
Increased taxon sampling reveals thousands of hidden orthologs in flatworms
- 1Sars International Centre for Marine Molecular Biology, University of Bergen, Bergen 5006, Norway;
- 2Whitney Laboratory for Marine Bioscience, University of Florida, St. Augustine, Florida 32080, USA
-
↵3 These authors contributed equally to this work.
Abstract
Gains and losses shape the gene complement of animal lineages and are a fundamental aspect of genomic evolution. Acquiring a comprehensive view of the evolution of gene repertoires is limited by the intrinsic limitations of common sequence similarity searches and available databases. Thus, a subset of the gene complement of an organism consists of hidden orthologs, i.e., those with no apparent homology to sequenced animal lineages—mistakenly considered new genes—but actually representing rapidly evolving orthologs or undetected paralogs. Here, we describe Leapfrog, a simple automated BLAST pipeline that leverages increased taxon sampling to overcome long evolutionary distances and identify putative hidden orthologs in large transcriptomic databases by transitive homology. As a case study, we used 35 transcriptomes of 29 flatworm lineages to recover 3427 putative hidden orthologs, some unidentified by OrthoFinder and HaMStR, two common orthogroup inference algorithms. Unexpectedly, we do not observe a correlation between the number of putative hidden orthologs in a lineage and its “average” evolutionary rate. Hidden orthologs do not show unusual sequence composition biases that might account for systematic errors in sequence similarity searches. Instead, gene duplication with divergence of one paralog and weak positive selection appear to underlie hidden orthology in Platyhelminthes. By using Leapfrog, we identify key centrosome-related genes and homeodomain classes previously reported as absent in free-living flatworms, e.g., planarians. Altogether, our findings demonstrate that hidden orthologs comprise a significant proportion of the gene repertoire in flatworms, qualifying the impact of gene losses and gains in gene complement evolution.
Understanding the dynamic evolution of gene repertoires is often hampered by the difficulties of confidently identifying orthologous genes, i.e., those genes in different species originated by speciation (Fitch 1970). Gene annotation pipelines and large-scale comparisons largely rely on sequence-similarity approaches for gene orthology assignment (Albà and Castresana 2007; Domazet-Lošo et al. 2007; Tautz and Domazet-Lošo 2011; Yandell and Ence 2012). These approaches depend on taxonomic coverage and the completeness of the gene databases used for comparisons. Although extremely useful in many contexts, sequence-similarity methods, such as Basic Local Alignment Search Tool (BLAST) (Altschul et al. 1990), can be confounded in situations in which a gene evolves fast, is short, has an abundance of indels, and/or exhibits similarity with other gene family members in only a small subset of residues (Moyers and Zhang 2015). These limitations, as well as intrinsic systematic errors of most algorithms (Liebeskind et al. 2016), can generate significant biases when studying the evolution of protein-coding gene families (Elhaik et al. 2006; Moyers and Zhang 2015, 2016). Accordingly, a proportion of the gene complement of an organism will be represented by genes that lack obvious affinity with orthologs in the gene sets of the best annotated genomes—thus mistakenly considered potential “taxonomically restricted,” or “orphan,” genes (Khalturin et al. 2009; Tautz and Domazet-Lošo 2011)—but actually representing fast evolving orthologs that we call hidden orthologs. This issue can potentially be overcome by more sensitive, although computationally intense, detection methods (e.g., profile HMMs, PSI-BLAST) (Altschul et al. 1997; Eddy 2011; Kuchibhatla et al. 2014), but also by increasing taxon sampling, which helps to bridge the long evolutionary gaps between hidden orthologs and their well-annotated, more conservative counterparts, e.g., via transitive homology (Fig. 1A; Pearson 1996; Yona et al. 1998).
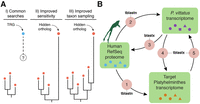
Hidden orthologs and the Leapfrog pipeline. (A) Taxonomically restricted genes (TRGs) are genes with no clear orthology relationship (dashed line and question mark) to other known genes (e.g., orthology group of red dots). Improved sensitivity in the detection methods and/or improved taxon sampling can help uncover hidden orthology relationships, thus referring to these former TRGs as hidden orthologs. (B) The Leapfrog pipeline performs a series of reciprocal BLAST searches between an initial well-annotated data set (e.g., human RefSeq), and a target and a “bridge” transcriptome. First, Leapfrog performs BLAST against the human RefSeq and the target (1) and the “bridge” transcriptome (2) and identifies reciprocal best-hit orthologs between the “bridge” and the human RefSeq proteins (3). These annotated genes of the “bridge” are then used to find orthologs in the target transcriptomes by reciprocal best BLAST hits (4 and 5). If these two pairs of reciprocal best BLAST hit searches are consistent between them, the gene in the target transcriptome is deemed a hidden ortholog. Colored shapes within green boxes represent different sequences of each data set.
Platyhelminthes (flatworms) exhibit significantly high rates of molecular evolution (Edgecombe et al. 2011; Struck et al. 2014; Laumer et al. 2015a). Gene loss and orphan genes have been attributed to evolutionary changes leading to Platyhelminthes morphology (Berriman et al. 2009; Martín-Durán and Romero 2011; Riddiford and Olson 2011; Tsai et al. 2013; Breugelmans et al. 2015). A prime example is the loss of centrosomes in planarian flatworms, where the apparent absence of genes critical to the functioning of animal centrosomes was used as evidence supporting the secondary loss of these organelles in Platyhelminthes (Azimzadeh et al. 2012). Recently, two phylogenomic analyses have provided an extensive transcriptomic data set for most platyhelminth lineages, in particular for those uncommon and less-studied taxa that otherwise occupy key positions in the internal relationships of this group (Egger et al. 2015; Laumer et al. 2015b). These important resources provide an ideal opportunity to address how increasing taxon sampling may improve the resolution of gene complement evolution in a fast evolving—and thus more prone to systematic error—animal group.
Results
The Leapfrog pipeline identifies hundreds of hidden orthologs in flatworms
To identify hidden orthologs in large transcriptomic data sets, we created Leapfrog, a simple pipeline that automates a series of BLAST-centric processes (Fig. 1B; Methods). We assembled a data set including 35 publicly available transcriptomes from 29 flatworm species and incorporated the transcriptomes of the gastrotrich Lepidodermella squamata, the rotifer Lepadella patella, and the gnathostomulid Austrognathia sp. as closely related outgroup taxa (Supplemental Table S1; Struck et al. 2014; Laumer et al. 2015a). Using a single-isoform version of the human RefSeq protein data set as initial queries, Leapfrog identified a total of 3427 putative hidden orthologs, 1217 of which were unique and 636 were species-specific (Fig. 2A,B; Supplemental Table S2). In 30 cases (<1% of the total recovered hidden orthologs), Leapfrog associated two or more human proteins, likely paralogs, ohnologs, or inparalogs, with the same hidden ortholog and “bridge” contig (Supplemental Table S3). Alignments of recovered hidden orthologs with their human and P. vittatus counterparts show that many amino acid positions that differ between the human and the hidden ortholog products are conserved between P. vittatus and one or the other sequences (e.g., Fig. 2C).
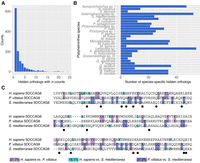
The Leapfrog pipeline recovers hundreds of hidden orthologs in Platyhelminthes. (A) Distribution of hidden orthologs according to their identification in one or more of the analyzed transcriptomes. Most of the hidden orthologs are unique to each lineage. (B) Distribution of species-specific hidden orthologs in each studied species. (C) Amino acid alignment of a fragment of the centrosomal protein SDCCAG8 of H. sapiens, P. vittatus, and S. mediterranea, and pairwise comparison of conserved residues. Positions that differ between the human and the hidden ortholog products are conserved between P. vittatus and one or the other sequences. Black dots indicate residues conserved among the three species.
The number of putative hidden orthologs recovered in each particular lineage ranged from 41 in the rhabdocoel Provortex sphagnorum to 198 in the planarian S. mediterranea and varied considerably between different species belonging to the same group of flatworms (Supplemental Fig. S1). However, completeness of each transcriptome and sequencing depth were also very irregular (Supplemental Fig. S1; Supplemental Table S1), suggesting that the number of putative hidden orthologs we recovered with Leapfrog is sensitive to the quality of the transcriptomes, perhaps influenced by the source tissue(s) used for transcriptome sequencing.
Accuracy of Leapfrog and impact of the ‘bridge’ transcriptome
In order to ensure that domain shuffling was not a source of false positives, we analyzed the domain content of 130 “bridge” proteins and their corresponding human sequences from the S. mediterranea analysis (Supplemental Table S4). In most cases (112; 86.15%), human and “bridge” proteins had identical domain content. In the other 18 cases, the “bridge” ortholog had a domain not reported in the human protein or vice versa. Nonetheless, many of these may be due to InterProScan 5 (Jones et al. 2014) sensitivity. For example, in the case of BATF2, InterProScan 5 identified the bZIP domain bZIP_1 (PF00170) in the human BATF2 and bZIP_2 (PF07716) in the “bridge” sequence; however, bZIP_1 and bZIP_2 are very similar, and both domains are recovered in an identical region of BATF2. In general, there was agreement between the domain architecture of the human and bridge sequences, and we found no evidence suggesting that these results were inflated due to homologous overextension (Gonzalez and Pearson 2010).
To recover even more putative hidden orthologs in our data set, we implemented an iterative approach that used each flatworm transcriptome with ≥85% CEGs as “bridge” and “target” in Leapfrog. This approach increased the number of recovered hidden orthologs 3.4 times (4216 versus 1240) with respect to the original Leapfrog with a constant, single “bridge” lineage (Supplemental Table S5). Each hidden ortholog was identified on average by 2.11 different “bridges,” which suggests that each “bridge” transcriptome is only suitable to retrieve by transitive homology a subset of the total amount of hidden orthologs present in a given “target” lineage. Noticeably, the “bridge” M. lineare, the flatworm species with the shortest evolutionary distance (Laumer et al. 2015b), recovered less hidden orthologs than P. vittatus used as “bridge,” suggesting that evolutionary rate is not necessarily the best criteria for choosing a “bridge” lineage.
Leapfrog identifies orthologs not detected by OrthoFinder and HaMStR
In order to be certain that we were identifying orthologs that would not be detected in a typical analysis, we compared our pipeline with the commonly deployed orthogroup inference algorithm OrthoFinder (Emms and Kelly 2015). To do this, we first performed an OrthoFinder analysis on human and the planarian S. mediterranea, and then we evaluated the impact of adding the “bridge” species P. vittatus to the calculation of orthogroups. Initially, OrthoFinder identified 5638 orthogroups containing at least one sequence of H. sapiens and S. mediterranea (Supplemental Fig. S2). The inclusion of the translated transcripts of P. vittatus led to an increase in orthogroups that included both human and planarian sequences (5816) (Supplemental Fig. S2). However, OrthoFinder only recovered 82.7% (62/75) of the putative hidden orthologs identified by the single “bridge” Leapfrog pipeline in the same S. mediterranea transcriptome (Brandl et al. 2016) with a similar E-value cutoff. Remarkably, the inclusion of the entire data set in OrthoFinder reduced the number of orthogroups shared by human and S. mediterranea (5213 orthogroups) and retrieved fewer putative hidden orthologs (56 of 75; 74.67%).
We also performed a HaMStR analysis (Ebersberger et al. 2009) by using S. mediterranea with default parameters, and we evaluated how many of the Leapfrog putative hidden orthologs it was able to recover. In the initial HMM step, only two of the 75 S. mediterranea Leapfrog hidden orthologs were present in the HMM outputs above the threshold, with an additional two below the threshold. Therefore, HaMStR is also not as sensitive to hidden orthologs as Leapfrog.
The number of hidden orthologs does not relate to the branch length of each lineage
To investigate the parameters that might influence the appearance and identification of putative hidden orthologs in our data set, we performed a principal component analysis (PCA) including variables related to the quality and completeness of the transcriptome, the mean base composition of the transcriptome, and the evolutionary rate of each lineage (Fig. 3A; Supplemental Table S6). The first principal component (PC1) was strongly influenced by the quality of the transcriptome, whereas the second principal component (PC2) mostly estimated the balance between evolutionary change (branch lengths and hidden orthologs) and transcriptome complexity (GC content). The two first principal components explained 67% of the variance of the data set, indicating that additional interactions between the variables exist. For example, the GC content can affect sequencing performance (Dohm et al. 2008; Benjamini and Speed 2012), and thus transcriptome quality and assembly.
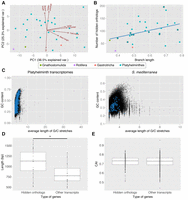
Hidden orthologs, evolutionary rates, and sequence composition analyses. (A) Principal component analysis of the analyzed data showing the eigenvectors for each variable. The first two principal components (PC1, PC2) explain together 67.6% of the observed variability. (B) Number of hidden orthologs in relation to the branch length of each lineage (linear regression in blue; dots with external black line indicate the taxa with highly complete transcriptomes). There is a low correlation between the two variables (R2= 0.124). (C) GC content of each transcript plotted against its average length of G/C stretches considering all studied flatworm transcriptomes (left) and only S. mediterranea (right). The transcripts corresponding to hidden orthologs are in blue. Hidden orthologs do not differentiate from the majority of transcripts. (D) Average length of hidden orthologs compared to the average length of the other genes in transcriptomes with ≥85% CEGs. Hidden orthologs are significantly longer than the rest (Mann-Whitney U test; P < 0.05). (E) Codon adaptation index (CAI) of the hidden orthologs of the planarian species B. candida, D. tigrina, and S. mediterranea compared with nonhidden orthologs. CAI index in hidden orthologs does not significantly differ from the rest of transcripts (Mann-Whitney U test; P < 0.05).
Despite the fact that the branch length of a given lineage and the number of putative hidden orthologs affected the dispersion of our data in a roughly similar manner, we did not detect a linear correlation (R2 = 0.131, P-value = 0.053) (Fig. 3B) between these two variables, not even when we considered those transcriptomes with similar completeness (≥85% CEGs identified; R2 = 0.331, P-value = 0.04). This result supported our previous observation that lineages with similar branch lengths could exhibit remarkably different sets of hidden orthologs (Supplemental Fig. S1).
Flatworm hidden orthologs do not show sequence composition biases
A recent report showed that very high GC content and long G/C stretches characterize genes mistakenly assigned as lost in bird genomes (Hron et al. 2015). In flatworms, however, hidden orthologs do not show a significantly different GC content and average length of G/C stretches than the majority of transcripts (Fig. 3C). We confirmed this observation for each particular transcriptome of our data set (Supplemental Fig. S3).
Systematic error in sequence-similarity searches is also associated with the length of the sequence and the presence of short conserved stretches (i.e., protein domains with only a reduced number of conserved residues). Short protein lengths decrease BLAST sensitivity (Moyers and Zhang 2015). We thus expected hidden orthologs to consist of significantly shorter proteins, as is seen in Drosophila orphan genes (Palmieri et al. 2014). When analyzed together, the length of the flatworm hidden ortholog transcripts is not significantly different from that of the rest of the transcripts (Supplemental Table S7). However, the putative hidden orthologs are significantly longer than the rest of the transcripts when only high-coverage transcriptomes (≥85% CEGs identified) are considered (Fig. 3D).
We next addressed whether the 1243 nonredundant human proteins homologous to the flatworm hidden orthologs were enriched in particular sequence motifs that could hamper their identification by common sequence similarity searches. We recovered a total of 1180 unique PFAM annotations, almost all of them present only in one (1016) or two (112) of the identified hidden orthologs (Supplemental Table S8). The most abundant PFAM domain (Table 1) was the pleckstrin homology (PH) domain (PFAM ID: PF00169), which occurs in a wide range of proteins involved in intracellular signaling and cytoskeleton (Scheffzek and Welti 2012). PH domains were present in 11 of the candidate hidden orthologs. Most other abundant domains were related to protein interactions, such as the F-box-like domain (Kipreos and Pagano 2000), the forkhead-associated domain (Durocher and Jackson 2002), and the zinc-finger of C2H2 type (Iuchi 2001). These more abundant domains vary significantly in average length and number of generally conserved sites (Table 1).
Most represented PFAM domains in flatworm hidden orthologs
Last, we looked to see if there were any patterns of codon usage associated with the putative hidden orthologs. We did not observe a statistically significant difference between the codon adaptation index of hidden orthologs of the planarian species B. candida, D. tigrina, and S. mediterranea and other open reading frames of these transcriptomes (Fig. 3E). Altogether, these analyses indicate that hidden orthologs do not show intrinsic properties that could cause systematic errors during homology searches.
The possible mechanisms driving hidden orthology in Platyhelminthes
To assess the contribution of duplication and divergence (Force et al. 1999) toward the generation of hidden orthologs, we looked for paralogs of the putative hidden orthologs in OrthoFinder orthogroups. Focusing on Tricladida (planarian flatworms), we observed that a putative hidden ortholog co-occurred in the same orthogroup with one or more sequences from the same species in 14%–30% of the cases (depending on the species) (Fig. 4A). These hidden orthologs can be interpreted as fast-evolving paralogs. For those one-to-one hidden orthologs of S. mediterranea, we calculated the number of nonsynonymous substitutions per nonsynonymous site (Ka) and the number of synonymous substitutions per synonymous site (Ks) in pairwise comparisons with their respective ortholog in the “bridge” transcriptome (Fig. 4B). Although for almost half of them the Ks value appeared to be saturated (Ks > 2), the Ka/Ks ratio for most of the rest was above or close to 0.5, which is often interpreted as a sign of weak positive selection or relaxed constraint (Nachman 2006).
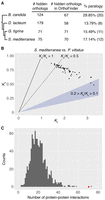
Level of paralogy and Ka/Ks values in triclad hidden orthologs. (A) Percentage of hidden orthologs identified by Leapfrog that are present in OrthoFinder and share an orthogroup with other sequences of the same species. We deem these cases as probable fast evolving paralogs (hidden paralogy). (B) Ka and Ks values of 53 one-to-one hidden orthologs of S. mediterranea compared with their respective homologs in the “bridge” species P. vittatus. Although in almost half of these hidden orthologs the Ks value suggested saturation (Ks > 2), for most of the rest the Ka/Ks value was above or around 0.5 (dotted line), which can be a sign of weak positive selection or relaxed constraint. (C) Number of predicted protein–protein interactions in S. mediterranea hidden orthologs (red dot) compared with a distribution of interactions observed in 1000 random samples of similar size (gray bars). Hidden orthologs show a significantly higher number of interactions, suggesting that complementary mutations between protein partners might drive hidden orthology in flatworms.
A gene ontology (GO) analysis of the nonredundant hidden orthologs identified in all flatworm transcriptomes and the planarian S. mediterranea revealed a wide spectrum of GO terms, with binding and catalytic activities being the most abundant (Supplemental Fig. S4). The statistical comparison of the GO categories of the putative S. mediterranea hidden orthologs revealed 248 significantly (P < 0.05) enriched GO terms (Supplemental Table S9). Interestingly, the putative hidden orthologs were enriched for biological processes and cellular compartments related to mitochondrial protein translation and the mitochondrial ribosome, respectively. Indeed, ribosomal proteins were among the most common hidden orthologs recovered from our data set (Supplemental Table S2). These findings suggest that mitochondrial genes show accelerated evolutionary rates (Solà et al. 2015), which might be causing nuclear-encoded proteins that are exported to the mitochondrion to adapt to this change (Barreto and Burton 2013).
To test if hidden orthologs are a result of rapid compensatory evolution, we investigated whether there were a disproportionate number of interactions between putative hidden orthologs in S. mediterranea. We used the BioGRID database (Chatr-Aryamontri et al. 2015) to count the number of physical interactions between S. mediterranea hidden orthologs. We identified 71 such interactions in S. mediterranea, which is statistically significant when compared with 1000 random sets of human genes (one-tailed Monte Carlo analysis with 100,000 iterations; P-value 1.9 × 10−4) (Fig. 4C). These findings suggest that compensatory mutations in binding partners and/or otherwise interconnected proteins are contributing to the origin of hidden orthologs.
The identified hidden orthologs fill out gaps in the flatworm gene complement
We used an expanded Leapfrog strategy to identify possible hidden orthologs for those centrosomal and cytoskeleton-related genes supposedly lost in the flatworms M. lignano, S. mediterranea, and S. mansoni (Azimzadeh et al. 2012). First, we used a reciprocal best BLAST strategy to identify orthologs of the human centrosomal proteins in each of our transcriptomes under study; thereafter, we used Leapfrog to identify any hidden member of this original gene set. We recovered at least one reciprocal best BLAST hit for 56 of the 61 centrosomal genes and identified fast-evolving putative orthologs in 19 of the 61 centrosomal genes (Fig. 5). In total, the number of hidden orthologs identified was 58 (counting only once those for the same gene in the different analyzed S. mediterranea transcriptomes). Most importantly, we found putative hidden orthologs for the genes CEP192 and SDCCAG8 in the planarian S. mediterranea (Fig. 5; Supplemental Figs. S5, S6), which were two of the five key genes essential for centrosome assembly and duplication (Azimzadeh et al. 2012). Noticeably, the expression patterns of these two genes (Supplemental Fig. S7) differ considerably from those reported for other centrosomal genes in S. mediterranea (Azimzadeh et al. 2012), which suggests that they might not be coexpressed on those planarian cells that assemble centrosomes and thus might have evolved alternative functions.
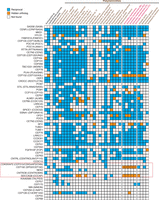
Hidden orthologs in the core set of centrosomal-related proteins. Presence (colored boxes) and absence (empty boxes) of the core set of centrosomal proteins (Azimzadeh et al. 2012) in all analyzed flatworm transcriptomes. Orthologs identified by direct reciprocal best BLAST hit are in blue boxes, and hidden orthologs are in orange. The asterisks indicate the CEP192 protein in the S. mediterranea transcriptomes (pink color code). These proteins were manually identified with the G. tigrina CEP192 sequence as “bridge” by reciprocal best BLAST hit. The five proteins essential for centrosomal replication are boxed in red.
Using as a “bridge” the orthologs found in the more conservative rhabditophoran species M. lignano and P. vittatus, we found hidden orthologs for dbx, vax, drgx, vsx, and cmp in the planarian S. mediterranea (Table 2; Supplemental Figs. S8, S9), which places the loss of these homeodomain classes most likely at the base of the last-common neodermatan ancestor. Importantly, the Hhex family was present in P. vittatus, but was not identified in M. lignano and S. mediterranea; the Prrx and Shox families were present in M. lignano, but absent from P. vittatus and S. mediterranea transcriptomes. These observations suggest that many of the losses of homeobox genes occurred in the ancestors to the Rhabitophora and Neodermata, with only a few losses of specific gene classes in particular lineages of free-living flatworms.
Presence or absence of hidden homeodomain genes in flatworms
Discussion
Our study implements a simple automated BLAST pipeline that uses increased taxon sampling and transitive homology (Pearson 1996; Yona et al. 1998) to overcome large evolutionary distances and identify putative hidden orthologs (Fig. 2). However, our results on an extensive data set of flatworm transcriptomes are likely an underestimation of the true number of hidden orthologs. First, we based our identification of hidden orthologs on reciprocal best BLAST hits, a valid and widely used approach (Tatusov et al. 1997; Overbeek et al. 1999; Wolf and Koonin 2012), but limited (Fulton et al. 2006; Dalquen and Dessimoz 2013). Second, an iterative Leapfrog recovers many more putative hidden orthologs (Supplemental Table S5). This indicates that there might be natural circumstances (e.g., presence of hidden orthologs and missing genes), even in more conservatively evolving lineages, which contribute to the suitability of a particular transcriptome to act as a “bridge.” Furthermore, we demonstrate that using hidden orthologs themselves as “bridge queries” on other lineages can help recover even more new hidden orthologs (Table 2). Finally, 16 of the 35 analyzed transcriptomes contain <80% of core eukaryotic genes (Supplemental Fig. S1) and can be regarded as fairly incomplete (Parra et al. 2009).
In our data set, putative hidden orthologs are not significantly shorter and do not exhibit either particular sequence composition biases (Fig. 3) or protein domains (Table 1) that could account for the difficulties in being detected by standard homology searches. Instead, hidden orthologs represent restricted fast evolving orthologs, which have been driven by either gene duplication events, weak positive selection and relaxed constraint, and/or by compensatory mutations between protein partners (Fig. 4). As observed in other organisms, whole or partial genomic duplications (Van de Peer et al. 2009a,b) and transposable elements (Feschotte 2008), which are common in flatworms (Benazzi and Benazzi-Lentati 1976; Garcia-Fernández et al. 1995; Sperb et al. 2009), could also contribute to generate sequence diversity, and thus hidden orthology, in Platyhelminthes. In some cases, however, hidden orthologs are apparently associated with divergent biological features of Platyhelminthes (Fig. 5; Table 2). The fact that most of them are species-specific indicates that the gene complement of an organism is in fact heterogeneous, composed of genes evolving at different evolutionary rates (Wolfe 2004), sometimes much higher or much lower than the “average” exhibited by that lineage.
Although orthology does not strictly imply functional conservation (Fitch 1970; Gabaldon and Koonin 2013), the “orthology conjecture” states that orthologous genes show a higher functional similarity than paralogs (Koonin 2005; Dolinski and Botstein 2007; Studer and Robinson-Rechavi 2009; Kryuchkova-Mostacci and Robinson-Rechavi 2016). Since only 14%–30% of the putative hidden orthologs identified in Tricladida are fast evolving paralogs (Fig. 4A), the orthology conjecture would imply that most of the putative hidden orthologs identified are indeed functionally conserved. In this regard, hidden orthologs are often protein partners (Fig. 4C), suggesting that compensatory mutations to maintain interaction, and thus probably functionality, could have promoted sequence divergence in some cases. However, a more detailed analysis of the two centrosomal hidden orthologs SDCCAG8 and CEP192 shows remarkably different expression patterns in the planarian S. mediterranea, making it unlikely that they still cooperate to assemble the centrosomes. This indicates that unraveling whether hidden genes are maintaining ancestral functions despite very high mutation rates, or are abandoning highly conserved ancestral functions but continuing to contribute to the biology of the organism, requires detailed individual analyses of each of the putative hidden orthologs.
The recovered hidden orthologs have an immediate impact on our understanding of gene complement evolution in Platyhelminthes, particularly those lineages that are the subject of intense research, such as the regenerative model Schmidtea mediterranea and parasitic flatworms (Berriman et al. 2009; Wang et al. 2011; Olson et al. 2012; Sánchez Alvarado 2012). The identification of fast-evolving orthologs for important centrosomal proteins in S. mediterranea and other flatworms lineages (Fig. 5) indicates that the evolutionary events leading to the loss of centrosomes are probably more complex than initially surmised (Azimzadeh et al. 2012). Similarly, the presence of presumably lost homeobox classes in S. mediterranea may affect our current view of gene loss and morphological evolution in flatworms (Tsai et al. 2013). The use of more molecularly conserved flatworm lineages, such as P. vittatus, can improve the identification of candidate genes, as well as help with the annotation of the increasingly abundant flatworm RNA-seq and genomic data sets (Berriman et al. 2009; Wang et al. 2011; Tsai et al. 2013; Robb et al. 2015; Wasik et al. 2015; Brandl et al. 2016). Therefore, we have now made available an assembled version of those highly complete nonplanarian flatworm transcriptomes in PlanMine, an integrated web resource of transcriptomic data for planarian researchers (Brandl et al. 2016). Importantly, the Leapfrog pipeline can also be exported to any set of transcriptomes/predicted proteins.
Methods
Leapfrog pipeline
We assembled a data set including 35 transcriptomes of 29 flatworm species, three outgroup taxa, and a single-isoform version of the human RefSeq proteome (for further details, see Supplemental Methods). Transcriptome completeness and quality analyses were performed with CEGMA (Parra et al. 2007) and TransRate (Supplemental Methods; Smith-Unna et al. 2015). All BLAST searches were conducted using BLAST+ version 2.2.31 (Camacho et al. 2009) using multiple threads (from two to 10 per BLAST). We first ran a TBLASTN search using the proteome HumRef2015 as a query against the untranslated Prostheceraeus vittatus transcriptome (tblastn -query HumRef2015 -db Pvit -outfmt 6 -out Hs_v_Pv). We next ran a BLASTX search using the Prostheceraeus vittatus transcriptome (DNA) as a query against the HumRef2015 protein data set (blastx -query Pvit -db HumRef2015 -outfmt 6 -out Pv_v_Hs). We ran a series of TBLASTX searches using the Prostheceraeus vittatus transcriptome (DNA) as a query against each of our target untranslated transcriptome database (e.g., tblastx -query “TRANSCRIPTOME” -db Pvit -outfmt 6 -out “TRANSCRIPTOME”_v_Pvit). Last, we ran a series of TBLASTX searches using our transcriptome databases (DNA) as queries against the untranslated Prostheceraeus vittatus transcriptome (e.g., tblastx -query Pvit -db Sman -out Pvit_v_Sman -outfmt 6). The tab-delimited BLAST outputs generated above were used as input to the Leapfrog program (https://github.com/josephryan/leapfrog) (Supplemental File 1). The default E-value cutoff (0.01) was used for all Leapfrog runs. The Leapfrog program identifies HumRef2015 proteins that fit the following criteria: (1) they have no hit to a target flatworm transcriptome; (2) they have a reciprocal best BLAST hit with a “bridge” (e.g., Prostheceraeus vittatus) transcript; and (3) the “bridge” transcript has a reciprocal best BLAST hit to the target flatworm transcriptome. The output includes the HumRef2015 Gene ID, the Prostheceraeus vittatus transcript, and the target flatworm transcript. The annotation of the hidden orthologs is provided in Supplemental Table S2.
OrthoFinder and HaMStR analyses
Single best candidate coding regions of all flatworm transcriptomes were predicted with TransDecoder v3.0.0 (Haas et al. 2013). The resulting flatworm proteomes, together with HumRef2015, were used to identify orthologous groups with OrthoFinder v0.7.1 (Emms and Kelly 2015). We ran HaMStR version 13.2.6 (Ebersberger et al. 2009) on S. mediterranea using the following command: (hamstr -sequence_file=Smed.dd.mod -taxon=Smed -hmmset=modelorganisms_hmmer3 -refspec=DROME -central). We used a Perl script to compare the list of putative hidden orthologs from Leapfrog to the HMM outputs of HaMStR (Supplemental File 1).
GC content, sequence length, CAI index, interaction analyses, and Ka/Ks values
Custom-made scripts were used to calculate the GC content of hidden orthologs and transcripts of our data set, the average length of the G/C stretches of each sequence, and the length of hidden orthologs and other transcripts (Supplemental File 1). We used the Codon Usage Database (Nakamura et al. 2000) and CAIcal server (Puigbò et al. 2008) to calculate “codon adaptation indexes” in hidden orthologs and three sets of transcripts of same size, randomly generated. Alignments of 53 one-to-one orthologs between S. mediterranea and P. vittatus were calculated with MAFFT v.5 (Katoh and Standley 2013), and PAL2NAL (Suyama et al. 2006) was used to infer codon alignments and trim gap regions and stop codons. Ka and Ks values were calculated with KaKs_Calculator 2.0 (Wang et al. 2010) with the default model averaging (MA) method. A custom Perl script identified physical interactions in S. mediterranea hidden orthologs. The same script built 1000 random gene sets (sampling genes from HumRef2015 that were current in Entrez Gene as of August 2016) and determined the number of physical interactions in each of these. The complete analysis can be repeated by running the biogrid.pl script (Supplemental File 1). All values were plotted in R (R Core Team 2015) using the ggplot2 package (Wickham 2009).
GO and InterPro analyses
All GO analyses were performed using the free version of Blast2GO v3 (Conesa et al. 2005). Charts were done with a cutoff value of 30 GO nodes for the analyses of all hidden orthologs, and 10 GO nodes for the analyses of S. mediterranea hidden orthologs. Resulting charts were edited in Illustrator CS6 (Adobe). InterProScan 5 (Jones et al. 2014) was used to analyze protein domain architectures.
Orthology assignment and gene expression analyses
Protein alignments (available in Supplemental File 2) were performed with MAFFT v.5 (Katoh and Standley 2013) using the G-INS-i option, and spuriously aligned regions were removed with gblocks 3 (Talavera and Castresana 2007). Orthology assignments were performed with RAxML v8.2.6 (Stamatakis 2014) with the autoMRE option. The genes SDCCAG8 and CEP192 were cloned using gene-specific primers (Supplemental Methods) on cDNA obtained from adult planarians and mixed regenerative stages. Whole-mount in situ hybridization was performed as described previously (Umesono et al. 1997; Agata et al. 1998). Representative specimens were cleared in 70% glycerol and imaged under a Discovery.V8 SteREO microscope equipped with an AxioCam MRc camera (Zeiss).
Software availability
The Leapfrog pipeline and all scripts used in this study can be found in Supplemental File 1. In addition, they are available in https://github.com/josephryan/leapfrog and https://github.com/josephryan/reduce_refseq.
Data access
Raw RNA-seq data produced in this study have been submitted to the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra/) under accession number SRX1343824.
Acknowledgments
We thank the members of the Hejnol's laboratory for support and discussions, particularly, Daniel Thiel and Anlaug Furu for taking care of the M. lignano and S. mediterranea cultures. We thank Gonzalo Giribet and Chris Laumer for the advance access given to platyhelmith transcriptomes at the beginning of the project. We thank Axios Review (https://axiosreview.org) editor, Titus Brown, and the six anonymous referees of this manuscript for their efficient and insightful reviewing. We also thank Francesc Cebrià and Susanna Fraguas for kindly providing S. mediterranea animals and cDNA, as well as Marta Iglesias for helping with planarian fixation and in situ hybridization. This research was funded by the Sars Centre core budget and the European Research Council Community's Framework Program Horizon 2020 (2014–2020) ERC grant agreement 648861 to A.H. J.F.R. was supported by startup funds from the University of Florida DSP Research Strategic Initiatives #00114464 and the University of Florida Office of the Provost Programs; J.M.M.D. was supported by the European Commission, Marie Curie IEF 329024 fellowship.
Author contributions: J.M.M.D. and J.F.R. designed the study. J.M.M.D., A.H., and K.P. collected material for the transcriptomes of M. lignano, P. vittatus, and L. squamata. J.F.R. wrote the code of Leapfrog. J.M.M.D., J.F.R., and B.C.V. performed the analyses. J.F.R., J.M.M.D., and A.H. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.216226.116.
-
Freely available online through the Genome Research Open Access option.
- Received September 30, 2016.
- Accepted April 10, 2017.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








