
Noncanonical secondary structures arising from non-B DNA motifs are determinants of mutagenesis
- Ilias Georgakopoulos-Soares1,
- Sandro Morganella1,
- Naman Jain2,
- Martin Hemberg1 and
- Serena Nik-Zainal1,3
- 1Wellcome Trust Sanger Institute, Wellcome Genome Campus, Hinxton CB10 1SA, United Kingdom;
- 2Department of Life Sciences, Imperial College London, London SW7 2AZ, United Kingdom;
- 3East Anglian Medical Genetics Service, Cambridge University Hospitals NHS Foundation Trust, Cambridge CB2 2QQ, United Kingdom
Abstract
Somatic mutations show variation in density across cancer genomes. Previous studies have shown that chromatin organization and replication time domains are correlated with, and thus predictive of, this variation. Here, we analyze 1809 whole-genome sequences from 10 cancer types to show that a subset of repetitive DNA sequences, called non-B motifs that predict noncanonical secondary structure formation can independently account for variation in mutation density. Combined with epigenetic factors and replication timing, the variance explained can be improved to 43%–76%. Approximately twofold mutation enrichment is observed directly within non-B motifs, is focused on exposed structural components, and is dependent on physical properties that are optimal for secondary structure formation. Therefore, there is mounting evidence that secondary structures arising from non-B motifs are not simply associated with increased mutation density—they are possibly causally implicated. Our results suggest that they are determinants of mutagenesis and increase the likelihood of recurrent mutations in the genome. This analysis calls for caution in the interpretation of recurrent mutations and highlights the importance of taking non-B motifs that can simply be inferred from the reference sequence into consideration in background models of mutability henceforth.
The canonical right-handed DNA double-helical structure, known as B-DNA, has been recognized since 1953. Although B-DNA is the predominant configuration inside the cell, more than 20 noncanonical secondary structures have been reported (Ghosh and Bansal 2003). These alternative structures include triple-helices, hairpins, cruciforms, and slipped structures, and they are more likely to form at particular repetitive sequences such as mirror repeats, inverted repeats, direct repeats, and short tandem repeats (Wells 2007). Noncanonical secondary structures are associated with increased mutability according to in vitro studies of prokaryotic (Todd and Glickman 1982; Hoede et al. 2006) and eukaryotic cells (Wang and Vasquez 2004; Wang et al. 2006, 2008; Voineagu et al. 2008; Lipps and Rhodes 2009; Biffi et al. 2013; Lu et al. 2015; Bacolla et al. 2016; Kaushik Tiwari et al. 2016; Del Mundo et al. 2017; Kouzine et al. 2017).
Here, we methodically explore the relationship between secondary structures and somatic mutability, focusing on seven common types of sequence motifs prone to forming noncanonical secondary structures, hereafter referred to as non-B DNA motifs for brevity: direct repeats (DR), G-quadruplexes (G4), inverted repeats (IR), mirror repeats (MR), H-DNA, short tandem repeats (STR), and Z-DNA (definitions of each of these can be found in Methods) (Fig. 1A–F). We investigate the contribution made by each type of non-B motif to mutability across many cancer types, including a thorough evaluation of the physical properties of the secondary structures that are formed by non-B motifs. We compare non-B motif predictive power relative to other predictors of mutability described previously and place these findings in context in terms of driver identification in cancer.
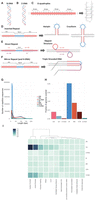
Noncanonical secondary structures arising from non-B DNA motifs in the human genome. (A) Normal configuration of human DNA. (B) Left-handed helical structure caused by Z-DNA. (C–F) Schematic representations of the primary sequence of various non-B motifs and their corresponding predicted secondary structures. (G) Length distribution of non-B DNA motifs. (H) Fraction of the human reference genome (hg19) covered by different non-B DNA motifs. (I) Enrichment of occurrences of non-B DNA motifs associated with various chromatin states (see Methods for calculation).
Results
Genomic characteristics of non-B DNA motifs
We systematically explored each of the seven non-B DNA motifs in the human reference sequence (Methods; Cer et al. 2013). Most motifs are <50 bp (Fig. 1G), and each category encompasses 0.07%–4% of the human genome (Fig. 1H), which may seem small fractionally, but absolute numbers of motifs are substantial (range 69,154–6,006,266). Non-B motifs show nonuniform distributions across the genome reflected by their variable enrichments at different chromatin-associated regions (Fig. 1I): G4 and Z-DNA are strongly enriched at GC-rich promoter regions; DR, H-DNA, and MR are modestly enriched in low complexity repetitive sequences (e.g., heterochromatin); and IR and STR are more uniformly distributed between gene-rich and gene-poor regions. Although some motifs are correlated with each other, there is limited overlap between distinct types of non-B motifs (Supplemental Fig. S1A,B).
Non-B DNA motifs are associated with increased mutability in cancer genomes
Genomic features such as histone epigenetic marks and replication time domains have been shown to be predictive of the variation in distribution of somatic mutations (Schuster-Böckler and Lehner 2012; Polak et al. 2015). We thus explored whether non-B motifs also had an impact on somatic mutagenesis. We used mutation catalogs derived from 560 whole-genome-sequenced (WGS) breast cancers (Nik-Zainal et al. 2016). The genome was binned, and mutations, non-B motifs, histone modifications, and replication time domains were counted for each bin (Methods; Supplemental Figs. S2, S3). Consistent with previous reports (Stamatoyannopoulos et al. 2009; Schuster-Böckler and Lehner 2012; Lawrence et al. 2013; Polak et al. 2015; Morganella et al. 2016), we find that genomic features linked to epigenetic modifications such as heterochromatin (H3K9me3, r = 0.31) and late replicating domains (r = 0.59) are associated with increased mutational density, while open chromatin (DNase I, r = −0.31), active cis-regulatory elements (H3K27ac, r = −0.52), and transcribed regions (H3K36me3, r = −0.57) are negatively associated with mutational density (Fig. 2A; Supplemental Fig. S4). Crude correlations for selected non-B DNA motifs, particularly IR (r = 0.28), STR (r = −0.33), G4 (r = −0.38), MR (r = 0.20), and Z-DNA (r = −0.19) (Fig. 2A; Supplemental Fig. S4) are observed. Partial correlation analysis reveals the association between somatic mutations and non-B motifs remains while controlling for epigenetic marks and replication timing (Supplemental Fig. S5), raising the possibility that non-B motifs are independent factors that contribute to mutability (De and Michor 2011; Du et al. 2013; Bacolla et al. 2016; Kamat et al. 2016). Negative correlations noted in the crude analysis of 500-kb bins are likely to be due to the relatively inflated bin-size, given the small proportion of genome that is covered by non-B motifs. Reinforcing this idea, we generated plots centered on substitutions or indels (Fig. 2B; Supplemental Fig. S10A,B), plotting the density of each type of non-B motif for a 2-kb window around each mutation. This showed distinctive peaks (of different heights for different non-B DNA motifs) implicating enrichment of non-B motifs at sites of somatic mutations. To ensure that this observation was not driven by GC content or sequence-specificity, we controlled for trinucleotide sequence and found that the enrichment remains largely unchanged (Supplemental Fig. S10C). For G4 motifs specifically, we observed peaks not only at the site of indels but also ∼150 nt away from it, for which we suggest a relationship with nucleosome positioning (Supplemental Fig. S10D).
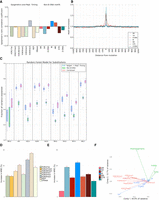
Non-B DNA motifs predict somatic mutability in human cancers. (A) Correlations between the number of non-B DNA motifs, and epigenetic features and replication timing, with the number of substitutions (Spearman's rank correlation coefficient). Please note interpretation is directional, e.g., a positive correlation with replication time would indicate increased mutability with early replication time domains, while a negative correlation denotes increased mutability in late replication time domains. (B) The distribution of different non-B DNA motifs in a window of 2 kb centered on substitutions across all tumor types. (C) Fraction of variance explained for predicting the number of mutations in 500-kb bins with random forest regression using non B-DNA motifs and epigenetic features/replication timing as predictors for multiple tumor types. (BRCA) Breast cancer, (LIRI) liver cancer, (OVCA) ovarian cancer, (ESAD) esophageal adenocarcinoma, (GACA) gastric cancer, (PBCA) pedriatic brain cancer, (PACA) pancreatic cancer, (RECA) renal cell carcinoma, (MALY) malignant lymphoma. Error bars represent standard error from 10-fold cross-validation. (D,E) Importance of the different predictors for the random forest regression. The y-axis shows the increase in mean square error (MSE) when the variable is excluded. (**) FDR < 0.01, as determined by a permutation test. (F) PCA. The first two principal components separate mutations (green), non-B DNA motifs (blue), and epigenetics and replication timing domains (red).
Non-B DNA motifs improve model predictions of cancer genome mutability in many cancer types
To explore this in more depth, we assessed the predictability of mutation density given the number of non-B motifs (as well as epigenetic features and replication time domains) by constructing models using linear regression and random forest regression (Fig. 2C; Supplemental Fig. S6). First, our analysis recapitulates previous studies showing that random forest regression explains a larger fraction of the variance than linear regression for base substitutions and also identifies H3K9me3 and replication timing as the most informative features for predicting mutability (Fig. 2D; Supplemental Fig. S7; Schuster-Böckler and Lehner 2012; Polak et al. 2015). Second, we find that IRs and G4s are relatively strong predictors of mutability, although other non-B motifs including MR, H-DNA, STR, and Z-DNA contribute predictive power (Fig. 2E; Supplemental Fig. S7). Third, although non-B motifs alone can explain 37% of observed variance in mutation density for base substitutions in breast cancer, regression models incorporating both epigenetic, replication time, and non-B motifs substantially improve the variance explained to 52%, performing better than either model separately (Fig. 2C). The enhanced model predictions featuring combined data is unsurprising in light of a principal component analysis biplot: Non-B motifs and epigenetic features are separated by the second component (Fig. 2F), suggesting that they contribute toward predicted mutability in different ways. Since non-B motifs can be computed from the reference genome alone, our results suggest a straightforward and cost-effective way of improving mutability predictions.
To validate our predictive model, we employed it across WGS cancer data sets from eight other tissue types, including liver, ovarian, esophageal, gastric, pancreatic, renal cell carcinoma, and pediatric brain cancers and malignant lymphoma (Supplemental Table S1; The International Cancer Genome Consortium 2010; Patch et al. 2015; Waddell et al. 2015; The Cancer Genome Atlas Research Network 2017; Fraser et al. 2017). The fraction of variance explained by the regression model varied by cancer type, with between 43% and 76% of the variance explained (Fig. 2C–E; Supplemental Figs. S6, S7). Consistently across all tumor types, non-B motifs made a smaller independent contribution toward predicting mutability but, in combination with epigenetic factors/replication timing, improved predictive ability overall. Regression analyses were performed for other mutation classes—indels and rearrangements—and predictive ability of the model similarly improved when the factors were combined (Supplemental Fig. S8). The model performed better for indels than for rearrangements, although the number of rearrangements is much lower than substitutions and indels (by orders of magnitude); hence, we cannot exclude the possibility that model performance is limited by sample size. Our findings bring together and reinforce previous observations of indel enrichment at disparate non-B motifs in experimental systems, e.g., IRs (Glickman and Ripley 1984; Sinden et al. 1991; Lu et al. 2015; Kamat et al. 2016), DRs (Schon et al. 1989; Wojcik et al. 2012), and G4s (Koole et al. 2014; Lemmens et al. 2015; Kamat et al. 2016).
We thus conclude that primary sequence features, as represented by non-B DNA motifs, are collectively informative for predicting local mutability across many tissue types, predominantly of substitutions and indels. Is it the physical presence of a noncanonical secondary structure that mechanistically drives the increased likelihood of mutagenesis? The evidence in favor of this possibility is described below.
Non-B motif-related increased mutability is dependent on physical properties that are optimal for secondary structure formation
First, we find that somatic mutations are not simply increased in the vicinity of non-B DNA motifs; they are elevated within non-B motifs themselves (Fig. 3A,B). H-DNA, STR, and Z-DNA motifs were most enriched for substitutions 1.7-, 1.6-, and 1.7-fold, respectively, while other motifs showed more modest enrichment: G4 (1.2-fold), IR (1.1-fold), DR (1.1-fold), and MR (1.1-fold) when compared to their immediate surrounding sequence (i.e., corrects for genomic GC variation). There is more striking enrichment of indels in general: Z-DNA (10.7-fold), H-DNA (sixfold), STR (5.8-fold), MR (2.5-fold), DR (2.3-fold), and G4 (1.5-fold), a finding that is not surprising given that most indels in human cancer occur at repeat tracts, which are present at a higher frequency particularly at Z-DNA, H-DNA, and STRs. For rearrangements, the absolute number per tumor type was low in comparison to substitutions and indels and the uncertainty higher; nevertheless, enrichment was observed within IRs in breast cancer (1.2-fold) (Fig. 3A), reinforcing observations in yeast and mammalian in vitro studies (Lu et al. 2015). Enrichment of mutagenesis within non-B motifs is remarkably consistent across all tumor types for some motifs (e.g., Z-DNA, STR, G4, H-DNA) (Supplemental Fig. S9). Essentially, we find that there is an excess of mutability not just associated with non-B DNA motifs but directly within them (Fig. 3A; Supplemental Fig. S9).
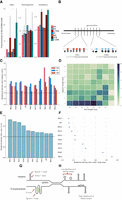
Non-B DNA motifs are mechanistically linked to mutability through formation of secondary structures. (A) Enrichment of mutagenesis for non-B motifs within their genomic bins, thus correcting for genomic GC variation. Error bars represent the standard error. (B) Depiction of enrichment per genomic bin, for results in A, demonstrating how mutations are enriched for non-B motifs. Red and blue boxes represent non-B motifs. (C) Mutational density in spacers compared to arms for direct repeats, inverted repeats, and mirror repeats across 10 tumor types. Error bars representing standard error are too small to visualize. A Wilcoxon signed-rank test was performed (P-value < 0.001 across all tumors for IR, MR, DR). (D) Heat map showing relative ratio of mutational density of spacers over arms for breast cancer at inverted repeats. (E) Enrichment of mutation density in loops: G-runs across ten cancer types. Error bars represent standard error from bootstrapping with replacement (n = 10,000). (F) Enrichment of mutation density at G-quadruplexes for small loop sizes (≤3 nt) relative to large loop sizes (>3 nt) across 10 cancer types. Error bars represent standard error from bootstrapping with replacement (n = 10,000). A Mann-Whitney U test was performed for each cancer type (P-value < 0.001 across all tumor types). (G) Depiction of two very different secondary structures that both have loop domains which are more mutable than their other components. (H) Some non-B motifs have characteristics such as arm or spacer lengths that increase the likelihood of stable hairpin formation. These perhaps can occur stably more frequently, and thus, their exposed regions are more likely to be damaged and mutated.
Second, we find that the elevated mutation densities in non-B motifs show domain-specificity. Selected non-B motifs have identifiable subcomponents–DR, IR, and MR consist of two symmetric “arms” flanking a stretch of “spacer” sequence (Fig. 1D–F). The arms can hybridize, forming a transiently stable structure, leaving the spacer sequence exposed to damage to potentially be more mutable (Fig. 1D–F). We find that spacer sequences are more enriched for substitutions than arm sequences (1.8-fold for DR, twofold for MR, and 1.7-fold for IR) (Fig. 3C; Supplemental Figs. S11, S12). This is in keeping with previous experimental reports demonstrating how the loop domain formed by the spacer sequence tended to mutate more frequently in hairpin structures (Saini et al. 2013; Vasquez and Wang 2013). It also reinforces a report that specifically explores a more conservative subset of IRs (with specific spacer and arm lengths), which suggests that mutability is an intrinsic property of these IRs, because nearly all mutational processes are elevated in IRs regardless of mutational process active in each tumor (Zou et al. 2017).
Third, non-B motifs do not have a uniform thermodynamic capacity to form secondary structures. Experimental and biophysical simulation studies suggest that hairpin formation (for example) is optimal at certain spacer and arm lengths (Nag and Petes 1991; Varani 1995; Goddard et al. 2000). If the physical formation of a secondary structure influenced mutability, then we would expect to observe elevated mutabilities particularly for spacer and arm lengths that are most favorable for hairpin/cruciform formation (Sinden et al. 1991; Lobachev et al. 1998). We find that spacer-to-arm mutation enrichment is indeed variable for different spacer sizes and various arm lengths (Fig. 3D; Supplemental Figs. S11C, S12). Heat maps of mutation enrichment demonstrate that, for IRs, which form hairpin and cruciform structures, mutability is greatest for spacer sequences of 1–3 nt and arm lengths of 10–14 nt (Fig. 3D) in keeping with previous reports highlighting physical specifications of in vitro IR mutability (Sinden et al. 1991; Lobachev et al. 1998). Also, DRs with short spacers and longer arms are more mutable (Supplemental Figs. S11C, S12), consistent with them being more likely to induce slipped structure misalignment (Pierce et al. 1991). In contrast, MRs exhibit more modest enrichment for particular spacer or arm lengths (Supplemental Fig. S13). However, a small subset of MRs are H-DNAs that have high AG content (>90%) and are more likely to form triple-helical structures held together by Hoogsteen bonds (Fig. 1F). H-DNAs are believed to be more mutable than MRs (Wang and Vasquez 2004), and we do observe an excess of mutability in H-DNA in our analysis (Fig. 3A). These observations across IRs, DRs, and MRs are recapitulated in other tumor types (Supplemental Fig. S14).
Fourth, our findings are reinforced by assessing noncanonical secondary structures with very different physical properties. Primary sequence comprising G-runs and interspersed loop elements can form a complex G4 structure (Fig. 1C). Experiments in yeast systems have shown that smaller loop elements confer greater thermodynamic stability to G4 formation where the exposed loops are prone to mutation (Fig. 1C; Tippana et al. 2014; Piazza et al. 2015; Kim et al. 2016). Indeed, our analysis supports these experiments showing that loops have a ∼1.15- to 1.8-fold enrichment in mutagenesis over G-runs (Fig. 3E; Supplemental Fig. S11A) and the subset of G-quadruplexes with average loop size of up to 3 nt is more mutable than their counterparts with larger loop elements (Fig. 3F).
In conclusion, the relationship between somatic mutation and non-B motifs is not simply an association—we find incriminating evidence to suggest that it is the physical formation of secondary structures that predispose to damage and mutagenesis: Not only are non-B motifs enriched for mutation (Fig. 3A), the enrichment is domain-specific for selected non-B motifs (Fig. 3G), and biophysical characteristics that predispose to stable secondary structure formation (such as loop size and stem length) appear to be associated with increased mutability (Fig. 3H).
Discussion
Our analyses suggest that noncanonical configurations are primary determinants of mutagenesis; potentially raising the prior probability of mutability to considerable levels in a highly localized way at specific locations. This has significant consequences for the biological interpretation of recurrent mutations.
A central tenet in cancer biology is the identification of driver mutations—those causally implicated mutations that are believed to drive tumorigenesis. Most drivers are found in protein-coding sequences, although recent WGS studies permit the exploration of noncoding sequences (Lovén et al. 2013; Fredriksson et al. 2014; Weinhold et al. 2014; Nik-Zainal et al. 2016). Due to the difficulties of interpreting non-protein-coding sequences, a useful criterion for identifying putative noncoding driver mutations is to focus on recurrently mutated loci (Weinhold et al. 2014; Nik-Zainal et al. 2016). We have demonstrated that non-B DNA motifs confer a marked propensity for increased mutability at local levels. Thus, we hypothesize that these motifs could be overrepresented among recurrently mutated loci. Indeed, one example of a statistically significant recurrently mutated locus is the promoter of the PLEKHS1 gene that has been shown to be an inverted repeat (Weinhold et al. 2014; Nik-Zainal et al. 2016). For the cancer types in our study, we first find that there are more recurrent substitutions than expected based on a truncated Poisson null model (Fig. 4A). Second, non-B DNA motifs are indeed overrepresented (fivefold) among recurrent substitutions (same site mutated two or more times) than nonrecurrent ones (Fig. 4B,C; Supplemental Fig. S15). Enrichment is variable from one motif to another, with short tandem repeats having 20-fold enrichment. Our finding that non-B DNA motifs are enriched for mutations, and in particular, recurrent mutations, due to the formation of secondary structures (Fig. 4D) has important implications; effectively obfuscating the interpretation of recurrently mutated loci. Consequently, the cautionary note is this: Statistical models of background mutability should consider the contribution to localized mutability provided by non-B DNA motifs in all future analyses.
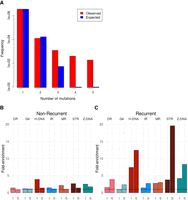
Non-B motifs contribute to locally elevated mutation rates resulting in recurrent mutations in the human genome. (A) Distribution of the number of recurrent events for 3,476,890 somatic mutations from 560 breast cancers (Nik-Zainal et al. 2016). The values do not fit a truncated Poisson distribution (χ2 test, P < 1 × 10−16) as there are more recurrent mutations than predicted by the null model. (B) Enrichment of nonrecurrent mutations overlapping non-B DNA motifs for indels (I) and substitutions (S). (C) Enrichment of recurrent mutations overlapping non-B DNA motifs for indels (I) and substitutions (S). Mann-Whitney U test for substitutions: P-value < 0.001 for all non-B DNA motifs. Mann-Whitney U test for indels: P-value < 0.001 for STR, H-DNA, Z-DNA, and MR, and P-value < 0.05 for DR and G4.
Methods
Somatic variants from cancer data were obtained from 1809 whole-genome-sequenced patients (The International Cancer Genome Consortium 2010; Nik-Zainal et al. 2016). All mutation calls were performed by the Wellcome Trust Sanger Institute's Cancer Genome Project whole-genome sequencing pipeline. Simulations were performed for 10% randomly selected substitutions for each tumor type, controlling for trinucleotide content and genomic location.
Genome-wide maps of each non-B DNA motif were derived from Cer et al. (2013). DNase and histone modification narrowpeak files were derived from Roadmap Epigenomics Consortium et al. (2015), and BAM files were derived from The ENCODE Project Consortium (2012) for the cell of origin of each tumor type. MNase data for K562 cell line were derived from The ENCODE Project Consortium (2012). Chromatin state annotations were defined as in Hoffman et al. (2012, 2013) using chromatin modifications from The ENCODE Project for six human cell lines. The enrichment of each non-B DNA motif at each chromatin state was subsequently calculated (Supplemental Materials and Methods). Reference coordinates for replication landmarks were inferred from Repli-Seq data of 14 cell-lines from The ENCODE Project Consortium (2012) and processed as described in Morganella et al. (2016). BEDTools utilities v2.21.0 were used to manipulate genomic files and intervals (Quinlan and Hall 2010).
The human genome (hg19) was partitioned in 500-kb segments, and the distributions of genomic and epigenomic features were calculated (Supplemental Materials and Methods). Partial correlations were applied to measure the relationship between mutations and non-B DNA motifs, controlling for the effect of epigenetic markers and replication timing.
To model the relationship between the number of mutations and a plethora of explanatory variables, we applied linear regression and random forest regression with 10-fold cross-validation. For the random forest regression model, feature importance was measured using the predictive measure of the original and the permuted data set.
Enrichment of each non-B DNA motif for somatic mutations was calculated across genomic bins (Supplemental Materials and Methods). The mutational density of spacers and arms for IRs, DRs, and MRs was calculated independently and was corrected for that expected based on the trinucleotide content of substitutions for each tumor type. Similarly, the mutational density at G-runs and loops for G-quadruplexes was measured independently and compared, also correcting for trinucleotide content of substitution.
To investigate the relationship between mutagenesis and the distribution of non-B DNA motifs, we generated a window of 2 kb centered at mutations and measured the distribution of non-B DNA motifs, from which we calculated the enrichment at each position. The signal profile and heat map plot for nucleosome occupancy around G4s was generated using deepTools (Ramírez et al. 2014).
The number of substitutions and indels at each genomic site was calculated per cancer type across patients using a Python script (Supplemental Script). The overlap between recurrently mutated sites for each mutation type and each non-B DNA motif was subsequently calculated. A truncated Poisson model was applied as the null model.
Competing interest statement
S.N.-Z. is an inventor on five patent applications with the UK IPO. S.N.-Z. is also a consultant for Artios Pharma Ltd.
Acknowledgments
This work has been performed on data that were previously published under the auspices of the International Cancer Genome Consortium and Breast Cancer Somatic Genetics Study (BASIS), a research project funded by the European Community's Seventh Framework Programme (FP7/2010–2014) under the grant agreement number 242006. M.H. is supported by the Wellcome Trust Sanger Institute core grant. S.N.-Z. was a Wellcome-Beit Fellow and personally funded by a Wellcome Trust Intermediate Clinical Research grant (WT100183MA) at the start of writing this manuscript and subsequently funded by a CRUK Advanced Clinician Scientist Award (C60100/A23916).
Author contributions: I.G.-S., M.H., and S.N.-Z. conceived the concepts and analytical framework, drove the intellectual exercise, and wrote the manuscript. I.G.-S., S.M., N.J., and M.H. wrote the code for analyzing and presenting the data.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.231688.117.
- Received October 27, 2017.
- Accepted July 12, 2018.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








