
Nanopore sequencing of complex genomic rearrangements in yeast reveals mechanisms of repeat-mediated double-strand break repair
Abstract
Improper DNA double-strand break (DSB) repair results in complex genomic rearrangements (CGRs) in many cancers and various congenital disorders in humans. Trinucleotide repeat sequences, such as (GAA)n repeats in Friedreich's ataxia, (CTG)n repeats in myotonic dystrophy, and (CGG)n repeats in fragile X syndrome, are also subject to double-strand breaks within the repetitive tract followed by DNA repair. Mapping the outcomes of CGRs is important for understanding their causes and potential phenotypic effects. However, high-resolution mapping of CGRs has traditionally been a laborious and highly skilled process. Recent advances in long-read DNA sequencing technologies, specifically Nanopore sequencing, have made possible the rapid identification of CGRs with single base pair resolution. Here, we have used whole-genome Nanopore sequencing to characterize several CGRs that originated from naturally occurring DSBs at (GAA)n microsatellites in Saccharomyces cerevisiae. These data gave us important insights into the mechanisms of DSB repair leading to CGRs.
Complex genomic rearrangements (CGRs) mixing together various genome alterations such as insertions, duplications, deletions, inversions, and translocations, are important contributors to genome variation in human disease. Loss of genes that protect the integrity of the genome in cancerous cells often results in an extreme degree of CGRs (Lee et al. 2016). Another example of CGRs called chromoanasynthesis (Carvalho and Lupski 2016), which combines chromosomal rearrangements with copy-number gains, leads to various severe congenital disorders, including MECP2 duplication syndrome (Carvalho et al. 2011) and Pelizaeus–Merzbacher disease (Beck et al. 2015). Several molecular mechanisms that could account for these CGRs were discussed in the literature. They include FoSTeS (fork stalling and template switching) (Zhang et al. 2009), BIR (break-induced replication) (Costantino et al. 2014), MMBIR (microhomology-mediated break-induced replication) (Zhang et al. 2009; Sakofsky et al. 2015), and others.
It was also noticed that DNA repeats that can form various non-B DNA structures (DNA cruciforms, triplex H-DNA, G4-DNA, etc.) were associated with the locations of break points of such CGRs (Bacolla et al. 2016; Carvalho and Lupski 2016). A particular class of repetitive sequences called trinucleotide repeats was implicated in hereditary human diseases known as repeat-expansion diseases, such as Huntington's disease, fragile X syndrome, and Friedreich's ataxia (Pearson et al. 2005; Mirkin 2007; Orr and Zoghbi 2007). The ability of trinucleotide repeats to form non-B DNA structures was shown to lead to polymerase stalling during DNA replication, transcription, and repair, ultimately resulting in their instability (expansions and contractions of the repeat tract) (Usdin et al. 2015; Neil et al. 2017; Polleys et al. 2017; Polyzos and McMurray 2017). We and others have also shown that trinucleotide repeat can induce mutagenesis at a distance (RIM- repeat induced mutagenesis) and trigger CGRs (Shah et al. 2012; Saini et al. 2013; Tang et al. 2013).
Although understanding the fine structure of CGRs can shed light on the origin and the mechanisms of human diseases, their detection has never been a straightforward affair. Visual analysis of karyotypes is limited to events that are very large, typically involving entire chromosome arms. Fluorescent in situ hybridization (FISH) allows detection of particular sequences that appear in unexpected locations (Aten et al. 2008). In Saccharomyces cerevisiae, the relatively short length of chromosomes allows their separation by size via contour-clamped homogeneous electric field (CHEF) gel electrophoresis (Vollrath and Davis 1987). Combined with Southern blotting, this approach allows estimation of medium to large-scale changes in chromosome size and can indicate whether particular regions have undergone translocation. However, the process is extremely laborious and limited in resolution.
Comparative genomic hybridization (CGH) arrays offer a vast improvement in resolution over visual methods and can detect specific copy number changes. This approach has been used to map structural variation in the human population (Iafrate et al. 2004; Sebat et al. 2004), as well as to uncover specific CGRs in human genomic disorders (Lee et al. 2007; Potocki et al. 2007; Carvalho et al. 2009). However, inversions and translocations do not appear as copy number changes, and extensive follow-up PCR and Sanger sequencing is required to map CGR junctions with base pair specificity. Even then, it is not always possible to map the boundaries of CGRs occurring in repetitive regions.
More recently, whole-genome and exome sequencing has been used to detect structural variation in model systems, human populations, cancer, and other settings (Kidd et al. 2008; Stephens et al. 2009; The 1000 Genomes Project Consortium et al. 2010; Macintyre et al. 2016; Jeffares et al. 2017). Copy number changes are represented by changes in read depth, and the sequences themselves can reveal junctions. However, analysis of CGRs has been hindered by the short sequencing reads that are inherent to the most commonly used sequencing platforms, such as Illumina. Typically <300 bp, relatively few reads will happen to fall on CGR junctions and may not be distinguishable as such if they fall within repetitive elements. Various experimental and computational approaches have been developed to overcome these hurdles to the extent possible, although many limitations remain (Alkan et al. 2011).
The latest developments in CGR detection have involved long-read sequencing technologies. Pacific Biosciences first developed a single-molecule sequencing approach capable of producing reads of >20 kb. This has been used to identify CGRs in patients with Potocki–Lupski syndrome and Pelizaeus–Merzbacher disease (Wang et al. 2015; Zhang et al. 2017). Due to the relatively high cost compared with Illumina sequencing, these studies used targeted sequence-capture approaches to focus on known regions of interest. Whole-genome sequencing has been feasible in S. cerevisiae, allowing detection of structural variation between different strains (Yue et al. 2017). Most recently, Oxford Nanopore Technologies has developed the MinION, a single-molecule sequencing approach in which DNA strands are unwound and passed through a protein pore. The shape of each nucleotide restricts the flow of ions through the pore to a different degree, allowing identification of the bases. Most importantly, there appears to be nearly no limit on the read length, aside from the length of the DNA polymer itself following purification. In practice, reads can reach hundreds of kilobases (Jain et al. 2016). These extremely long reads have already proved useful in genome assembly and structural variation detection (Loman et al. 2015; Jain et al. 2016, 2017; Norris et al. 2016; Debladis et al. 2017; Istace et al. 2017; Jansen et al. 2017).
Here, we decided to explore the potential of Nanopore sequencing as a method for characterizing the DNA repair pathways involved in CGRs caused by unstable microsatellite repeats. Our laboratories have used S. cerevisiae to study the length instability and CGRs caused by (GAA)n repeats, which are responsible for Friedreich's ataxia, as well as interstitial telomeric sequences (ITS) (Shishkin et al. 2009; Shah et al. 2012; Aksenova et al. 2013). Previously, these CGRs were identified using a combination of CGH arrays, CHEF gels, Southern blotting, PCR, and Sanger sequencing (Kim et al. 2008; Shishkin et al. 2009; Aksenova et al. 2013; Tang et al. 2013). It appeared that a number of the events were truly complex, involving various combinations of chromosomal arm inversions, BIR responsible for arm duplications, and/or nonallelic homologous recombination (HR) mediated by microsatellites and transposable elements (Kim et al. 2008; Aksenova et al. 2013). However, these approaches were extremely laborious, limited in resolution, and hindered by the repetitive elements involved. The present study is dedicated to CGRs triggered by (GAA)n repeats. We evaluated whether the ultralong reads of Nanopore sequencing could effectively identify spontaneous (GAA)n-mediated CGRs in a single step. Because of the potential for CGRs involving chromosome-scale changes, we chose a whole-genome sequencing approach, as opposed to targeted sequence capture. Our results demonstrate that Nanopore sequencing is an effective and efficient method of identifying novel CGRs in S. cerevisiae, which provided important insights into the mechanisms of DNA repair.
Results
Initial characterization of CGRs
To generate strains with CGRs, we used a previously characterized selectable system for repeat instability in S. cerevisiae (Shishkin et al. 2009; Shah et al. 2012), in which (GAA)n repeats are located within an intron inside of the counter-selectable marker gene URA3. Selecting for inactivation of the URA3 gene most frequently turns up expansions of the repeat tract, which is the type of mutation most commonly associated with the inheritance of Friedreich's ataxia (Pandolfo 2002). However, the same process also selects for large deletions and CGRs that remove or separate the two halves of the split URA3 gene. Because the selectable cassette is located in a region on Chromosome III (Fig. 1A) that contains essential genes both centromere-proximal and distal to the repeats, this precludes simple chromosomal arm loss, leading to more complex DNA repair events. In this system, probable CGR events are detected by the lack of a PCR product that typically amplifies the repetitive cassette. Twenty-three strains with probable CGRs mediated by (GAA)n repeats in the URA3 cassette were analyzed by CHEF gels combined with Southern hybridization (characteristic results are shown in Supplemental Fig. S1) followed by CGH analysis as previously described (Aksenova et al. 2013). Using this course of analysis, 16 of the strains showed a likely gene conversion event between our UR-(GAA)100-A3 cassette on Chromosome III and the ura3-52 allele, an inactive copy of the URA3 gene remaining on Chromosome V. This appears similar to what was previously observed as a rare event for ITS (Aksenova et al. 2013). The remaining strains showed evidence of more complex rearrangements that were not fully resolvable from the initial analysis.
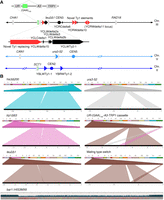
Known deletions and rearrangements in SMY502 versus S288C. (A) Map of Chromosomes II, III, and V, indicating positions of genes, centromeres, and Ty elements. Diagonal lines represent contiguous sequences not in display, such that the displayed portions are pictured to scale. Genes are shown by gray boxes with points indicating the orientation. Centromeres are marked by circles. Ty elements are indicated by triangles, with black indicating Ty elements found in the S288C reference genome, and red indicating previously unannotated Ty elements. The point of the triangle indicates orientation. Small black triangles represent solo LTR sequences, also known as delta elements. Zoomed-in portions of Chromosome III show a cluster of Ty elements, as well as the UR-(GAA)100-A3 selectable cassette. Bright green portions represent the location of (GAA)n repeats. (B) Ribbon single-read views highlighting known large-scale genomic changes in the SMY502 parent strain, mapped to the S288C reference genome. For each panel, the top bar contains a color-coded list of chromosomes, whereas the bottom black bar displays the full sequencing read. Windows connect the portion of the read that maps to the chromosomal position. his3Δ200, trp1Δ63, and leu2Δ1 are 1- to 2-kb deletions and are observed as split reads in which part of the reference sequence (top) is missing from the read (bottom). Ura3-52 is an insertion of an ∼6-kb Ty element, observed as a split read in which sequence not in the reference (top) is found in the read (bottom). Because of high sequence similarity among Ty elements, the insertion is not always associated with a particular part of the reference sequence in each individual read. Our UR-(GAA)100-A3-TRP1 selectable cassette also maps as an insertion, but the 5′ portion of URA3 and the TRP1 gene are both matched to their respective genomic locations in the reference sequence. The difference in mating types between S288C and our strain is also observed as a split read, due to the peculiar control of yeast mating type, in which one of two inactive regions on either end of Chromosome III is copied via HR into the active mating loci, located near the middle of Chromosome III. One allele, bar1::HIS3MX6, was not apparent in the Ribbon analysis, because the BAR1 gene was replaced with a similarly sized marker gene. The HIS3MX6 marker was aligned to the reference with a number of mismatches and short gaps, which was readily apparent in the UGENE alignment. In this view, gray boxes indicate bases that match the reference sequence, and colored boxes indicate bases that do not match the reference sequence: (blue) G; (green) C; (yellow) A; (light red) T; (dark red) deletion. The blue bars above represent read depth at each position.
Nanopore sequencing approach
To unambiguously characterize the observed CGRs, the CGR strains together with our starting strain SMY502 (Shah et al. 2012) were subjected to Nanopore sequencing. DNA from each strain was purified and used to construct barcoded sequencing libraries. The libraries were then pooled and sequenced together on a single flow cell resulting in roughly 30× coverage per strain. Nearly three gigabases of total sequence were generated, largely by reads with a length of 20–30 kb and above (Supplemental Fig. S2).
Genomic alterations in the parent yeast strain
Our parent yeast strain is closely related, but not identical to S288C, the extremely well-characterized laboratory strain used in the initial systematic sequencing of S. cerevisiae. In order to identify CGRs in the Nanopore sequences, it was first necessary to examine the parent strain for changes relative to the S288C reference genome available from the Saccharomyces Genome Database (SGD; https://www.yeastgenome.org). To do this, reads were aligned to S288C and examined for potential structural variants. The alignment/variant-calling approach was chosen, as opposed to genome assembly, because it involved significantly fewer computing resources, and because the S288C genome is extremely well-characterized and closely related to our parent strain. Alignments were visualized using Ribbon, a sequence visualization tool specializing in split reads, or reads that map to multiple genomic locations (Nattestad et al. 2016). In addition, the alignments were visualized using the bioinformatics software UGENE, which can display a pileup of reads for each chromosome (Okonechnikov et al. 2012).
Using this approach, we confirmed the presence of a number of known structural variants in our parents strain, including alterations in selectable marker genes, as well as the insertion of our UR-(GAA)100-A3 selectable cassette, and a mating type switch (Fig. 1B). This demonstrates a high success rate in finding relatively simple structural variations. We also found four unexpected Ty element insertions that were not present in the S288C reference genome, three of which appear on Chromosome III (Fig. 1A; Supplemental Fig. S3).
The reference genome was altered to reflect these observed changes and used as the reference to which the remaining strains were aligned. We discuss here three independent CGR events observed in strains 101, 118, and 105, which were analyzed independently by CHEF/CGH and by Nanopore sequencing.
Strain 101: gene conversion involving Ty retrotransposon elements
The CHEF/CGH analysis identified strain 101 as containing a ura3-52 gene conversion event. Specifically, CHEF analysis showed that strain 101 had only one change: Chromosome III was slightly smaller than observed in the starting strain (Fig. 2). This smaller chromosome hybridized to three probes specific to genes on Chromosome III (CHA1, LEU2, and RAD18) as expected (Figs. 1A, 2). By CGH arrays (Fig. 3A), strain 101 had a deletion with a left endpoint located between SGD coordinates 75,142 and 75,758, which overlaps with the location of the UR-(GAA)100-A3 cassette (replacing SGD coordinates 75,594–75,641). The right end of the deletion had a breakpoint between SGD coordinates 82,646 and 84,263. This region overlaps with a cluster of Ty retrotransposon elements, including an unannotated Ty1 element replacing YCLWdelta15 (SGD coordinates 82,700–83,036) (Supplemental Fig. S3), a Ty2 element (YCLWTy2-1 at SGD coordinates 84,811–90,769), and multiple delta sequences (long terminal repeats [LTRs] left behind by ancestral Ty elements). Note that although strain 101 represents a frequently observed ura3-52 gene conversion event (Supplemental Fig. S1), it is likely that similar CGRs vary in their interactions with the particular Ty elements in this cluster.

CHEF gel analysis. CHEF gel electrophoresis was used to separate whole chromosomes by size. The left panel shows the gel stained with ethidium bromide, displaying all chromosomes. Lane 1 is a size ladder of S. cerevisiae chromosomes. The following lanes contain DNA prepared from the strains as indicated. Black triangles point to chromosomes with an altered size. The four right panels display Southern blot hybridizations using probes to the indicated genes.
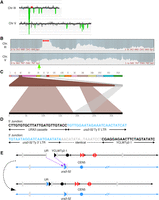
Identifying genomic rearrangements in strain 101. (A) CGH microarray analysis displaying results for Chromosomes III and V. The large green region corresponds to the deletion surrounding the repeats. By examining the hybridization values for individual oligonucleotides on the microarrays, we found that the small red and green regions depicted in this figure do not represent true duplications and deletions, respectively. (B) Nanopore sequencing coverage maps of Chromosomes III and V, generated via UGENE, with a red arrow highlighting the deletion boundaries and a green arrow highlighting the duplication at ura3-52. (C) Ribbon single-read view highlighting a read mapping the entirety of the gene conversion event in which a Ty element was inserted in place of the 3′ half of the UR-(GAA)100-A3 cassette on Chromosome III. (D) Single base pair resolution of the 5′ and 3′ breakpoints of the deletion. The 5′ junction connects the 5′ portion of the UR-(GAA)100-A3 cassette with ura3-52 on Chromosome V. Note that the crossover could have occurred anywhere in the 341 bp of identity between the cassette and ura3-52. The 3′ junction consists of the 3′ LTR region of ura3-52 and YCLWTy2-1 on Chromosome III. The gray region represents an 80-bp window of identity between ura3-52 and YCLWTy2-1 in which the gene conversion occurred. SNPs are visible in the alignment on each side of this window (Supplemental Fig. S4C). (E) Diagram of the CGR event resulting in a gene conversion. Chromosome maps have the same format as in Figure 1A. Relevant features are labeled. Purple arrows indicate sites of HR invasion. The top portion displays the broken Chromosome III, processed to expose ends for HR, and the donor Chromosome V. The bottom portion displays the final chromosome products. See main text for details.
The Nanopore sequencing analysis of strain 101 arrived at the same conclusion and was able to narrow the breakpoints to single base pair resolution (Fig. 3D; Supplemental Fig. S4C). In particular, the right end of the deletion was shown to extend into the 5′ LTR of YCLWTy2-1 (Fig. 3B,D). Ribbon displayed an ∼6 kb insertion at these breakpoints (Fig. 3C; Supplemental Fig. S4A,B), which is the length of a typical Ty element. Although the inserted sequence could not be mapped with high confidence in each individual read, the ura3-52 gene was the most commonly identified (Supplemental Fig. S4B). The ura3-52 gene could also be identified by SNPs in the consensus sequence (Supplemental Fig. S4C). Thus, the rearrangement hypothesized in the CGH and CHEF gel-electrophoresis analyses was confirmed and refined through our analysis of Nanopore whole-genome sequence.
Based on these observations, we suggest that a double-strand break (DSB) occurred within the (GAA)n tract. The centromere-distal side of the break was resected into the 5′ end of URA3, and the centromere-proximal side of the break was processed into or near YCLWTy2-1 (Fig. 3E). These broken ends initiated HR with the ura3-52 gene on Chromosome V, and repair of the resulting gap produced a gene conversion event.
Strain 118: gene conversion involving (GAA)n repeats
The CHEF analysis of strain 118 showed that Chromosome III appeared ∼10 kb longer, and no other changes were observed (Fig. 2; Supplemental Fig. S1). Since Chromosome III is 365-kb long, a difference in size of 10 kb is often difficult to visualize; this small difference in size is more obvious in Supplemental Figure S1. This chromosome hybridized to probes for all three genes along Chromosome III (Fig. 2). By CGH, strain 118 showed a deletion of the second half of the UR-(GAA)100-A3 cassette, similar to strain 101 (Fig. 4A). Additionally, CGH analysis showed a duplication of a 15- to 21-kb portion of Chromosome II (corresponding to SGD coordinates 205,204–205,992 and 220,575–226,877) (Fig. 4A). The right end of this duplication overlaps with the Ty element YBLWTy1-1. This implied the possibility of a gene conversion event, similar to strain 101, but involving Chromosome II as the donor. Subsequent CHEF analysis (Fig. 2) showed that the longer Chromosome III did not hybridize to a probe for the Chromosome II centromere (CEN2) and confirmed that Chromosome II did not change in size, eliminating the possibility of a reciprocal translocation.
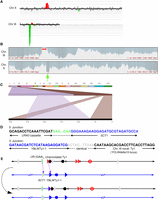
Identifying CGRs in strain 118. (A) CGH microarray analysis, displaying results for Chromosomes II and III. The large green region corresponds to the deletion surrounding the repeats, whereas the red region corresponds to the duplication surrounding the SCT1 locus. By examining the hybridization values for individual oligonucleotides on the microarrays, we found that the small red and green regions depicted in this figure do not represent true duplications and deletions, respectively. (B) Nanopore sequencing coverage maps of Chromosomes III and II, generated via UGENE, with a red arrow highlighting the deletion boundaries, and a green arrow indicating a duplication. (C) Ribbon single-read view highlighting a long read that captured the entire gene conversion event, showing a ∼20-kb insertion of Chromosome II in place of the deleted region on Chromosome III. (D) Single base pair resolution of the 5′ and 3′ junctions between Chromosomes III and II. The 5′ junction shows that the break and repair occurred within the (GAA)n repeats. The 3′ junction shows that the recombination event occurred within Ty elements on Chromosomes II and III. The gray region represents a 23-bp window of identity, with SNPs on either side identifying the specific Ty element (Supplemental Fig. S5D). The unannotated Ty1 element is adjacent to YCLWTy2-1 (Supplemental Fig. S3). (E) Diagram of the CGR event resulting in a gene conversion. Chromosome maps have the same format as in Figure 1A. Relevant features are labeled. Purple arrows indicate sites of HR invasion. The top portion displays the broken Chromosome III, processed to expose ends for HR, and the donor Chromosome II. The bottom portion displays the final chromosome products. See main text for details.
Nanopore sequencing of strain 118 arrived at the same conclusion as the CHEF/CGH analysis. We see a deletion on Chromosome III and a duplication on Chromosome II consistent with CGH analysis (Fig. 4B). Further, sequencing revealed that the centromere-distal junction of this gene conversion links the (GAA)100 repeat in the cassette with an imperfect (GAA)n repeat inside the SCT1 gene on Chromosome II (Fig. 4D; Supplemental Fig. S5C). Nanopore sequencing also revealed single-base resolution of the centromere-proximal junction between the Chromosome II YBLWTy1-1 and the unannotated Ty1 element located ∼11 kb downstream from the UR-(GAA)100-A3 cassette (Fig. 4D; Supplemental Fig. S5D). Multiple reads were found crossing each of these junctions (Supplemental Fig. S5A,B); indeed, at least two reads were able to capture the complete ∼17-kb insertion, unambiguously identifying the event as a gene conversion (Fig. 4C). Note that a gene conversion consisting of an ∼17-kb insertion and an ∼11-kb deletion results in a ∼6-kb longer Chromosome III, which is qualitatively consistent with the CHEF analysis.
Altogether, these results indicate that the CGR in strain 118 likely resulted from a DSB within the (GAA)100 repeats in the cassette. The centromere-distal broken end was processed only a short distance, or not at all, depending on the exact location of the DSB within the repeats, and then recombined with the imperfect (GAA)n repeat within SCT1. The centromere-proximal broken end was processed to the unannotated Ty1 element adjacent to YCLWTy2-1, which then interacted with the homologous YBLWTy1-1 on Chromosome II. Invasion of the broken ends into Chromosome II, followed by gap repair, produced the gene conversion (Fig. 4E).
Strain 105: highly complex rearrangement involving chromosome-scale duplications
Strain 105 was the most complex strain predicted from the CGH/CHEF analysis. By CHEF analysis, Chromosomes II and III appeared to be replaced by three novel chromosomes with approximate lengths of 700, 480, and 440 kb (Fig. 2). The ∼700-kb chromosome hybridized to the CEN2 probe and the CHA1 probe from Chromosome III, whereas the ∼480-kb chromosome hybridized to the Chromosome III probes LEU2 and RAD18, and the ∼440-kb chromosome hybridized to the CEN2 and RAD18 probes (Fig. 2). By CGH analysis (Fig. 5A), we observed a deletion centromere-proximal to the UR-(GAA)100-A3 cassette, similar to those deletions in strains 101 and 118. In addition, sequences on Chromosome III were duplicated from SGD coordinates around 123,000–168,000, and triplicated from around 168,000 to the end of the chromosome. These approximate breakpoints overlap with a solo LTR (YCRCdelta6; SGD coordinates 124,134–124,465) and an unannotated pair of Ty elements replacing YCRWdelta11 (SGD coordinates 169,573–169,888) (Supplemental Fig. S3; Lemoine et al. 2005). In addition, the CGH analysis showed a duplication of a centromere-containing ∼50-kb region of Chromosome II (SGD coordinates around 205,000–260,000). The left end of this duplication again overlaps the SCT1 gene, whereas the right end overlaps a Ty1 element (YBRWTy1-2).

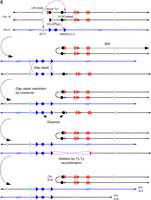
Identifying complex genomic rearrangements in strain 105. (A) CGH microarray analysis, displaying results for Chromosomes II and III. Large green and red areas show regions of the chromosome that are deleted and duplicated, respectively. By examining the hybridization values for individual oligonucleotides on the microarrays, we found that the small green regions depicted in this figure do not represent true deletions. (B) Nanopore sequencing coverage maps of Chromosomes III and II, generated via UGENE. Positions of relevant sequence features and large-scale copy number changes are indicated above and below the coverage maps. Positions of observed split reads are overlayed on the coverage map, and are labeled i–iii. (C) Ribbon single-read view corresponding to the indicated split reads. The X-shaped window in the third panel indicates that this portion of the read maps to an inversion of the chromosome. (D) Single base pair resolution of the indicated junctions. Junction i shows that the break and repair occurred within the (GAA)n repeats. Junction ii shows that the split read joining YBRWTy1-2 and the novel Ty1 on the right arm of Chromosome III actually contains sequences matching to YCLWTy2-1 on the left arm of Chromosome III, suggesting an intermediate step involving a dicentric chromosome (see main text for details). The gray region represents a 46-bp window of identity, with SNPs on either side identifying the specific Ty element (Supplemental Fig. S6E). The unannotated Ty1 element is the second of two Ty1 elements inserted in place of YCRWdelta11 (Supplemental Fig. S3). Junction iii shows the inverted left arm of Chromosome III joining to the beginning of the YCRCdelta6 LTR on the right arm of Chromosome III. (Figure continues on following page.)(E) Diagram of the CGR event. Chromosome maps have the same format as in Figure 1A. Relevant features are labeled. Purple arrows indicate sites of HR invasion. Purple dashed lines indicate sites of Holliday junctions. The top portion displays the broken Chromosome III following duplication and processing to expose ends for HR, as well as the donor Chromosome II. The second portion displays an intermediate step in which two new chromosomes have been formed, one by intrachromosomal BIR and another by gap repair using Chromosome II as a donor, resulting in a dicentric. The third portion shows the previous gap repair resolved as a crossover, resulting in a reciprocal translocation. In the fourth portion, a DSB in the dicentric chromosome is processed to expose homology between the two Ty elements, recombination between which results in deletion of CEN3; this recombination event could be a crossover (as shown) or a single-strand annealing event. The bottom portion displays the final chromosome products. See main text for details.
Nanopore sequencing of strain 105 revealed the same copy number changes as observed in the CGH analysis, with the ratio of the read depth corresponding to the duplication and triplication regions as predicted (Fig. 5B). Three groups of split reads were observed (Fig. 5C). The first group consists of the left half of the UR-(GAA)100-A3 cassette joined to the SCT1 gene on Chromosome II (Fig. 5C,D; Supplemental Fig. S6A,B). This junction is nearly identical to that in strain 118, except that a larger portion of the (GAA)100 repeat appears intact (Supplemental Fig. S6C). The second group of split reads map from YBRWTy1-2 on Chromosome II to the second of the two unannotated Ty1 elements located at YCRWdelta11 on Chromosome III, making up the duplication/triplication border (Fig. 5C,D; Supplemental Fig. S6A,B). Interestingly, SNPs located at this junction indicate the presence of a small portion of YCLWTy2-1 located in between YBRWTy1-2 and the unannotated Ty1 (Fig. 5D; Supplemental Fig. S6E). Nonsplit reads map to the same breakpoint on Chromosome III in an approximate 2:1 ratio with the split reads (Supplemental Fig. S6A,B). Both ends of the Chromosome II duplication also show nonsplit reads mapping across breakpoints in an approximate 1:1 ratio with the split reads (Supplemental Fig. S6A,B). Finally, the third group of split reads maps the centromere-proximal broken end from YCLWTy2-1 to YCRCdelta6 on the opposite side of the Chromosome III centromere (Fig. 5C,D; Supplemental Fig. S6A,B). These two loci are linked in an inverted orientation, and this junction corresponds to the single-copy/duplication border on Chromosome III (Fig. 5D; Supplemental Fig. S6A,B). Nonsplit reads map to the YCRCdelta6 loci in an approximate 1:1 ratio with the split reads (Supplemental Fig. S6A,B). There is no evidence for any other consistent group of split reads elsewhere in the genome.
The CGH/CHEF analysis combined with the Nanopore sequencing reveal key features of this complex genomic rearrangement with little ambiguity. The main limitation of our Nanopore sequencing analysis is due to the lack of reads spanning the entire ∼50-kb duplicated region of Chromosome II. This unfortunately prevents the distinction between gene conversion and reciprocal translocation involving the duplicated portion of Chromosome II. In the scenario in which a reciprocal translocation occurred between Chromosomes II and III, the predicted chromosome sizes from the Nanopore analysis match the novel chromosomes observed in the CHEF gel.
Given the sheer complexity of CGRs in 105, one could have imagined multiple possible pathways. The most plausible scenario based on the combination of our results is presented in Figure 5E. We suggest that after a DSB originated within the (GAA)100 tract, the broken chromosome was duplicated, resulting in two copies of Chromosome III with four broken ends. One copy underwent the following rearrangements: Its centromere-proximal end was processed to the unannotated Ty1 element adjacent to YCLWTy2-1, and this end invaded YCRCdelta6, initiating a BIR event that duplicated the whole right arm of Chromosome III distal to YCRCdelta6. This intrachromosomal BIR event generated the ∼480-kb-long chromosome (III-III in Fig. 5E). The centromere-distal acentric arm of Chromosome III was likely lost during cell division. The second broken Chromosome III was repaired as follows: The centromere-distal end crossed with the imperfect (GAA)n tract in SCT1 on Chromosome II. The centromere-proximal end was processed to the YCLWTy2-1 element and crossed with the YBRYTy1-2 of Chromosome II. After the gap repair event and crossover resolution, two translocated chromosomes were formed: an ∼700-kb-long hybrid chromosome (III-II in Fig. 5E) and an unstable II-III dicentric chromosome. CEN3 was subsequently lost from this dicentric chromosome by recombination between the YCLWTy2-1 element and the Watson-oriented unannotated Ty element that replaces YCRWdelta11 on the right arm of the chromosome, resulting in the ∼440-kb product (II-III in Fig. 5E). Loss of one centromere in dicentric chromosomes as a consequence of recombination between flanking repeats has been reported previously (Brock and Bloom 1994; Lemoine et al. 2005).
Discussion
Our study is the first to directly compare the use of Nanopore whole-genome sequencing to traditional methods used to identify and map CGRs in S. cerevisiae. We show that this approach was able to replicate results from the established but more laborious techniques used in prior studies and was further able to uncover novel observations of CGRs that were not easily resolvable through prior methods. In addition, this study represents the first extensive investigation of complex genomic rearrangements resulting from spontaneous breakage of a microsatellite sequence located in an essential chromosome region. In a previous study, we examined genetic alterations in a strain in which the (GAA)n tract was inserted in a nonessential region and in which we selected events that resulted in loss of sequences distal to the tract (Kim et al. 2008). Thus, this analysis was biased toward the recovery of nonreciprocal BIR events. Our current analysis allows an exploration of a more varied spectrum of events, both reciprocal and nonreciprocal.
In strain 101, a DSB generated within the (GAA)100 repeat was able to be processed to expose homology in the 5′ end of the URA3 gene, allowing recombination with the inactive ura3-52 gene. Events of this type were previously observed in experiments in which the URA3 reporter gene had interstitial telomeric sequences (ITS) instead of (GAA)n repeats (Aksenova et al. 2013). In contrast, in strains 118 (Fig. 4) and 105 (Fig. 5), HR was initiated directly from the broken (GAA)n repeats on Chromosome III, invading an imperfect (GAA)n repeat within the SCT1 gene on Chromosome II. (GAA)n and other homopurine repeats have previously been observed to promote CGRs, both in our previous studies in S. cerevisiae, as well as in cancer genomes (Kim et al. 2008; Bacolla et al. 2016). Interestingly, in strain 105, nearly the full (GAA)100 repeat is present following the CGR, whereas strain 118 contains only approximately 25 repeats following the rearrangement (Supplemental Figs. S5C, S6C,D). These differences may reflect the tendency of the DSB to form close to the 5′ end versus the 3′ end of the repeat or may reflect variability in end-processing efficiency at (GAA)n repeats. This observation also demonstrates the ability of Nanopore sequencing to measure the length of long (GAA)n microsatellites. This use of long-read sequencing technologies to uncover difficult-to-measure variations in microsatellite length is another important area of focus that is relatively unexplored (Liu et al. 2017).
Another novel observation is of the numerous and varied rearrangements that occurred in strain 105 as the result of a single originating DSB. Three recombinant chromosomes were produced, likely involving four broken DNA ends (Fig. 5E). One of these ends initiated an intrachromosomal BIR that generated a new chromosome in which the right arm of Chromosome III was duplicated. Two other broken ends of Chromosome III interacted with Chromosome II at loci ∼50 kb apart. Repair of this gap required extensive DNA synthesis, possibly involving DNA repair or a BIR-like mechanism originating from both broken ends. The meeting of these two synthesis events would result in the formation of a double-Holliday junction that could be processed into two translocated chromosomes. Finally, one of the translocated chromosomes appeared to be an unstable dicentric, which subsequently lost one centromere via an intrachromosomal recombination between two Ty elements. The presence of SNPs from a third Ty element at the junction of this recombination (Fig. 5D; Supplemental Fig. S6E) is strong evidence that this event indeed took place. Altogether, different nonreciprocal mechanisms involving both intra- or inter-chromosomal interactions are involved in the repair of broken DNA ends resulting from a single DSB. Note that this scenario could explain complex de novo rearrangements called chromoanasynthesis that were observed in several human congenital disorders (Carvalho and Lupski 2016). Importantly, unraveling this CGR required a whole-genome sequencing approach, which was able to identify chromosome-scale duplication and triplication events that would not have been observed via targeted sequencing of the (GAA)n region.
The question arises of how much sequencing coverage is needed to determine the nature of various CGRs. Ultimately, multiple factors must be considered. In the example of strain 118, our lowest-covered sample, two reads unambiguously showed that the ∼20-kb insertion was resolved via gene conversion. Although a number of additional reads map separately to each of the III-II and II-III junctions, ultimately our interpretation relies on these two reads that spanned the entire event. Thus, we might consider about 20× coverage to be nearly the minimum coverage required to map an ∼20-kb insertion, given a ∼25-kb average read length. However, in the example of strain 105, we did not find any reads covering the entire ∼50-kb insertion end-to-end, despite a higher coverage of about 40×, which limited our ability to distinguish between gene conversion and crossover resolution. Thus, the ability to unambiguously identify an event is a function of the coverage, the average read length, and the size of the insertion in question. With a greater average read length, less coverage may be required. Nanopore sequencing is capable of ultralong reads in excess of ∼800 kb when careful DNA extraction techniques are used (Liu et al. 2017). One strategy to reduce the sequencing burden would be to sequence a large number of samples at low coverage, then identify ambiguous CGRs and sequence those samples to greater depth.
Without the crucial component of long-read sequencing, the intricacies of these CGRs would not have been uncovered. Nanopore sequencing brings a rapid and effective new method of analysis for CGRs, allowing single base pair resolution of breakpoints and long reads that span repetitive regions. This analysis can be applied to whole-genome sequences for the identification of previously uncharacterized CGRs without a priori knowledge of the regions involved. Importantly, the level of detail obtained through this method is sufficient to extensively interrogate the mechanisms of DNA repair involved in CGR formation. For analyzing CGRs in S. cerevisiae, this technology is already capable of sequencing large numbers of genomes at relatively low cost. The techniques used here can be performed in a small laboratory with minimal specialized equipment and a modest level of expertise. As the output and accuracy of this developing technology continues to improve, similar analysis of human genomes, including cancer genomes with numerous complex rearrangements, will surely be possible.
Methods
Generation and isolation of yeast strains containing CGRs
The parent strain, SMY502, is a haploid strain of S. cerevisiae derived from FY1679 (ura3-52, his3Δ200, leu2Δ1, trp1Δ63, bar1::HIS3MX6, mat a). It also contains the UR-(GAA)100-A3 selectable cassette, located ∼1 kb downstream from the replication origin ARS306. Fluctuation tests were performed with SMY502 as previously described (Shah et al. 2012). Briefly, strains are grown in the presence of 5-FOA, which selects against an active URA3 gene. Inactivation of the UR-(GAA)100-A3 selectable cassette results in 5-FOA resistant colonies, which were then categorized for mutation type as previously described (Shishkin et al. 2009), via PCR primers located just outside of the repeats. Those colonies that showed a lack of this PCR product were tested with a further PCR primer pair that amplifies the entire selectable cassette in order to distinguish CGRs from short deletions. Strains with possible CGRs were saved as frozen stocks at −80°C.
CHEF gel and CGH array analysis
Experiments and analysis were performed as previously described (Aksenova et al. 2013).
DNA extraction
DNA was extracted via ethanol precipitation (for details, see Supplemental Methods). This method of DNA preparation resulted in an average fragment size of 24–48 kb (Supplemental Fig. S2A). DNA quantity was measured via Qubit (Qubit dsDNA BR Assay kit, Thermo Scientific).
Nanopore sequencing
Using the ONT (Oxford Nanopore Technologies) Ligation Sequencing Kit 1D (SQK-LSK108) in combination with the Native Barcoding Kit 1D (EXP-NBD103), 1.5 µg of purified DNA was used to construct barcoded sequencing libraries. All procedures recommended in the ONT-provided protocol were followed, including nick repair (NEBNext FFPE Repair mix, New England Biolabs). The libraries were pooled and sequenced together on a single SpotON Flow Cell Mk I R9.4 (FLO-SPOTR9) for 48 h.
Bioinformatics
Raw current traces generated by ONT sequencing were basecalled via the Albacore basecalling software (ONT version 2.02). For the parent strain, reads were then aligned to the S288C reference genome (R64-1-1, obtained from https://www.ensembl.org) using NGM-LR (Sedlazeck et al. 2017). The output was imported to Ribbon (Nattestad et al. 2016), as well as the bioinformatics software UGENE (Okonechnikov et al. 2012), for visualization. This analysis of the parent strain identified various deletions, insertions, and SNPs (Fig. 1B; Supplemental Fig. S3), which were then incorporated into the reference genome. Following this, the preceding analysis pipeline was repeated for each strain. Single base pair resolution of breakpoints within Ty elements was determined by analysis of SNPs within each Ty element of origin (for more details, see Supplemental Methods).
Data access
The sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra) under accession number SRP111355.
Acknowledgments
We thank Oxford Nanopore Technologies for providing the MinION device and necessary reagents under the MinION Access Program. This work was supported by the NIH (National Institute of General Medical Sciences) grants R01GM60987 and P01GM105473 to S.M.M. and R35GM118020 to T.D.P.
Author contributions: Conceptualization: R.J.M., T.D.P., and S.M.M. Methodology: R.J.M., T.D.P., and S.M.M. Software: R.J.M. Investigation: R.J.M., R.G.R., A.J.N., M.D., and D.K. Writing (original draft): R.J.M. and S.M.M. Writing (review and editing): R.J.M., T.D.P., and S.M.M. Visualization: R.J.M. and T.D.P. Supervision: T.D.P. and S.M.M. Funding acquisition: T.D.P. and S.M.M.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.228148.117.
-
Freely available online through the Genome Research Open Access option.
- Received July 21, 2017.
- Accepted October 26, 2017.
This article, published in Genome Research, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








