standard positives and gold-standard
negatives (i.e., 93 in this case). This simulation was repeated 100 times.
where T is the number of identified gold-standard positives, F is the number of identified gold-standard negatives, TP and FP are the total numbers of the gold-standard positives and gold-standard
negatives (i.e., 93 in this case). This simulation was repeated 100 times.
The second method is based on the CPTAC colon data set (Zhang et al. 2014). The variant peptides reported in the original study were taken as input to PepQuery. The variant peptide-sample pairs reported in the original study were used as gold-standard positives, whereas all other variant peptide-sample pairs were used as gold-standard negatives. TPR and FPR were calculated similar to the above description.
Software availability
The PepQuery web application can be accessed at http://www.pepquery.org. The stand-alone application and scripts for reproducing the work can be downloaded at the same website. The PepQuery source code and scripts for reproducing the work are also available in the Supplemental Material as Supplemental Code and Supplemental Scripts, respectively.
Acknowledgments
This study was supported by the National Cancer Institute (NCI) CPTAC award U24 CA210954, the Cancer Prevention and Research Institutes of Texas (CPRIT) award RR160027, and funding from the McNair Medical Institute at The Robert and Janice McNair Foundation. We thank Dr. Daniel Liebler for proofreading the manuscript and providing useful suggestions.
Author contributions: B.Z. conceived the study. B.W. implemented the algorithm and developed the software. B.W., B.Z., and X.W. analyzed data. B.W. and B.Z. wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.235028.118.
- Received January 24, 2018.
- Accepted December 28, 2018.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








