
Solid-phase reverse transfection for intracellular delivery of functionally active proteins
Abstract
Delivery of large and functionally active biomolecules across cell membranes presents a challenge in cell biological experimentation. For this purpose, we developed a novel solid-phase reverse transfection method that is suitable for the intracellular delivery of proteins into mammalian cells with preservation of their function. We show results for diverse application areas of the method, ranging from antibody-mediated inhibition of protein function to CRISPR/Cas9-based gene editing in living cells. Our method enables prefabrication of “ready to transfect” substrates carrying diverse proteins. This allows their easy distribution and standardization of biological assays across different laboratories.
Numerous physical, chemical, and biological methods are available for the transfer of foreign material into cells (Kaestner et al. 2015). Diverse molecules pass the cellular membrane with varying efficiency. Transfection of short nucleic acids (e.g., siRNAs or miRNA mimics) is a well-established procedure, whereas cell entry of large molecules (e.g., proteins) is inefficient due to its low membrane penetrating ability or low efficiency of the endosomal escape. The majority of available methods rely on diffusion of large cargo through transient openings in the cell membrane that can be caused by electroporation, laser pulsing (Wu et al. 2015), or microfluidic-based cell squeezing (Sharei et al. 2013). Alternative techniques enhance endocytosis by packing cargo into nanoparticles or nanocapsules (Slowing et al. 2007; Yan et al. 2010), complexing with cell-penetrating peptides (CPPs) (Morris et al. 2001; Ramakrishna et al. 2014), fusing with supercharged proteins (Thompson et al. 2012), or induced transduction by osmocytosis and propanebetaine (iTOP) (D'Astolfo et al. 2015). However, these approaches bear a number of limitations such as the need for special equipment, tedious translational fusion, conjugation to a carrier molecule, or low post-transfection cell viability. Microinjection does not have these pitfalls but is applicable only in low throughput. Viral vector and plasmid-based delivery is limited to nucleic acids, packing of large cargo is challenging, and integration into the host genome may occur in an uncontrolled manner (Ramakrishna et al. 2014; Wang et al. 2016). Therefore, more convenient, versatile, scalable, and less detrimental delivery methods of large cargo are urgently needed for basic research and therapeutic applications.
Lipofection is based on cationic lipids that interact with negatively charged nucleic acids. It became one of the most popular ways for cellular delivery of nucleic acids due to its applicability to many cell types and up to 100-fold higher efficiency in comparison to other chemical transfection methods (Kaestner et al. 2015). Recently, even less charged and chemically diverse macromolecules, like proteins, were transfected by lipofection. To increase the efficiency of membrane transfer, the proteins were fused to negatively charged carriers (e.g., supernegative GFP) before complexation with the cationic lipids (Zuris et al. 2015). Furthermore, the net anionic nature of the Cas9–gRNA complex allows its intracellular delivery via lipofection, resulting in targeted gene editing without additional fusion to polyanionic carriers (Liang et al. 2015; Zuris et al. 2015). So far, protein lipofection has been performed for only a few example molecules under the conditions of direct liquid transfection (liquid-phase).
Here, we present a solid-phase reverse transfection method for proteins and demonstrate its utility for diverse application areas. Our approach can be applied for gene engineering and high-content assays in multiwell plates and cell microarrays (Ziauddin and Sabatini 2001; Erfle et al. 2007, 2008).
Results
Optimization of the method
In the beginning, we tested how varying concentrations of transfection reagent and protein influence the efficiency of solid-phase reverse transfection and, as a result, cellular response. Based on those results, we introduced a transfection “optimization matrix” comprising four different concentrations of transfection reagent and two concentrations of the corresponding protein, resulting in eight different conditions (Supplemental Table S1). The transfection reagents Lipofectamine RNAiMAX and Lipofectamine 3000 showed the best performance in HeLa and HEK293T cells. At least three replicates were performed for each experiment, and a total of approximately 1400–4000 cells per replicate were counted. We applied the matrix for analyzing the transfection efficiency and localization of a small protein, namely green fluorescent protein (GFP), with a size of 27 kDa. In agreement with the literature (Llopis et al. 1998), the intracellular-delivered GFP was localizing to the cytoplasm and nucleus (Fig. 1A). The highest transfection efficiency, of more than 73% with the expected localization, was obtained by using 0.5 µL Lipofectamine RNAiMAX and 1 µg GFP, whereas other reaction compositions either reduced the transfection efficiency and/or induced excessive accumulation of GFP in punctate structures (Supplemental Fig. S1). Then we tested solid-phase reverse transfection of antibodies as an example case for large proteins (an average size of ∼150 kDa). We initially tested the efficiency of antibodies to enter living cells via solid-phase reverse transfection. For this, we used several antibodies that work well in conventional immunofluorescent staining (IF) in fixed cells and target proteins localizing to diverse subcellular organelles. One of them was a fluorescently labeled antibody recognizing Golgin subfamily A member 2 (GOLGA2; further in the text GM130), a matrix protein of the Golgi complex. We also included the molecular motor kinesin family member 11 (KIF11) and an anti-alpha tubulin antibody. Following the transfection with the antibodies and incubation for 2–4 h, cells were detached from the coated surface by trypsinization and replated on a mock-coated surface to remove the excess of the antibody that did not enter the cells. HeLa cells were fixed after an additional 4 h of incubation and imaged (Methods). We reached a high percentage of transfected cells for all antibodies and counted all cells with correct localizations plus cells with correct and endosomal localizations: 98% with anti-GM130 antibody, 86% with anti-KIF11 antibody, and 62% with anti-alpha tubulin antibody (Fig. 1E). Here, the highest concentration of the antibody and transfection reagent (Supplemental Table S1, condition VIII) led to a more efficient delivery. Lipofectamine RNAiMAX turned out to be efficient for the delivery of the anti-KIF11 and anti-alpha tubulin antibodies as well as for GFP, whereas Lipofectamine 3000 was more suitable for anti-GM130 antibody delivery (Supplemental Methods).
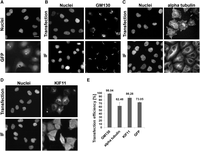
Comparison of protein localization after solid-phase reverse antibody transfection and immunofluorescent staining in HeLa cells. (A) The intracellular distribution of transfected GFP. The upper images show the localization of transfected anti-GM130 (B), anti-alpha tubulin (C), and anti-KIF11 (D) antibodies. The lower images show the results of the IF staining using the same antibodies. (E) Transfection efficiency of antibodies and GFP. Bars represent average transfection efficiency derived from six independent replicas with every antibody shown and 14 independent replicas with GFP; error bars represent the standard deviations. GFP, anti-KIF11, and anti-alpha tubulin antibodies were transfected with Lipofectamine RNAiMAX, using reaction condition I for GFP and condition VIII for antibodies. Anti-GM130 antibody was transfected with Lipofectamine 3000 using condition VI (Supplemental Table S1). The transfections were performed in 384-well plates. Nuclei were counterstained with Hoechst 33342 dye. (Scale bar) 20 µm.
Observation of the subcellular localizations of the transfected antibodies showed different localization patterns, which resembled the localizations obtained by IF (Fig. 1B–D; Supplemental Fig. S2). All tested antibodies and GFP also accumulated in punctate intracellular structures but at a varying degree (Fig. 1). As the intracellular delivery of proteins is expected to exploit endocytic pathways, the structures likely present various populations of endosomes and lysosomes. Indeed, we could detect a partial colocalization of the transfected anti-KIF11 antibody and early endosomes (Supplemental Fig. S3). A thorough spatial and temporal analysis of distribution of the transfected protein through the endocytic pathway was out of scope of this study. At any rate, when optimizing the transfection conditions, we considered not only the efficiency of protein delivery, but also the pattern of the expected localization and minimal occurrence of punctate intracellular structures (Supplemental Fig. S1).
Functional knockdown induced by a transfected antibody
We investigated whether a transfected antibody could inhibit the function of its target protein. For this we focused on the mitotic spindle regulator KIF11, whose down-regulation by RNAi has a clearly detectable phenotype—arrest of prometaphase via formation of a monoastral microtubule array (Neumann et al. 2010)—and the same phenotype was obtained by antibody microinjection (Blangy et al. 1995). Here, we show that prometaphase arrest can be induced by the transfection of anti-KIF11 antibody (Fig. 2). The optimization matrix was applied to identify the transfection conditions leading to the highest phenotype frequency (Supplemental Table S2). Up to 60% of cells showed a prometaphase arrest 24 h after antibody transfection, thus approaching the frequency obtained in RNAi experiments at the same time point (Fig. 2B). For the negative control (anti-rabbit IgG antibody), we obtained only a 0.25% prometaphase arrest frequency (Supplemental Table S3), and this result was confirmed by microinjection. An antibody showing a correct localization after delivery might fail to induce a cellular change. For example, both anti-KIF11 antibodies tested in this study localized to the cytoplasm (Fig. 1D; Supplemental Fig. S2), but only one of them induced the appearance of monoastral microtubule arrays when transfected. In agreement with previous data (Blangy et al. 1995), the antibody that targeted C-terminal residues of KIF11 induced a mitotic arrest. Binding of the functional antibody possibly impaired phosphorylation and association of KIF11 to the mitotic spindle (Sawin and Mitchison 1995), causing the observed effect.
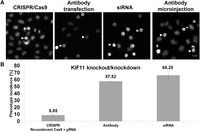
Comparison of KIF11 depletion by gene editing, RNAi, and antibody transfection. (A) Prometaphase arrest was obtained after solid-phase reverse transfection of HeLa cells: (left to right) with recombinant Cas9 + gRNA targeting KIF11, anti-KIF11 antibody, siRNA targeting KIF11, and anti-KIF11 antibody microinjection. Two example cells showing the prometaphase arrest phenotype are indicated by arrows in every image. (Scale bar) 20 µm. (B) Phenotype incidences after KIF11 knockout and knockdown. Bars represent average values derived from six independent replicas, and error bars represent the standard deviations. Phenotype analysis and quantification were performed as described in Methods. Transfection of the anti-KIF11 antibody was performed under the conditions described in Figure 1. The transfections were performed in 384-well plates and the microinjection was performed in a 35-mm petri dish.
Functional knock-in of Cas9 for CRISPR-mediated gene editing
Finally, we explored the efficiency of solid-phase reverse transfection for gene editing using recombinant Cas9 endonuclease and gRNA of three endogenous genes. Similar to RNAi-based and antibody-based deactivation of KIF11, the appearance of prometaphase arrest was obtained following solid-phase reverse cotransfection of Cas9 recombinant protein and gRNA targeting KIF11. In contrast to the previous experiments (Supplemental Fig. S4), up to 50 h incubation time was necessary to achieve the expected cellular phenotype (Fig. 2A). To confirm these results, we tested the knockout consequences of two other genes, namely PLK1 (polo like kinase 1) and INCENP (inner centromere protein), both of which are involved in cell division and cytokinesis (Neumann et al. 2010). Solid-phase reverse cotransfection of Cas9 and a gRNA targeting PLK1 resulted in the same prometaphase arrest phenotype as functional removal of KIF11. Solid-phase reverse cotransfection of Cas9 and a gRNA targeting INCENP-induced chromosome segregation defects resulting in the appearance of poly-lobed nuclei (Supplemental Fig. S5). We performed gene editing experiments in two commonly used cell lines, HeLa and HEK293T. The phenotype frequency for PLK1 and KIF11 knockout reached up to 10% in HEK293T cells (negative control, a nontargeting gRNA: 0.18% + −0.04%) (Supplemental Table S3), whereas knockout of INCENP resulted in <3% of phenotypic cells (negative control, a non-targeting gRNA: 0.23% + −0.03%) two days after Cas9–gRNA transfection (Supplemental Table S4). Besides the phenotypic analysis, we determined the indel incidence (insertions or deletions of bases in the DNA) by a genomic cleavage detection assay (Supplemental Fig. S6) and obtained, as expected, about twofold to fivefold higher indel incidences than those of the phenotypes after the same incubation time (Supplemental Table S4). The overall low occurrence of INCENP indel incidences and the respective phenotypes can potentially be attributed to a less efficient gRNA. We compared the results obtained by Cas9 and gRNA cotransfection to transfection of cells with an all-in-one cDNA-vector containing sequences for Cas9 and the respective gRNA (both performed by solid-phase reverse transfection method) (Fig. 3), as well as to a lentiviral-based transduction in HeLa cells (Supplemental Fig. S6; Supplemental Table S4). Similar to solid-phase reverse cotransfection of the Cas9–gRNA complexes, the phenotype frequencies were, for both methods, significantly lower than that of the indel cleavage efficiency. All in all, solid-phase reverse cotransfection of Cas9–gRNA complexes is more efficient in comparison to the other two methods and, importantly, does not require tedious cloning or viral particle production. The overall high degree of fluctuations between indel and phenotype incidences comparing different CRISPR-mediated gene editing methods might arise from still not optimized incubation times for cellular phenotypes (Neumann et al. 2010) or stochastic gene expression (Munsky et al. 2012). Additionally, we performed standard liquid-phase cotransfection of Cas9 and gRNA (Methods) to compare the performance of both transfection methods. In HeLa cells, solid-phase reverse cotransfection outperformed liquid-phase cotransfection in terms of phenotype frequency and indel rates using the same reagents and incubation times (Fig. 3A; Supplemental Table S4), whereas in HEK293T cells, both methods were equally efficient.
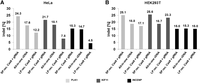
Comparison of solid-phase reverse and liquid-phase transfection for CRISPR/Cas9-mediated gene editing. The bar chart shows gene editing efficiencies (produced indels) after solid-phase reverse and liquid-phase cotransfection of recombinant Cas9 with gRNAs in HeLa (A) and HEK293T cells (B). gRNAs targeting PLK1 (light gray), KIF11 (dark gray), and INCENP (black) were analyzed. For comparison reasons, we included all-in-one cDNAs encoding the respective gRNAs and delivered by solid-phase reverse transfection. For clarity, we summarized the performance of the negative controls in Supplemental Table S3. All transfection reactions were performed in 384-well plates. Each target was knocked out in triplicates. The cell lysates of the triplicates were collected in one single tube for the indel analysis.
Finally, we tested the scalability of solid-phase reverse transfection. For this, we coated 384-well plates with transfection mixtures containing GFP (Methods) by using robotics. The robustness of the approach is demonstrated by a high delivery rate of GFP in every individual well, which was documented by imaging with an automated screening microscope (Fig. 4).

Scalability of solid-phase reverse transfection of proteins. (A) A schematic representation of a 384-well plate coated with GFP containing transfection complexes using condition I (Supplemental Table S1) (gray colored wells): (X) wells that were imaged. (B) The robustness of solid-phase reverse transfection of GFP. HeLa cells were seeded on the precoated wells, incubated for 24 h, fixed, and imaged. Nuclei were stained with Hoechst 33342. (Scale bar) 20 µm.
Discussion
In this study, we developed a novel solid-phase reverse transfection method that allows cellular delivery of large biomolecules such as proteins. The method is based on surface coating with mixtures containing transfection reagent, protein, and the carrier molecules. Precoated surfaces can be overlaid with cells, taking up the protein and modifying their features (Supplemental Fig. S7). We are the first to show the suitability of solid-phase reverse transfection for large cargo.
In contrast to previous work that largely focuses on liquid-phase transfection either of proteins or protein–nucleic acid complexes for CRISPR/Cas9-mediated gene editing, we demonstrate the versatility of our method in both of these applications (Liang et al. 2015; Zuris et al. 2015). We show the potential of solid-phase reverse transfection for gene editing using recombinant Cas9 endonuclease and gRNAs of three endogenous genes: PLK1, KIF11, and INCENP. Comparable results were obtained in HEK293T cells, and a higher gene editing efficiency was reached in HeLa cells (Fig. 3; Supplemental Table S4) than what has been published with liquid-phase cotransfection of Cas9–gRNA complexes.
Next, we are the first to show that a solid-phase reverse transfection of antibodies disrupts function of its intracellular target. For example, transfection of anti-KIF11 antibody and incubation for 24 h induces appearance of monoastral microtubule arrays as RNAi and CRISPR/Cas9-mediated gene editing (Fig. 2; Supplemental Fig. S4). So far, similar data have been published only with a microinjected antibody (Blangy et al. 1995). Disabling function of the antibody can manifest by multiple phenotypes, particularly if the targeted protein acts in different pathways. KIF11 is known to play a role in interphase as a plus-end microtubule molecular motor (Wakana et al. 2013). Loss-of-function of KIF11 might interfere with the trafficking of post-Golgi carriers that are dependent upon the activity of this motor. In this case, inhibition of the secretory cargo from the trans-Golgi network (TGN) toward the cell periphery might occur, in a similar manner as it is described for the overexpression of a catalytically inactive mutant of KIF11 (T112N) (Wakana et al. 2013). Indeed, transfected anti-KIF11 antibody following short, 2–4 h of incubation time was found in perinuclear punctate structures (Fig. 1D). Finally, transfected anti-alpha tubulin antibody might impair microtubule polymerization and, as a result, the integrity of the microtubule network. Indeed, we observe an increased fraction of cytoplasmic alpha tubulin and disorganized microtubules in agreement to the results obtained with microinjected anti-beta tubulin antibody (Füchtbauer et al. 1985). Similar effects were also reported when treating cells with microtubule destabilizing agents, such as nocodazole or paclitaxel (Ebneth et al. 1998; Putnam et al. 2003). In summary, antibody transfection allows validation of gene knockdown and knockout experiments, resulting in reduced off-target results, work, costs, and time when choosing an appropriate cellular assay.
Solid-phase reverse transfection of antibodies additionally has the potential to detect subcellular localization of their epitopes. Following the transfection of all three antibodies used in this study, differential localizations were obtained, which are, importantly, resembling the patterns of IF (Fig. 1; Supplemental Fig. S2). In the majority of cells, transfected antibodies and GFP (Fig. 1) localize besides the expected localization pattern to perinuclear punctate structures. Presumably, that results from protein accumulating in the endosomal–lysosomal pathway, as we could show by colocalization experiments using an early endosome marker EEA1 (Supplemental Fig. S3). In some cases, as proposed by KIF11, these structures might be phenotypic consequences of targeted loss of function. Therefore, interpretation of the localization pattern might require pattern filtering via image processing. The ever growing knowledge on protein localization and function is highly supportive for our method, e.g., for recognizing the native epitopes that are destroyed under the denaturing conditions of IF (http://www.proteinatlas.org/; locate.imb.uq.edu.au). For instance, it is known that the dynamic nature of post-Golgi carriers makes it difficult to observe them by IF in fixed cells.
Solid-phase reverse transfection of proteins (Supplemental Fig. S7) is not only suitable to address specific research questions, but is also a scalable method for simultaneous analysis of numerous molecules. The application for CRISPR/Cas9-mediated gene editing is straightforward and scalable to high-throughput applications. The same can be said about solid-phase transfection of small proteins, like GFP (Fig. 4), that can be used for target-based applications without the need of creating stable cell lines. Alternatively, loss-of-function experiments can be performed by using diverse transfection conditions for the same antibody in one experiment. By this, we could contribute to initiatives for standardizing antibody production (Bradbury and Plückthun 2015) and antibody-mediated biomedical applications (Vaishya et al. 2015).
In contrast to physical methods (Sharei et al. 2013; Wu et al. 2015), our approach can be applied in any standard cell biology laboratory and enables more standardized experimentation across various laboratories with the possibility of obtaining better comparable data.
Methods
Cell culture
HeLa cells (ATCC CCL-2) were cultivated in the recommended medium (DMEM, Gibco by Life Technologies) supplemented with 10% fetal bovine serum (Biochrom), 2 mM L-glutamine (Gibco by Life Technologies), and 100 units/mL, 100 µg/mL penicillin/streptomycin.
HEK293T cells (ATCC CRL-11268) were cultivated in the recommended medium (DMEM, Gibco by Life Technologies) supplemented with 10% fetal bovine serum (Biochrom), 1× nonessential amino acids (#11140050, Gibco by Life Technologies), and 100 units/mL, 100 µg/mL penicillin/streptomycin.
Coating of transfection solution in multiwell plates
The biomolecules (antibody, siRNA, cDNA, or a mixture of gRNA and Cas9 protein) and the transfection reagent were added to OptiMEM, containing 1.82 M trehalose dihydrate in a single well of a 384-well plate (Thermo Fisher Scientific) (Erfle et al. 2008). Then the solution was incubated for 5 min at RT to allow complex formation. Collagen IV solution (1 mg/mL in ddH2O) was added into each well, and the mixture was diluted at a ratio of 1:11 with ddH2O. Of this solution, 7.0 µL was transferred to each well of 384-well plates (Becton Dickinson). From each transfection solution, three wells were filled (triplicates) and dried (e.g., lyophilization or speed-vac) (Erfle et al. 2008). Detailed information for the preparation of the transfection solutions and the different biomolecules can be found in Supplemental Methods.
Coating of transfection solution in 384-well plates by robotics
The coating protocol was the same as described above using optimized transfection conditions for GFP (Supplemental Methods). Pipetting of the reagents was carried out using the automated liquid handling workstation Microlab Star (Hamilton) with a 96-probe head. The transfection mixtures were prepared in 20 wells of a 384-well plate simultaneously and then each mixture was transferred to four wells of a 384-well plate.
Lentiviral transduction
On day one, 3 × 102 HeLa cells were seeded per well in a 384-well plate in 80 µL culture medium. On day two, the medium was removed and culture medium that contained 8 µg/mL polybrene (#H9268, Sigma-Aldrich) and 3.3 × 105 TU of the lentiviral particles was added. On day three, the medium was exchanged with fresh culture medium. On day five, the cells were either fixed for the image acquisition or lysed for the genomic cleavage detection assay.
Cell seeding and fixation
The cells were seeded in 80 µL complete culture medium per well in a 384-well plate and then incubated at 37°C with 5% CO2. For fixation, 30 µL of a 3% PFA solution with 1 µg/mL Hoechst 33342 was added to the medium and then stored at 4°C until image acquisition. The total number of cells per experiment and the incubation times can be found in Supplemental Methods.
Liquid-phase cotransfection of recombinant Cas9 endonuclease and gRNA
HeLa (1600 cells/well) and HEK293T (8000 cells/well) cells were seeded in 100 µL complete culture medium per well in a 96-well plate and then incubated at 37°C with 5% CO2. The following day, the transfection complexes were prepared and added to the cells. In one tube, 36.57 ng Cas9 endonuclease and 9.14 ng gRNA were added to 5 µL OptiMEM serum-free medium and incubated for 10 min at RT; in another tube, 0.50 µL Lipofectamine RNAiMAX was added to 5 µL OptiMEM serum-free medium and incubated for 5 min at RT. Then the tubes were mixed together to allow complex formation for 15 min at RT. Of this transfection solution, 5 µL was added to each well, and the cells were then incubated for another 48 h until fixation or cell lysis. The total amounts of Cas9 and gRNA that were used in one well of a 384-well plate were adapted to the growth area of one well of a 96-well plate.
Cell lysis and genomic cleavage detection assay
After medium removal from the cells, lysis was performed directly in 384-well plates according to the manufacturer's protocol using DirectPCR lysis reagent (#31-301-C, PEQLAB). The cell lysates of the triplicates were collected in one single tube. PCR amplification was done, according to protocol, using Q5 Hot Start High-Fidelity 2X Master Mix (#M0494, New England Biolabs). The enzyme digest of mispaired dsRNA was done using T7E1 endonuclease (#M0302, New England Biolabs) according to the manufacturer's protocol. The DNA was loaded on a 2% agarose gel and run with a 50-bp marker (#D3812, Sigma-Aldrich) for 60 min at 50 mV. The target sequences of the gRNAs and the primer sequences for the amplification can be found in Supplemental Methods.
Immunofluorescent staining
For staining microtubules, cells were fixed in methanol for 5 min at −20°C. For all other stainings, cells were fixed with 3% paraformaldehyde solution and permeabilized with 0.1% Triton X-100 for 5 min. Blocking was performed with 3% BSA in PBS for 60 min. All primary antibodies were added in blocking buffer in dilutions of 1:200 and incubated overnight at 4°C. All secondary antibodies were added in blocking buffer in a dilution of 1:1000 and incubated for 2 h at RT. Nuclear counterstaining was performed with Hoechst 33342 (#H3570, Molecular Probes by Life Technologies) with a working solution of 1 µg/mL in PBS for 2 min at RT. Between all steps, the cells were washed three times for 5 min with 1× PBS at RT. A list of all antibodies can be found in Supplemental Methods.
Immunofluorescent staining after antibody transfection
HeLa cells, which were transfected with anti-alpha tubulin and anti-KIF11 antibody, were trypsinized 2 h post-transfection, whereas cells that were transfected with anti-GM130 antibody were trypsinized 20 h post-transfection. The cells were then seeded to a mock-coated (Supplemental Methods) 384-well plate to reduce signal from antibodies that remained in the coating and were not taken up by the cells. After another 4 h, the cells were fixed, and IF was performed. The following procedure, i.e., permeabilization, blocking, and secondary antibody incubation was done as described above.
Phenotype analysis and quantification
The total number of cells was counted using a self-made workflow with the KNIME software. The phenotypes that are described in Supplemental Figure S5 and the transfection efficiency of GFP and all antibodies were counted manually.
Fluorophore labeling of primary antibody anti-KIF11
Prior to the labeling using Alexa Fluor 647 Antibody Labeling Kit (#A20186; Molecular Probes, Thermo Fisher Scientific) the Tris buffer needed to be exchanged by PBS. For this, the Amicon Pro Purification System with an Ultra-0.5 Device (#ACS501002, Merck Millipore) was used. Both steps were performed as described in the manufacturer's protocol.
Acknowledgments
This project is funded by a grant (RNA-Code: FKZ 031A298) by the Federal Ministry of Education and Research (BMBF). The Viro-Quant-CellNetworks RNAi Screening Facility is supported by the CellNetworks-Cluster of Excellence (EXC81) and by the Baden-Württemberg Stiftung. We thank Dr. Matthew Betts (Heidelberg University) for carefully reading the manuscript and helpful comments, Dr. Rainer Saffrich (Heidelberg University) for the introduction to microinjection, and Jürgen Beneke for support with robotics.
Author contributions: R.B. designed and performed the experiments. R.B., H.E., and V.S. planned the experiments, discussed the results, and wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.215103.116.
- Received August 24, 2016.
- Accepted August 1, 2017.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








