
HD-Marker: a highly multiplexed and flexible approach for targeted genotyping of more than 10,000 genes in a single-tube assay
- Jia Lv1,2,4,
- Wenqian Jiao1,4,
- Haobing Guo1,4,
- Pingping Liu1,
- Ruijia Wang1,
- Lingling Zhang1,2,
- Qifan Zeng1,2,
- Xiaoli Hu1,3,
- Zhenmin Bao1,3 and
- Shi Wang1,2
- 1MOE Key Laboratory of Marine Genetics and Breeding, College of Marine Life Sciences, Ocean University of China, Qingdao 266003, China;
- 2Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266237, China;
- 3Laboratory for Marine Fisheries Science and Food Production Processes, Qingdao National Laboratory for Marine Science and Technology, Qingdao 266237, China
-
↵4 These authors contributed equally to this work.
Abstract
Targeted genotyping of transcriptome-scale genetic markers is highly attractive for genetic, ecological, and evolutionary studies, but achieving this goal in a cost-effective manner remains a major challenge, especially for laboratories working on nonmodel organisms. Here, we develop a high-throughput, sequencing-based GoldenGate approach (called HD-Marker), which addresses the array-related issues of original GoldenGate methodology and allows for highly multiplexed and flexible targeted genotyping of more than 12,000 loci in a single-tube assay (in contrast to fewer than 3100 in the original GoldenGate assay). We perform extensive analyses to demonstrate the power and performance of HD-Marker on various multiplex levels (296, 795, 1293, and 12,472 genic SNPs) across two sequencing platforms in two nonmodel species (the scallops Chlamys farreri and Patinopecten yessoensis), with extremely high capture rate (98%–99%) and genotyping accuracy (97%–99%). We also demonstrate the potential of HD-Marker for high-throughput targeted genotyping of alternative marker types (e.g., microsatellites and indels). With its remarkable cost-effectiveness (as low as $0.002 per genotype) and high flexibility in choice of multiplex levels and marker types, HD-Marker provides a highly attractive tool over array-based platforms for fulfilling genome/transcriptome-wide targeted genotyping applications, especially in nonmodel organisms.
Recent advances in next-generation sequencing (NGS) technologies now allow rapid and affordable generation of extensive genomic resources in numerous less-studied organisms and offer opportunities to address many scientific questions with unprecedented power and precision (Levy and Myers 2016). During the last decade, the scientific community has witnessed the rapid development of a variety of high-throughput genotyping-by-sequencing (GBS) methods and their huge success in numerous and diverse genomic applications (for reviews, see Davey et al. 2011; Andrews et al. 2016). The common feature of these GBS methods is the utilization of restriction enzymes for genome complexity reduction, which provides an effective way to genotype a large number of samples at an affordable cost (e.g., Elshire et al. 2011; Wang et al. 2012, 2016). Because they sample the genome approximately at random, GBS methods are mostly competent for de novo marker discovery and genotyping, but not for interrogating genomic regions or loci that are of particular interest to researchers. More recently, several target region–oriented GBS methods have been developed (Rife et al. 2015; Ali et al. 2016; Schmid et al. 2017), but genomic loci that can be targeted by these methods are still limited to small regions around restriction sites.
Gene-related molecular markers (e.g., genic single-nucleotide polymorphisms [SNPs] or microsatellites), which are derived from the transcribed regions of the genome, are particularly valuable for genetic, ecological, and evolutionary studies (Andersen and Lübberstedt 2003; De Wit et al. 2015). Such “functional” markers have great potential for quickly identifying causal genes responsible for genetic traits or adaptive evolution (Namroud et al. 2008; Liu et al. 2012; Jiao et al. 2014; Lek et al. 2016). Although discovering a large set of gene-related markers can now be readily achieved through transcriptome sequencing (Ekblom and Galindo 2011; De Wit et al. 2015), targeted genotyping of these markers on a large scale (e.g., thousands to tens of thousands of markers) and in a cost-effective manner remains a major challenge especially for laboratories working on nonmodel organisms. Highly multiplexed array-based genotyping platforms (e.g., Affymetrix arrays), although very powerful and widely applied in human and model organisms, are largely inaccessible to a majority of nonmodel organisms due to the lack of inexpensive standardized commercial arrays (otherwise requiring large investments in custom array fabrication) (Thomson 2014; Jiang et al. 2016). Highly multiplexed PCR-based approaches, for example, microdroplet PCR (Tewhey et al. 2009) and AmpliSeq (Damiati et al. 2016), and sequence capture approaches like hybrid capture (Hodges et al. 2007; Gnirke et al. 2009) and molecular inversion probe capture (MIP-seq) (Turner et al. 2009), take advantage of NGS technologies and allow thousands to tens of thousands of target sequences to be captured simultaneously in a single assay. However, these approaches are more suited for targeting broad genomic regions of interest (e.g., searching for both known and unknown variants) than for examining specific loci of particular interest (Mamanova et al. 2010; Mertes et al. 2011), and/or they may suffer from remarkable nonuniformity of capture efficiency (e.g., MIP-seq) (Mamanova et al. 2010; Boyle et al. 2014), making them a less than ideal choice in genotyping applications focusing on user-predefined markers. In addition, most of these approaches remain very costly for large-scale genotyping applications due to either a requirement for expensive, specialized instruments (e.g., RainStorm or Ion Proton systems) or high library preparation cost prior to capture (Mertes et al. 2011; Ali et al. 2016).
GoldenGate technology is based on the high specificity and accuracy of oligo extension and ligation assay and is well known for its high marker multiplexity and high flexibility of marker selection (Syvänen 2005; Fan et al. 2006; Perkel 2008; Paux et al. 2012). GoldenGate was once recognized as one of the key technologies that revolutionized the SNP genotyping field (Perkel 2008). To date, GoldenGate has been widely adopted in numerous and diverse applications (Shen et al. 2005; Bibikova and Fan 2009; Chao and Lawley 2015), with the most prominent example being the generation of approximately 250 million genotypes for the International HapMap Project (The International HapMap Consortium 2003). Despite these striking features, GoldenGate has been less favorable in the NGS era, largely because it builds on the BeadArray platform (Shen et al. 2005; González-Neira 2013) and suffers from several array-based limitations, including relatively high genotyping costs for custom-built SNP panels, attainable capacity, and flexibility limited by available array formats (e.g., 96 or 384–3072 loci per assay and 12, 32, or 96 samples per array), the inability to assay marker types other than SNPs (e.g., microsatellites, insertions/deletions [indels]), a highly complicated and labor-intensive experimental procedure, and the reliance of genotype retrieval on an expensive, specialized instrument (e.g., Illumina iScan System). With the rapid development of sequencing technologies, it is conceivable that switching GoldenGate from BeadArray to NGS platforms would revive this powerful methodology by addressing array-related issues and achieve the goal of cost-effective targeted genotyping of transcriptome-scale markers, which is highly attractive for researchers working on nonmodel organisms.
Here, we develop a high-throughput, NGS-based GoldenGate approach (called HD-Marker) that allows highly multiplexed, targeted genotyping of user-defined markers and is flexible in terms of both multiplex levels and marker types. We demonstrate the performance of HD-Marker on various multiplex levels (296, 795, 1293, and 12,472 SNPs) across two sequencing platforms (Illumina and SOLiD) in two nonmodel species (scallops Chlamys farreri and Patinopecten yessoensis). The potential for high-throughput genotyping of alternative marker types (e.g., microsatellites and indels) is also explored.
Results
Overview of the HD-Marker methodology
The HD-Marker technique, as shown in Figure 1, allows for highly multiplexed, targeted genotyping of user-defined markers (SNPs, microsatellites, or indels) in a single reaction through highly specific extension, ligation, and amplification steps. The whole procedure takes place in a single tube and can be finished within 3 d. In HD-Marker, only two probes are needed to genotype a locus, in contrast to the three probes used in the Illumina BeadArray-based GoldenGate assay (Fan et al. 2003). Probes can be either column- or array-synthesized, with the latter allowing for extremely high multiplex levels (up to 12,000 markers) at a very low cost per probe (Fig. 1A). Each probe consists of a locus-specific 3′ portion used to recognize the genomic target regions and a 5′ portion that incorporates a universal PCR primer-binding sequence (Fig. 1B). The universal primer-binding sequences allow the same probe panel to be used for different NGS platforms (e.g., Illumina or SOLiD) through two rounds of PCR during library preparation. Preparation of an HD-Marker library (Fig. 1C) begins with attaching the genomic DNA to a solid support and then hybridizing two locus-specific probes (LSPs) to the immobilized DNA. This enables stringent washing to remove excess and incorrectly hybridized probes. The gap between the two LSPs covers the locus of interest, which is filled in by a polymerase, followed by ligation of the extended LSP to the downstream LSP, creating a molecule that can be amplified by PCR. In effect, probe hybridization provides specificity for finding the correct locus in the genome, and ligation confers additional specificity because incorrectly hybridized probes are unlikely to be adjacent. Finally, two rounds of PCR amplification are performed on the created molecules, and desirable NGS platform-specific adaptor and barcode sequences can be incorporated during the second round of PCR. The libraries prepared from different samples can be pooled for high-throughput sequencing on the preferred NGS platform.
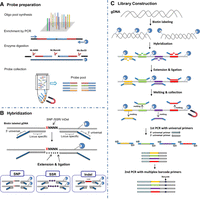
Overview of the HD-Marker approach. (A) Probe preparation: Probes can be either column- or array-synthesized, with the latter allowing extremely high multiplex levels (up to 12,000 markers) at a very low probe cost (∼$0.001 per base). The diagram shows the preparation of single-stranded probes from an array-synthesized oligo pool through the steps of PCR amplification, enzyme digestion, and isolation by magnetic beads. (B) In-solution hybridization: Highly parallel probe hybridization is achieved in a single tube, with each probe consisting of a locus-specific portion and a universal PCR primer-binding portion. The gap between two probes covers the locus of interest (e.g., SNPs, microsatellites/SSRs or indels), which is filled in by a polymerase, followed by ligation of the extended LSP to the downstream LSP, creating a molecule that can be amplified by PCR. (C) Preparation of an HD-Marker library begins with attaching the genomic DNA to a solid support and then using two locus-specific probes (LSPs) to hybridize to the immobilized DNA. Through highly specific extension, ligation, and amplification steps, the libraries prepared from different samples can be pooled for high-throughput sequencing on a preferred NGS platform.
Benchmarking the HD-Marker technique
We benchmarked the HD-Marker technique using two nonmodel species (the scallops C. farreri and P. yessoensis), which are among the best molecularly characterized bivalves with abundant SNP and microsatellite marker resources (Zhan et al. 2009; Hou et al. 2011; Wang et al. 2013, 2017; Jiao et al. 2014; Li et al. 2017) and would benefit greatly from the development of a cost-effective, high-throughput targeted genotyping approach for genetic, genomic, and breeding studies.
SNP panel choice and library setup
Two large SNP panels were chosen for technical evaluation of HD-Marker. The first panel (Supplemental Table S1) contained 1293 SNPs that were previously discovered from various transcriptome data sets of C. farreri (Hou et al. 2011), and the validity of these SNPs had been primarily verified using the high-resolution melting (HRM) method (Jiao et al. 2014). The HD-Marker probes targeting these SNPs were column-synthesized and pooled at three multiplex levels (296, 795, and 1293). To enable across-level comparisons, we stipulated that all SNP loci in a given multiplex level must be present in all higher levels (e.g., 1293-plex contains all loci in 296- and 795-plexes, and 795-plex contains all loci in 296-plex). The second panel (Supplemental Table S2) contained 12,472 SNPs that were identified from 11,771 genes in the P. yessoensis genome (Wang et al. 2017), with 60%, 28%, and 12% of these SNPs being distributed in the exonic, intronic, and 3′/5′ UTR regions, respectively (Supplemental Table S3). In order to reduce the total cost of probe synthesis, the HD-Marker probe set for the second SNP panel was synthesized by using an array-based technology (CustomArray, Inc.; ∼$0.001 per base). In total, eight HD-Marker libraries were prepared at four multiplex levels (296, 795, 1293, and 12,472) with two technical replicates per multiplex level for Illumina sequencing.
Specificity, capture rate, and uniformity
In total, 1.0, 2.4, 5.2, and 21.3 million raw Illumina reads were obtained for 296-plex, 795-plex, 1293-plex, and 12,472-plex, respectively (Table 1), of which 94.9%–99.9% were retained as high-quality (HQ) reads. The high specificity of the HD-Marker assay was observed for 296-plex, 795-plex, and 1293-plex, with ∼92%–97% of HQ reads aligned to target regions (Table 1), whereas a relatively lower rate (∼80%) was observed for 12,472-plex, likely resulting from the incorporation of low-quality and/or incorrectly generated probes as expected for the high-throughput array-based approach for oligo synthesis and probe preparation. The capture rate of target loci reached 97.6%–98.8% with unnoticeable difference observed for different multiplex levels (Table 2; Fig. 2). For all multiplex levels, the large majority (98.6%–99.7%) of detected loci were covered by eight or more reads—the coverage threshold for genotype calling (Supplemental Table S4). The locus detection was highly reproducible between technical replications (Table 2), and even across multiplex levels (Supplemental Table S5). For three column-synthesized probe sets, the loci detected in the 296-plex and 795-plex were almost all detected in higher multiplex levels (99.3%–99.6%) (Supplemental Table S5). Quantification of capture uniformity for 296-plex, 795-plex, 1293-plex, and 12,472-plex showed that the sequencing coverage of target loci varied within two to four orders of magnitude (Supplemental Fig. S1), with 98.1%, 97.4%, 97.8%, and 95.1% of loci falling within in a 100-fold range, and 95.0%, 90.4%, 90.7%, and 70.3% of loci falling within a 10-fold range. The high capture uniformity seemed to be unaffected by the GC content of target regions (Pearson's r: 0.058–0.097) (Supplemental Fig. S2), and the distributions of sequencing depths were largely comparable among exonic, intronic, and UTR regions (Supplemental Fig. S3). The sequencing coverage of target loci that were commonly detected at different multiplex levels showed high correlation across technical replicates (r = 0.97–0.99) and multiplex levels (r = 0.95–0.99) (Supplemental Fig. S4). The performance of allelic sampling by HD-Marker was largely comparable to that of whole-genome sequencing (WGS) (Supplemental Fig. S5).
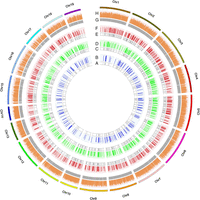
Chromosomal distribution (A,C,E,G) and sequencing coverage (B,D,F,H) of target SNP markers for four multiplex levels. Extremely high capture rate (∼98%–99%) and even sequencing coverage across loci are observed for all multiplex levels. (A,B) 296-plex; (C,D) 795-plex; (E,F) 1293-plex; (G,H) 12,472-plex.
Illumina data processing and alignment to target regions
Summary of loci detection, genotype calling, and concordance between replicates based on Illumina data
Genotype calling and accuracy
Genotype calling rates were extremely high for all multiplex levels, with 99.7%, 98.8%, 99.3%, and 99.2% for 296-plex, 795-plex, 1293-plex, and 12,472-plex, respectively (Table 2). Notably, the distribution of allelic sampling closely matched the expectations both within and across multiplex levels, converging to 0.5 for heterozygous loci and to 1 for homozygous loci (Fig. 3; Supplemental Fig. S6; Supplemental Table S6). This provides an excellent basis for accurate calling of homozygotes and heterozygotes. Genotyping accuracy was evaluated in three aspects. First, genotype calls were compared between technical replicates, revealing ∼98% genotype concordance for all multiplex levels (Table 2). Second, genotype calls of common loci were compared across different multiplex levels, revealing 99.7%–100% genotype concordance (Supplemental Table S5). Third, 128, 154, and 173 SNP loci from 296-plex, 795-plex, and 1293-plex, respectively, were subjected to genotype validation by Sanger sequencing, revealing 98.3%–99.2% genotype concordance across all multiplex levels (97.2%–98.6% for homozygotes and 100% for heterozygotes) (Table 3). For 12,472-plex, genotype validation was conducted based on the WGS sequencing of the same assayed individual, revealing 97.2% genotype concordance (96.9% for homozygotes and 97.8% for heterozygotes) (Table 3). The small proportion of inconsistently genotyped loci was mostly due to the differential power of the two methods in detecting rare somatic mutations (high for HD-Marker but low for WGS) (Supplemental Figs. S7A, S8), as well as biased allelic sampling by WGS for some loci with low sequencing coverage (Supplemental Fig. S7b). Further evaluation of HD-Marker on five well-known gene families revealed that the rates of loci detection (97.2%–100%), genotype calling (99.1%–100%), and genotyping accuracy (96.9%–100%) were all comparable to those calculated from all loci in the 12,472-plex (Supplemental Table S7), suggesting the high performance of HD-Marker for targeting multigene families when unique probes can be designed.
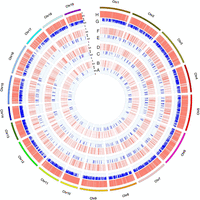
Performance of allelic sampling for four multiplex levels. Allelic sampling closely matches the expectations for all multiplex levels, converging to 0.5 for heterozygous loci (A,C,E,G) and to 1 for homozygous loci (B,D,F,H). (A,B) 296-plex; (C,D) 795-plex; (E,F) 1293-plex; (G,H) 12,472-plex.
Genotype validation by Sanger-based amplicon sequencing (296-plex, 795-plex, and 1293-plex) and genome resequencing (12,472-plex)
Rarefaction and cost analysis
Rarefaction analysis was conducted for each multiplex level based on the combined data set from two replications. For 296-plex, 795-plex, 1293-plex, and 12,472-plex, locus detection is saturated at 0.08, 0.2, 0.6, and 2 million reads, respectively, with 96.2%–97.8% of target loci genotyped at corresponding depths and less than 0.3%–0.5% gains obtained by doubling the sequencing effort (Fig. 4A–D). At the optimal sequencing depths, 98.9%, 98.6%, 98.5%, and 97.5% of genotyping accuracy can be achieved for 296-plex, 795-plex, 1293-plex, and 12,472-plex, respectively. Genotyping cost including library preparation and NGS sequencing was estimated for each multiplex level based on the optimal sequencing depths inferred from the rarefaction analysis (Table 4). As the number of samples increased, the cost per sample or per genotype significantly decreased especially for three column-synthesized probe sets, because the cost of expensive probe synthesis could be attributed to more samples (e.g., for 1293-plex, ∼$150 per sample for the scale of 100 samples in contrast to $14 per sample for the scale of 10,000 samples). Genotyping is highly cost-effective with an array-synthesized probe set, and costs only $25–$44 per sample for 12,472-plex, which makes for an extremely low cost per genotype ($0.002–$0.004) in contrast to $0.01–$0.14 in three column-synthesized probe sets (Table 4).
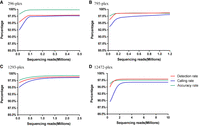
Rarefaction analysis of HD-Marker libraries at different sequencing scales. For 296-plex (A), 795-plex (B), 1293-plex (C), and 12,472-plex (D), loci detection is saturated at 0.08, 0.2, 0.6, and 2 million reads, respectively, and 98.9%, 98.6%, 98.5%, and 97.5% of genotyping accuracy can be achieved at the optimal sequencing depths.
Genotyping costs estimated for different multiplex levels at different sample scales
Cross-platform application
A special feature of HD-Marker is that the same probe panel can be used for different NGS platforms through two rounds of PCR during library preparation (Fig. 1C; for details, see Methods), which provides another level of flexibility and eliminates the need for creation of additional probe panels to suit different sequencing platforms. To evaluate the cross-platform applicability of HD-Marker, we generated six additional data sets based on the ABI SOLiD platform with the same three probe sets (296-plex, 795-plex, and 1293-plex) and the same assayed individual used in previous analyses. For all multiplex levels, 73.6%–75.8% of HQ reads were aligned to target regions (Supplemental Table S8), which was substantially lower than what was observed for Illumina platform, and the difference might be largely due to the overall low quality of SOLiD reads (65%–73% HQ reads in contrast to 95%–99% for Illumina reads). Nevertheless, high rates of locus detection (96.6%–98.0%) were revealed for all multiplex levels (Supplemental Table S9). When a stringent genotyping approach was adopted, high genotype concordance was observed between technical replicates (98.8%–99.6%) (Supplemental Table S9), between HD-Marker and Sanger-based validations (96.4%–97.6%) (Supplemental Table S10), and between SOLiD and Illumina data sets (95.1%–95.9%) (Supplemental Table S11), demonstrating the cross-platform applicability of HD-Marker.
Microsatellite and indel analysis
To explore the applicability of HD-Marker in genotyping alternative marker types, we chose 50 microsatellites (Supplemental Table S12) and 15 indels (Supplemental Table S13) and evaluated them in HD-Marker assays. For microsatellite markers, two Illumina sequencing data sets (representing two technical replicates) were generated using column-synthesized probes, and ∼98% of HQ reads were aligned to target regions (Supplemental Table S14). Approximately 92% of target microsatellite loci were detected, of which ∼84% had genotype calls (Supplemental Table S15). Genotyping concordance between technical replicates was 90.3%. When 10 loci with consistent calls between replicates were chosen for genotype validation by capillary electrophoresis, we found that all loci had the correct genotype calls (Fig. 5; Supplemental Table S15). This suggests that preparation and sequencing of two replicate HD-Marker libraries for microsatellite genotyping would be ideal in practice to ensure high genotyping accuracy. Because of the small number of indel markers, we mixed them with three levels (296, 795, and 1293) of SNP plexes for library preparation and sequencing, with each level having two technical replicates. A locus detection rate of 93.3%–100% and a calling rate of 92.9%–93.3% were observed, with high genotype concordance (100%) between replicates (Supplemental Table S16).
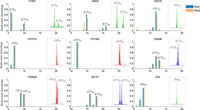
Comparison of microsatellite genotyping between HD-Marker and capillary-based analysis (in small window) for nine selected loci. Highly consistent results were observed between the two methods.
Discussion
Platform switching: from BeadArray to NGS
Illumina's GoldenGate assay, which is built on the BeadArray platform for low to moderate multiplex SNP genotyping, has been widely adopted in numerous and diverse applications. The BeadArray platform enables the production of randomly assembled universal arrays and the analysis of any custom-built panel of SNPs, thus providing more flexibility than conventional microarrays (Oliphant et al. 2002). This feature facilitates researchers to create GoldenGate assays tailored directly to their specific genotyping needs, e.g., focusing on targeted regions, candidate genes, or pathways. Despite its advantage over conventional microarrays, however, the GoldenGate assay has become less favorable in the NGS era, largely due to inherent array-based limitations.
Through the adoption of the NGS platform, our HD-Marker approach inherits the main advantages of GoldenGate assay but also addresses its array-based limitations. First, as built on the highly cost-effective NGS platform (∼$0.01 per Mb; see www.genome.gov/sequencingcostsdata), HD-Marker allows for affordable targeted genotyping of thousands to tens of thousands of user-defined SNPs, which currently remains a major challenge especially for laboratories working on nonmodel organisms. Second, adopting the NGS platform removes the technical restrictions imposed by array formats and enhances the capacity and flexibility of HD-Marker in both SNP multiplexity (up to ∼12,000 SNPs per assay) and sample throughput (e.g., ∼1500–6000 samples per Illumina X Ten sequencing run when targeting from about 1000 to 10,000 SNPs). Third, our HD-Marker protocol is substantially simpler than Illumina GoldenGate protocol, because it eliminates multiple experimental steps involving array preparation (e.g., PCR product immobilization and ssDNA preparation) and hybridization (e.g., array preconditioning, hybridization, and multiple washing). Fourth, HD-Marker can be easily adopted by ordinary laboratories for routine genotyping applications, because it does not require any expensive, specialized instrument and the NGS platform can be widely accessed in most core facilities or commercial biotech companies. Finally, HD-Marker allows flexible choice among a variety of marker types (e.g., SNPs, microsatellites, or indels) and sequencing platforms, providing great potential to meet diverse research needs.
Targeted genotyping on the transcriptome scale
Targeted genotyping of transcriptome-wide markers is highly attractive for genetic, ecological, and evolutionary studies. However, current genotyping methods are either impossible or suboptimal for fulfilling such a task, especially in nonmodel organisms. Commonly used genotyping methods such as TaqMan and high-resolution melting (HRM) assays are mostly suitable for low- or medium-throughput genotyping and would incur a prohibitively high cost when targeting a very large marker panel (e.g., thousands to tens of thousands of SNPs). Array-based genotyping is a popular and viable option, but building custom arrays remains highly expensive for nonmodel organisms, and fixed arrays also lack the flexibility for loci rearrangement. Sequence capturing approaches are better suited for targeting broad genomic regions of interest rather than specific loci and would waste a substantial amount of sequencing effort if used for targeted genotyping. In particular, MIP-seq technique is methodologically analogous to GoldenGate assay (Porreca et al. 2007; Turner et al. 2009), but to date, has been mostly adopted for sequence capture-based resequencing applications (Mamanova et al. 2010; Mertes et al. 2011; Niedzicka et al. 2016). The key limitation of the MIP methodology is the remarkable nonuniformity of capture efficiencies within probe sets (Mamanova et al. 2010; Boyle et al. 2014), which is likely related to the thermodynamics of padlock formation (Deng et al. 2009; Diep et al. 2012). As an improved and commercialized version of MIP for human disease–related applications, HEAT-seq (offered by Roche) addresses the key limitation of MIP by configuration of probes in optimal concentrations based on the preknown empirical information of probe performance; however, such preknown information of probe performance is usually unavailable for nonmodel organisms. HD-Marker is distinct from MIP in that it uses two separate probes to recognize target regions and does not involve with padlock formation, giving it the potential to alleviate the problem of nonuniform capture efficiencies. Consistent with this, HD-Marker exhibits the improved uniformity of capture efficiencies over MIP-seq, for example, ∼92%–96% column-synthesized and ∼70% array-synthesized probes falling within a 10-fold range in our study, in contrast to <80% (without probe rebalancing) and <58% in previous MIP-seq studies (Porreca et al. 2007; Li et al. 2009b; Turner et al. 2009; Teer et al. 2010; O'Roak et al. 2012; Niedzicka et al. 2016).
Among existing targeted genotyping methods, Illumina's GoldenGate assay is prominent for its high marker multiplexity (Paux et al. 2012). However, its current capacity is limited to approximately 3000 loci per assay based on the BeadArray platform (Chao and Lawley 2015), making it less favorable for transcriptome-wide targeted genotyping. It remains unclear whether much higher multiplexity is achievable for this methodology. Our study demonstrates for the first time that this methodology, when switched to the NGS platform, allows more than 12,000 loci (corresponding to approximately 11,700 genes) to be genotyped simultaneously in a single tube. Through the construction of multiple 12,000 libraries for pooled sequencing, our current protocol allows the user to target a vast number of loci that could be comparable to those of conventional microarrays (e.g., 100,000–500,000 loci). Further expansion of the single-tube level of multiplexity may also be expected, because the capacity of current array-based oligo synthesis can reach up to 90,000 unique sequences per pool for as low as $0.0004 per base (offered by CustomArray, Inc.).
Targeting highly homologous genomic regions can present a challenge for HD-Marker, and for multigene families, only SNPs with adjacent regions that allow the design of unique probe sequences for a family member can be currently interrogated by HD-Marker. In the present study, we demonstrate the feasibility and high performance of HD-Marker assay when targeting multigene families. Although not the focus of this study, targeting highly homologous sequences may represent an interesting direction that is worthy of further exploration, for which comprehensive experimental designs (for different homology levels of sequences under different levels of probe specificity and multiplexity) would be needed.
Compared with targeted genotyping, whole-genome sequencing (WGS), although very appealing, remains a costly choice particularly for nonmodel organisms. The cost comparison of HD-Marker and WGS is provided in Supplemental Table S17 for different scales of sample/locus number, sequencing coverage, and genome size. Although the application of WGS may be advantageous for model organisms with small genomes, HD-Marker does have its special advantages over WGS in several aspects. First, when only a small number of loci are targeted, sequencing the whole genome would be unnecessary and costly even at low sequencing coverage. Second, HD-Marker does not require haplotype information, which is crucial for achieving cost-efficient low-coverage WGS (Le and Durbin 2011) but remains unavailable for most nonmodel organisms. Third, resequencing of very large genomes (e.g., 32 Gb for Mexican axolotl) (Nowoshilow et al. 2018) would be prohibitively expensive, but this issue would not apply to HD-Marker because its cost does not depend on genome size. Last but not least, a high-quality reference genome would be required for applying WGS, but such a prerequisite may not be met by many nonmodel organisms for which transcriptome data and associated SNPs are instead often available for HD-Marker probe design.
High-throughput targeted microsatellite genotyping
One of the striking features of HD-Marker is its ability to target diverse marker types, with microsatellites of particular interest. Microsatellites are short tandem repeated sequences (typically <100 bp) that are ubiquitous and highly polymorphic in eukaryotic genomes (Tóth et al. 2000). They, together with SNPs, have been the markers of choice in genetic, ecological, and evolutionary studies over the last 20 yr (Abdul-Muneer 2014; Vieira et al. 2016). The major advantage of microsatellites over SNPs is the higher statistical power per locus owing to their higher mutation rates and polyallelic nature (Liu et al. 2005; Haasl and Payseur 2011). Traditional microsatellite genotyping based on the gel- or capillary-based detection methods are generally laborious, costly, and low throughput. Recent studies suggest the feasibility and reliability of NGS for high-throughput microsatellite genotyping (Cao et al. 2014; Zavodna et al. 2014; Willems et al. 2017), but only a few NGS-based tools have yet been developed for targeted microsatellite genotyping, for example, capture-based approaches (Guilmatre et al. 2013; Duitama et al. 2014) or the MIP-based approach (MIPSTR) (Carlson et al. 2015). We demonstrated the feasibility of HD-Marker for targeted microsatellite genotyping, with the genotyping accuracy (90%–100%) comparable to existing methods (88%–98%) (Guilmatre et al. 2013; Duitama et al. 2014; Carlson et al. 2015). HD-Marker is potentially advantageous over existing methods, because it has higher targeting specificity than capture-based methods and could be more efficient than MIPSTR for capturing long-stretch microsatellites due to the use of two separate probes. The high flexibility of marker type choice makes HD-Marker very promising for meeting diverse research needs. For example, high-resolution genome scanning using ultradense SNPs would be appropriate for identifying the loci responsible for phenotypic variation or evolutionary adaptation, whereas high-throughput genotyping of highly polymorphic microsatellites would be appropriate for detecting subtle population substructure or differentiation (Tian et al. 2008; Slavov et al. 2010).
Methods
DNA samples
Adult individuals of the scallop species, Chlamys farreri and Patinopecten yessoensis, were used for evaluation of HD-Marker assays. High-quality genomic DNA was extracted from scallop adductor muscles by using the conventional phenol/chloroform extraction method (Sambrook et al. 1989).
SNP, microsatellite, and indel markers
Two large SNP panels (1293 and 12,472 SNPs) that were discovered from various transcriptome data sets of C. farreri (Hou et al. 2011) and the P. yessoensis genome assembly (GenBank accession no. GCA_002113885.2) (Wang et al. 2017) were chosen for technical evaluation of HD-Marker (Supplemental Tables S1, S2) at different multiplex levels (296, 795, 1293, and 12,472). Fifty microsatellites (with repeat units of 2–4 bp) (Supplemental Table S12) and 15 1-bp indels (Supplemental Table S13) were chosen for evaluation of HD-Marker applicability in genotyping alternative marker types. These loci were either retrieved from our previous studies (Zhan et al. 2007, 2009; Wang et al. 2013) or identified through the mining of the genome assembly of scallop C. farreri (available from http://mgb.ouc.edu.cn/cfbase/html/download.php) (Li et al. 2017).
Probe design and preparation
In the HD-Marker assay, two probes (LSP1 and LSP2) are designed for genotyping a locus. Probes were designed by meeting three criteria: (1) Each probe contains a ∼20 bp locus-specific sequence, while keeping a certain-size gap (e.g., SNP + 4Ns or Microsatellite + 4Ns; here 4Ns are set for checking hybridization specificity during reads mapping) between LSP1 and LSP2; (2) the GC content and melting temperatures of probes are in the range of 40%–60% and 55°C–65°C, respectively; and (3) probes have unique locations in the reference genome/transcriptome, i.e., no sequence similarity to nontarget regions by allowing up to two mismatches. The unique molecular identifier (UMI) was not included in our probe design, but when PCR duplicates are of significant concern, the use of UMI can offer an accurate estimation of allele frequency.
For three lower levels (296, 795, and 1293) of SNP probe sets (Supplemental Table S1), 50-microsatellite probe set (Supplemental Table S12), 15-indel probe set (Supplemental Table S13), column-synthesized probes were obtained from Sangon Biotech. For each multiplex level, LSP1 and LSP2 probes were separately mixed in equal molar ratio to a final concentration of 50 nM per probe. The 5′-end phosphorylation reaction was performed for LSP2 probe pools to allow for subsequent ligation. The reaction was set up in a 50-µL volume containing 1.25 µM of each LSP2, 10 units T4 Polynucleotide Kinase (NEB) and 1× T4 Polynucleotide Kinase Reaction Buffer (NEB). The reaction was incubated for 30 min at 37°C and then heat inactivated for 20 min at 65°C.
To prepare the 12,472-plex SNP probe pool (Supplemental Table S2), a mixture of 12,472 array-synthesized oligos was obtained from CustomArray, Inc. Each oligo is ∼126-bp long with a common sequence containing a Nt.BsmAI site and connecting the two probes (LSP1\LSP2) in the middle, flanked by two universal primer sequences (containing Nt.AlwI and Nb.BsrDI sites) for oligo amplification. The LSP1 and LSP2 probes were obtained from these array-synthesized oligos through the steps of PCR amplification, enzyme digestion, and isolation by magnetic beads described as follows.
PCR amplification
Amplification of the 12,472-plex oligo pool was performed in a 30-µL reaction containing 1 µL of 1/500th dilution of the oligo pool, 0.4 µM forward primer (Oligo_F), and 0.4 µM biotinylated-reverse primer (Oligo_R), 0.3 mM dNTPs, 1× Phusion HF buffer, and 0.5 units Phusion high-fidelity DNA polymerase (NEB). PCR was carried out using the following conditions: 26 cycles of 5 sec at 98°C, 20 sec at 60°C, and 10 sec at 72°C, and then a final extension of 10 min at 72°C. Six PCR runs were conducted, and the products were combined and purified using a QIAquick PCR purification kit (Qiagen). The purified products were dissolved in 30 µL of pure water and then used for enzyme digestion to generate usable probe pools.
Enzyme digestion
The probe pool for hybridization was isolated from the oligo pool by using three nicking enzymes. Digestion was set up in a 100-µL volume composed of 60 µL of PCR product (∼2.5 µg in total), 3 µL of Nt.AlwI, and 1× CutSmart buffer. The reaction was incubated for 3 h at 37°C, followed by 20 min at 80°C. Then, 3 µL of Nb.BsrDI was added to the tube and was incubated for 3 h at 65°C, followed by 20 min at 80°C. Finally, 3 µL of Nt.BsmAI was added to the tube and was incubated for 3 h at 65°C, followed by 20 min at 80°C.
Probe isolation by magnetic beads
Streptavidin magnetic beads were used to remove the biotin-labeled complementary strand of the target probe. In total, 60 µL of streptavidin magnetic beads (NEB) was used and separated into three tubes. Magnetic beads were suspended using 50 µL of washing buffer (0.5M NaCl, 20 mM Tris-Cl, 1 mM EDTA) and then a magnet was applied to discard the supernatant. The 35 µL of digested product was added to each tube, and the mixture was incubated at room temperature for 20 min, followed by heating for 5 min at 95°C to denature the double-stranded DNA. Then, the denatured product was quickly chilled in an ice bath for 5 min, and the supernatant was transferred into a new tube after applying a magnet. A total of 120 µL of supernatant containing LSP1 and LSP2 was collected in such way, which was purified using a Nucleotide Removal Kit (Qiagen). The probe pool was dissolved using 30 µL of elution buffer (10 mM Tris-Cl, pH 8.5), which is then ready for hybridization.
Library preparation and sequencing
Preparation of biotin-labeled genomic DNA
HD-Marker assay began with the preparation of biotinylated genomic DNA for in-solution hybridization. For genotyping of SNPs, 1 µg genomic DNA was labeled using a PHOTOPROBE biotin labeling kit (Vector Laboratories) by following the manufacturer's instructions for thermal coupling. For genotyping of microsatellites, genomic DNA was labeled in an alternative way to avoid dense biotin labeling resulting from thermal coupling, which may interfere with long-range polymerase extension. The restriction enzymes were chosen because their restriction sites did not occur across the targeting regions of microsatellites. The DNA sample was first digested in a 20-µL reaction containing 10 units EcoRI (NEB) and 10 units MseI (NEB) for 3 h at 37°C and then heat inactivated for 20 min at 65°C. Then, 25 µL of a ligation master mix containing 0.2 µM EcoRI-adaptor, 0.2 µM MspI-adaptor,1 mM ATP (NEB), and 800 units T4 DNA ligase (NEB) was added to each digestion product and incubated for 8 h at 16°C. Ligation products were amplified in a 20-µL reaction composed of 7 µL ligated DNA, 0.1 µM EcoRI biotin-labeled primer, 0.1 µM MseI biotin-labeled primer, 0.3 mM dNTP, 1× Phusion HF buffer, and 0.4 units Phusion high-fidelity DNA polymerase (NEB). PCR was conducted in a MyCycler thermal cycler (Bio-Rad) for 5 min at 98°C; 8 cycles of 5 sec at 98°C, 20 sec 56°C, and 60 sec at 72°C; and then a final extension of 10 min at 72°C. PCR products from three independent amplifications were combined and purified using a QIAquick PCR purification kit (Qiagen) for subsequent use.
Hybridization
In-solution hybridization was performed in a 50-µL reaction composed of ∼200 ng biotinylated genomic DNA, 5–10 µL each of two LSP (LSP1/LSP2) probe mixtures, 5 µL streptavidin magnetic beads (NEB), and ULTRAhyb-Oligo hybridization buffer (Ambion). Hybridization was carried out in a MyCycler thermal cycler (Bio-Rad) by ramping temperature from 70°C to 30°C over ∼8 h.
Extension and ligation
After hybridization, magnetic beads were washed twice using washing buffer 1 (2× SSC, 0.5% SDS) and washing buffer 2 (2× SSC), respectively, to remove excess and mishybridized LSPs. Then, 25 µL master mix containing 0.4 units Phusion high-fidelity DNA polymerase (NEB), 40 units Taq DNA ligase (NEB), 1 mM NAD (NEB), 0.1 mM dNTPs, 1× Phusion HF Buffer was added to the beads. The reaction was incubated for 20 min at 45°C to allow upstream LSP1s to extend and ligate to downstream LSP2s. After extension and ligation, the beads were washed once with elution buffer (10 mM Tris-Cl, pH 8.5), then resuspended in 35 µL elution buffer and heated for 1 min at 95°C to release the ligated products.
Library preparation and sequencing
Sequencing libraries were constructed based on two rounds of PCR amplification of the ligated products. The first-round PCR was set up in a 50-µL reaction composed of 30 µL of the ligated products, 0.1 µM each of two universal PCR primers (1st-UP1 and 1st-UP2), 0.3 mM dNTPs, 1× Phusion HF buffer, and 0.8 units Phusion high-fidelity DNA polymerase (NEB). PCR was conducted with 20 cycles of 5 sec at 98°C, 20 sec at 60°C, and 10 sec at 72°C, and then a final extension of 10 min at 72°C. The target band (for SNPs) or smear (for microsatellites) was excised from an 8% polyacrylamide gel, and the DNA was diffused from the gel in nuclease-free water for 6–12 h at 4°C. Desirable NGS platform-specific adaptor sequences and barcodes were introduced by the second-round PCR using platform-specific barcode-bearing primers. The second-round PCR was set up in a 20-µL reaction composed of 25 ng of the purified first-round PCR product, 0.1 µM of each primer (Slx-2nd-Primer and Slx-2nd-Barcode for Illumina; SLD-2nd-Primer and SLD-2nd-Barcode for SOLiD), 0.3 mM dNTP, 1× Phusion HF buffer, and 0.4 units Phusion high-fidelity DNA polymerase. Seven cycles of the PCR profile as described above were performed. PCR products from two independent amplifications were combined and purified using QIAquick PCR purification kit (Qiagen). The prepared libraries using column-synthesized probes were subjected to Illumina HiSeq 2000 sequencing (PE100 for SNPs, microsatellites, and indels) and SOLiD4 sequencing (SE50 for SNPs). The 12,472-plex SNP library was subject to Illumina HiSeq 2500 sequencing (SE50). All primer and adaptor sequences used in the HD-Marker library preparation are provided in Supplemental Table S18.
Data processing and analysis
Illumina and SOLiD raw reads were first preprocessed to remove any sequences with ambiguous base calls (N), long homopolymer regions (>10 bp), or excessive low-quality positions (>20% of positions with quality score less than 10). Besides, for SOLiD sequencing reads, the terminal 15-bp positions were trimmed from each read to eliminate low-quality positions that might interfere with accurate mapping. The trimmed, high-quality reads formed the basis for all subsequent mapping and genotyping.
Illumina high-quality reads were aligned to the reference target sequences (i.e., a set of roughly 50-bp sequences surrounding the target loci) by using BWA (Li and Durbin 2009).The output alignment files were sorted and converted into mpileup files using the SAMtools pipeline (Li et al. 2009a) for subsequent analysis. The detection of a locus requires the support of at least one read, and the high reliability is ensured even with a single read, because the correct construct is unlikely formed if any step of probe hybridization, extension, and ligation is wrongly performed. VarScan (Koboldt et al. 2009) was used to genotype SNP and indel markers with parameters “--min-coverage 8 --min-reads2 2 --min-var-freq 0.01 --min-freq-for-hom 0.99 --p-value 99e-2”. Genotypes in positions with coverage less than three were considered as a missing genotype. HipSTR (Willems et al. 2017) was used to genotype microsatellite with def-stutter-model and at least five reads were required to genotype a locus. RMSE and R2 values were calculated for quantitatively evaluating the concordance between replicative assays.
High-quality reads generated from SOLiD platform were mapped to the reference target sequences in color space (gmapper-cs) using the SHRiMP software package (Rumble et al. 2009). Read mapping adopted stringent parameters for enhanced specificity, with a spaced seed of 111100111, penalties for mismatches, and gap opening (-i -9 -g -250-q -25); penalties for crossover and gap extension (-x -35 -e -10 -f -10); and Smith-Waterman thresholds for the full and vector searches (-h 260 -v 100). The resulting matches were filtered to eliminate statistically weak matches (P > 0.001) and ambiguous matches (reads matching more than one site equally well) using the probcalc program (-n 0.8 -p 0.001). Final alignments were produced for each read and its matching reference site using prettyprint and prettyprint-cs programs. To account for relatively low quality of SOLiD reads, genotypes were determined from the sequence alignments using a stringent maximum likelihood approach, for which posterior probability was calculated for two possible genotypes (i.e., homozygote or heterozygote) at a given locus, and then a likelihood ratio test was performed to determine the most likely genotype. A coverage threshold (≥20× for a site and ≥10× for a minor allele) was applied to eliminate low-coverage sites for which homozygote and heterozygote could not be reliably called. In addition, to ensure the confidence of allele assignment, any loci with alleles that conflicted with known polymorphisms were considered as undetermined (no genotype assigned).
Genotype validation
In total, 173 SNPs were randomly chosen for validation by amplicon-based Sanger sequencing by basically following the procedure described in a previous study (Fu et al. 2013). All primer sequences that amplified ∼100–200 bp fragments flanking each target loci are provided in Supplemental Table S19.
To validate the genotyping data obtained from the 12,472-plex assay, genome resequencing was conducted using the same individual. Whole-genome shotgun libraries were constructed in duplicate using the Next-Ultra DNA Library Prep Kit for Illumina (NEB) by following the manufacturer's recommendations. Sequencing was performed on the Illumina HiSeq X Ten platform with a total coverage of about 35×. Resequencing data were aligned to the P. yessoensis reference genome (GenBank accession no.GCA_002113885.2) using BWA (Li and Durbin 2009). SNPs were genotyped using VarScan with parameters “--min-coverage 3 --min-reads2 1 --min-var-freq 0.01 --min-freq-for-hom 0.99 --p-value 99e-2”. The consistent genotypes from replicate libraries were used for validation of HD-Marker genotypes.
Validation of microsatellite genotypes was conducted by basically following a fluorescence-based capillary electrophoresis protocol of Schuelke (2000). Primer pairs for 10 microsatellite markers were shown in Supplemental Table S20. The fluorescence-labeled PCR products were subject to the fragment analysis on an ABI 3730XL genetic analyzer (Applied Biosystems) by following the manufacturer's instructions.
Data access
The sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession numbers SRP115866, SRP115869–SRP115871, and SRP115873.
Competing interest statement
The authors filed a patent application based on the described work.
Acknowledgments
We thank Thomas Willems (MIT, Cambridge) for help on microsatellite genotyping using HipSTR software. We acknowledge the grant support from the National Key Research and Development Program of China (2018YFC0310802), the National Natural Science Foundation of China (U1706203), the major basic research projects of Shandong Natural Science Foundation (ZR2018ZA0748), the Blue Life Breakthrough Program of LMBB (MS2018NO01) and AoShan Talents Program (2015ASTP-ES02) of Qingdao National Laboratory for Marine Science and Technology, and the Fundamental Research Funds for the Central Universities (201762001, 201841001).
Author contributions: S.W. and Z.B. conceived and designed the study; J.L., W.J., H.G., and P.L. performed the experiments; J.L., W.J., S.W., P.L., R.W., and Q.Z. participated in data analysis; S.W., J.L., L.Z., X.H., and Z.B. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.235820.118.
-
Freely available online through the Genome Research Open Access option.
- Received March 11, 2018.
- Accepted October 25, 2018.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








