
Single-cell-level spatial gene expression in the embryonic neural differentiation niche
- Yi Huang1,2,
- Xiaoming Yu1,3,
- Na Sun1,
- Nan Qiao1,
- Yaqiang Cao1,2,
- Jerome D. Boyd-Kirkup1,
- Qin Shen4,5 and
- Jing-Dong J. Han1,6
- 1Chinese Academy of Sciences Key Laboratory of Computational Biology, Chinese Academy of Sciences-Max Planck Partner Institute for Computational Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai 200031, China;
- 2University of Chinese Academy of Sciences, Beijing 100049, China;
- 3Center for Molecular Systems Biology, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China;
- 4Tsinghua-Peking Center for Life Sciences, Tsinghua University, Beijing 100084, China;
- 5Center for Stem Cell Biology and Regenerative Medicine, School of Medicine, Tsinghua University, Beijing 100084, China;
- 6Collaborative Innovation Center for Genetics and Developmental Biology
- Corresponding author: jdhan{at}picb.ac.cn
Abstract
With the rapidly increasing availability of high-throughput in situ hybridization images, how to effectively analyze these images at high resolution for global patterns and testable hypotheses has become an urgent challenge. Here we developed a semi-automated image analysis pipeline to analyze in situ hybridization images of E14.5 mouse embryos at single-cell resolution for more than 1600 telencephalon-expressed genes from the Eurexpress database. Using this pipeline, we derived the spatial gene expression profiles at single-cell resolution across the cortical layers to gain insight into the key processes occurring during cerebral cortex development. These profiles displayed high spatial modularity in gene expression, precisely recapitulated known differentiation zones, and uncovered additional unknown transition zones or cellular states. In particular, they revealed a distinctive spatial transition phase dedicated to chromatin remodeling events during neural differentiation, which can be validated by genomic clustering patterns, epigenetic modifications switches, and network modules. Our analysis further revealed a role of mitotic checkpoints during spatial gene expression state transition. As a novel approach to analyzing at the single-cell level the spatial modularity, dynamic trajectory, and transient states of gene expression during embryonic neural differentiation and to inferring regulatory events, our approach will be useful and applicable in many different systems for understanding the dynamic differentiation processes in vivo and at high resolution.
With worldwide initiatives towards image-based analysis of the transcriptome of the developing mouse embryo, and developing mouse and human brains, in situ RNA hybridization (ISH) images are being rapidly generated in a high-throughput manner using automated sample preparation and microscopy image acquisition. For example, the Eurexpress project has generated ISH images for more than 18,000 mRNAs and 400 miRNAs in mouse prenatal samples, many of which are from Theiler Stage 23 (or embryonic day 14.5 [E14.5]) (Diez-Roux et al. 2011). More recently, the Allen Institute for Brain Science has generated a large number of ISH images for mouse and human brains, including the Allen Mouse Brain Atlas (Lein et al. 2007), Allen Human Brain Atlas (Hawrylycz et al. 2012), BrainSpan Atlas of the Developing Human Brain (Miller et al. 2014), and Allen Developing Mouse Brain Atlas (Thompson et al. 2014).
These resources provide detailed visualization of the spatial pattern of gene expression at the cellular level in vivo. However, currently these images can only be visually examined one at a time or automatically annotated at a coarse level (Diez-Roux et al. 2011; Thompson et al. 2014), limiting their broad usage by the research community. How to effectively analyze these image data at high spatial resolution for global patterns and testable hypotheses has thus become an urgent challenge. Here we use the cerebral cortex development system as a model to develop a semi-automated image analysis pipeline.
The neocortex is a distinctive feature of mammals and is critical to many cognitive functions. Due to the complexity of its establishment, accuracy of its function, and vulnerability during aging, the neocortex is under intensive investigation (Bystron et al. 2008). Understanding the mechanism of the establishment process will not only explain the design of such an integrated functional system, but will also be important to the development of regenerative strategies for treating neurodegenerative diseases.
Many embryonic stem cell (ESC)-derived CNS cells and their precursors have been partially induced in vitro (Vazin and Freed 2010). However, the mechanisms of such procedures are largely unknown, and whether they reflect in vivo differentiation trajectories is unclear. This is at least partly due to the lack of comprehensive mapping of the in vivo neural differentiation trajectory. Recent studies have tried to address this through either microdissecting a few known cell layers or sorting with labeled proliferative/differentiating cell-type markers, and analyzing the gene expression profiles in each layer or stage (Ayoub et al. 2011; Belgard et al. 2011; Aprea et al. 2013; Miller et al. 2014). These approaches, although very helpful in understanding global gene expression changes during in vivo neural differentiation, are unable to capture precise and detailed spatial information or to identify gene expression features in undissected layers or unrecognized stages. Such limitations can be circumvented by examining spatial gene expression profiles, such as ISH images, which provide signals at very high resolution, even at the single-cell level.
During mouse and human embryonic cerebral cortex development, neural stem cells (NSCs) and intermediate progenitor cells proliferate in the telencephalon ventricular zone (VZ) and the subventricular zone (SVZ). Their differentiating daughter cells migrate through the intermediate zone (IMZ) and finally differentiate into neurons in the cortical plate (CP) (Bystron et al. 2008). In E14.5 mouse embryos, neurogenesis has reached its peak and early-born neurons have partly migrated to form the deeper layers, while migratory processes for the upper layer neurons have not yet started (Li and Jin 2010; Diez-Roux et al. 2011). This period is, therefore, an ideal in vivo system for studying the developmental microenvironment and neuronal lineage determination in the cerebral cortex.
By computationally analyzing the image-based expression profiles at the single-cell level and further analyzing the relationships between network modules, we recapitulated the known cellular layers of the E14.5 cortex that can be validated by commonly used cell/lineage marker gene expression profiles in microdissected tissue (Bystron et al. 2008). Based on these well-layered cellular states, we identified potential genes and functional interactions that regulate neurogenesis. In particular, we found that functional interactions connecting transcriptionally anti-correlated gene expression modules are enriched for NSC differentiation regulators. These analyses revealed a distinctive spatial transition phase for chromatin remodeling during neural differentiation. The critical role of chromatin remodeling is supported by the key regulatory circuitry uncovered through network analysis and the identification of chromatin organization centers during the spatial gene expression state transition. With our digitized single-cell-level transcriptome map, we were also able to detect cell cycle stage, especially the role of mitotic checkpoints during neurogenesis.
Results
Image analysis of the Eurexpress data
The Eurexpress database provides RNA ISH images of 19,411 assays (as of January 9, 2012), corresponding to more than 18,000 protein-coding genes and 400 miRNAs on ∼24 sagittal sections per E14.5 murine embryo (Supplemental Fig. S1A). Among them, 6410 assays have been annotated with EMAP IDs as identifiers of anatomical regions of gene expression (Diez-Roux et al. 2011; Hayamizu et al. 2013) according to the definitions from the EMAP eMouse Atlas Project (http://www.emouseatlas.org). We selected images labeled by 10 EMAP IDs specific to the telencephalon (Supplemental Table S1) and additionally included images of commonly used neural development markers (Supplemental Table S2) and miRNAs. Altogether, 3966 images of the middle-most sagittal section (Section 11 or 14) at stage E14.5 were selected (Supplemental Fig. S1B) and sequentially displayed with the MATLAB interface for visual inspection of their quality. If an image exhibited clear boundaries and detectable signals without contamination, we manually cropped three radial lines (as three repeats) in the cerebral cortex region to represent the expression pattern of a gene across the entire thickness of the cerebral wall, as well as one line over the non-embryo area to measure the background (Fig. 1A,B; Supplemental Methods). After this semi-automated curation, we digitized the raw gray-scale intensities (automatically smoothed for each pixel with its 3 × 3 neighbors to avoid harsh intensity changes) along the lines on 1816 images, corresponding to 1598 Entrez genes and 53 miRNAs (Fig. 1C; Supplemental Fig. S1B; Supplemental Table S3). As different samples have slightly different scales and background, and to make image profiles comparable, we removed image background noise, standardized spatial depth by scaling each radial line to 20 bins, and applied log-transformation. In this way, we obtained 60-bin profiles for these 1816 images (Fig. 1D; Methods; Supplemental Table S3). We observed that after these scaling and binning steps, the different image profiles and their repeats were well aligned with each other, which allowed for clearly separated radial regions and a high consistency between repeats (Supplemental Fig. S1C–F). Furthermore, 106 assays were curated twice for both Section 11 and 14 images to evaluate the consistency of the method, by examining the Pearson correlation coefficients (PCC) between profiles of these paired images. The significantly positive PCCs (mean ± standard deviation 0.65 ± 0.14, all P-values ≤ 0.00074) among these pairs demonstrated the high consistency and robustness of our image process pipeline.
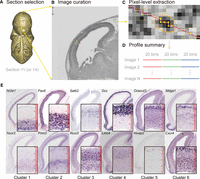
Workflow to extract and digitize expression profiles. (A) The middle-most sagittal section (Section 11 or 14) was selected for expression profile measurements. The Eurexpress 3D online mouse embryo model is here used to illustrate the relative position of Section 11. (B) A MATLAB graphical interface was used for manually curating three radial lines across the cerebral cortex. Another line at the top left of the image was cropped for background correction. (C) Mean intensity values of every nine neighboring pixels (shown as dotted 3 × 3 squares) on the cropped line were extracted as a vector of smoothed expression intensities. The region of approximately a single cell (marked by the orange eclipse) can be captured with smoothed intensities in six pixels—the average length of a bin. (D) Each line was scaled to a 20-bin profile to represent the expression profile of a gene across the radial axis from CP to VZ. Then, the profiles for all telencephalon-expressed genes were summarized on a matrix of genes versus their expressions in 3 × 20 bins. (E) The representative Eurexpress ISH images with different expression patterns. In the zoomed-in panels (bottom to top, VZ to CP), the high-resolution ISH images display the signals and scales for single cells relative to the size of each radial bin as indicated by the 20-bin red rulers. Image sources are euxassay_007249_11.jpg, euxassay_009400_11.jpg, euxassay_017942_14.jpg, euxassay_003376_11.jpg, euxassay_018949_14.jpg, euxassay_009808_14.jpg, euxassay_009545_11.jpg, euxassay_004815_14.jpg, euxassay_019619_11.jpg, euxassay_008979_11.jpg, euxassay_011139_11.jpg, and euxassay_006007_11.jpg, respectively, from top to bottom and left to right.
The E14.5 cerebral wall has been estimated to be 200–300 μm (Takahashi et al. 1995a,b), and for a 100 × 4 μm (tangential width × sample depth) section during E14-E15, there are ∼300 cell nuclei (Takahashi et al. 1995b). In our curation, the area covered by a radial line is around 2 × 20 μm (tangential width × sample depth). Thus, the expected number of nuclei covered by a radial line is ∼30. This estimation is consistent with the fact that, based on ISH images with clear cell outlines, in early E14 there are ∼20 cells across the cerebral wall (Takahashi et al. 1992), while in E15 there are ∼40 (Takahashi et al. 1995a). Since a curated radial line was divided into 20 bins, the intensity within each bin is approximately at single-cell resolution (Fig. 1C,E).
ISH spatial gene expression clusters identify known and unknown cell layers
During curation, we observed several radial expression patterns across the cerebral cortex (Fig. 1E) and hypothesized that these different expression patterns may represent different cellular states or layers. In order to identify such patterns, we applied image-wise z-score normalization (Methods) and used our recently developed Super k-means algorithm (Liu et al. 2013). Compared to the routine k-means algorithm, the Super k-means algorithm is orders-of-magnitude faster, and the solution from Super k-means clustering is both unique and optimum (it would require an infinite number of iterations of the routine k-means algorithm to obtain a similar solution). This allows us to rapidly scan a large range of k to determine the optimum number of clusters represented by these expression profiles. Based on both the “elbow” method (Thorndike 1953), which compares the intra- versus inter-sum-of-squared Euclidean distances (SSD), and the adaptive Bayesian information criterion (BIC) method (Zhang et al. 2013), the gene clusters reached an optimum modularity when k = 6 (Methods; Supplemental Fig. S2A,B). These gene clusters align remarkably well with respect to the cell layers from CP to VZ (Fig. 2A; Supplemental Table S3; Takahashi et al. 1995a,b). Visual examination in the Allen Developing Mouse Brain Atlas further indicates that these gene clusters have similar patterns on E13.5 and E15.5 (Supplemental Fig. S2C).
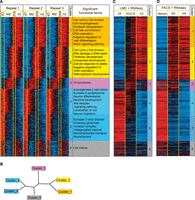
VZ to CP ISH profile-based spatial gene expression clusters. (A) Spatial expression profiles of telencephalon-expressed genes were clustered using the Super k-means algorithm with k = 6 and laid out according to their highest expression location, from the ventricle side moving outward. Cortical layers (Takahashi et al. 1995a,b) can be approximately separated into four zones, the CP, IMZ, SVZ, and VZ, as delimited by the yellow vertical lines. Gene expression profiles were extracted based on gray-scale pixel intensities as illustrated in Figure 1. Each column represents one of the 60 bins from the three neighboring lines marked on each image. Each row represents one of the 1816 images (for the 1598 Entrez genes and 53 miRNAs, all digitized expression intensity values are provided in Supplemental Table S3). The functional GO or KEGG terms enriched within each cluster of genes are listed next to the heatmap. (B) Neighbor-joining tree of the six clusters based on their average expression profiles. (C) RNA-seq expression levels of panel A genes (1561 detected RefSeq genes) in the six microdissected samples from the CP, SVZ-IZ (SVZ-IMZ), and VZ, respectively (two samples from each region and two technical repeats for each sample). (D) RNA-seq expression levels of panel A genes (1521 detected RefSeq genes) in three cell types (three samples from each cell type). (PP) Proliferative progenitor, (DP) differentiating progenitor.
We identified the GO/KEGG terms that were enriched in the Super k-means derived gene clusters using the DAVID package (Fig. 2A; Supplemental Table S4; Huang da et al. 2009). We have also listed all the transcriptional factors (TFs), miRNAs, and neural lineage markers included in each cluster (Supplemental Table S3).
The innermost cluster (Cluster 1) is enriched for cell cycle, cell morphogenesis, forebrain development, and cell fate commitment genes, and most importantly, the Notch signaling pathway, which is a key pathway active in NSCs (Bolos et al. 2007). The next most inner cluster (Cluster 2) is enriched for cell cycle genes, in particular, DNA damage/repair and chromosome condensation genes, which underlines the heavy replicative stress and epigenetic changes that are underway prior to differentiation. The middle cluster (Cluster 3) is specifically enriched for genes related to “chromosome” events, suggesting a distinctive spatial phase dedicated to chromatin remodeling events between the VZ to CP transition. The second most outer cluster (Cluster 4) is involved in axonogenesis, cell motion, synapse function, and dendrite development and is enriched for genes of the Wnt receptor-signaling pathway. This cluster may therefore mark the process of migration and functional establishment. The outermost cluster (Cluster 5) is enriched for ion channel and neurotransmitter genes, which represent neuron maturation. Interestingly, in addition to the above-mentioned clusters that have the highest expression in one location across the radial axis, we also identified Cluster 6 that has a bimodal expression pattern, with high expression at both the outermost layer and inner SVZ-VZ boundary, and is enriched in cell motion-related genes. To better illustrate the lineage relationship, a neighbor-joining tree was constructed based on the average profiles (Methods; Fig. 2B). This tree clearly shows that Clusters 1 and 2 are of similar lineage, Clusters 4 and 5 are also close, and Clusters 1, 2 and Clusters 4, 5 are farthest apart from each other with Clusters 3 and 6 in the middle, yet diverging in different directions. This suggests that chromatin events and cell motion are most active at different spatial phases, which is consistent with a previous finding that actin reorganization and chromatin assembly also occur at distinct times (Iyer et al. 2012).
ISH spatial gene expression has higher spatial resolution than the gene expression profiles of prenatal samples
To further corroborate the association of the ISH spatial gene expression cluster with the defined cell lineages, we analyzed a recent RNA-seq data set of laser-microdissected (LMD) samples from the CP, SVZ-IMZ, and VZ in the E14.5 murine cerebral cortex (Ayoub et al. 2011; Methods). We found that the clusters identified based on ISH images agree remarkably well with the expression profiles of the LMD samples (Fisher's exact test, P < 0.05 after Bonferroni correction) (Supplemental Table S5). Compared with the RNA-seq profiles of LMD samples, the ISH profiles displayed a higher spatial resolution in gene expression. For example, as the SVZ and IMZ layers were not separated in the LMD samples, the genes expressed only in the SVZ or IMZ (e.g., Cluster 3 genes) cannot be distinguished, and hence the spatial expression patterns are lost (cf. Fig. 2, A and C; note the difference between two biological repeats of SVZ-IMZ samples). As a consequence, these profiles from LMD samples missed the distinctive transition phase of chromatin remodeling during the neural differentiation process.
Recent array-based profiles of LMD human prenatal cortex (Miller et al. 2014) assayed more neocortex layers and identified the separation of germinal zones and post-mitotic zones, whose patterns are similar to homologous genes in the ISH clusters (Supplemental Fig. S2D). Nonetheless, the chromatin remodeling genes were again undetected in their study.
Alternative to microdissection, fluorescence activated cell sorting (FACS) based on cell surface markers was utilized to profile transcriptome changes during E14.5 mouse cortex differentiation (Aprea et al. 2013). Compared to FACS, high-resolution ISH profiles have the advantage of directly capturing and visualizing the trajectory and the transient states from differentiating progenitors to neurons (Fig. 2D; Supplemental Table S5).
Chromatin remodeling as a distinctive transition phase between NSC and neurons
As described above, Cluster 3 is specially identified by high-resolution ISH profiles and particularly enriched for chromatin regulators, such as Hmgn1, Rec8, and Trp53bp1 (Supplemental Table S4). Therefore, we hypothesized that there is strong epigenetic regulation at the VZ-CP transition interface during neural differentiation, especially during the period marked by Cluster 3, when cells migrate through the IMZ. As epigenetic modification often regulates stretches of chromatin regions composed of multiple genes or genomic clusters, we first examined whether the VZ-side and CP-side expressed genes tend to locate separately in different chromatin domains (Fig. 3A). Indeed, compared to random expectation, genes from the two transcriptionally anti-correlated modules (Cluster 1 and 2 versus Cluster 4 and 5) tend to be located on different but clustered chromosomal regions, especially on Chromosome 5, 10, and 19 (overall permutation P = 0.0398 to obtain at least three significantly clustered chromosomes out of 20) (color-coded panel in Fig. 3A; Supplemental Methods). For example, on Chr 10 (clustering P = 0.033) there is a domain that contains a significantly consecutive segment of Cluster 4/5 genes (permutation P = 9.2 × 10−4) (Supplemental Methods; Fig. 3A).
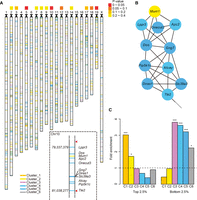
Role of chromatin remodeling in neurogenesis. (A) The ideogram of all genes curated by the image analysis pipeline. No genes on Chr Y were detected. Other chromosomes in mouse are acrocentric; centromeres are indicated with black triangles. The color-coded panel indicates P-values for chromosomal clustering of genes from anti-correlated modules. (Dashed box) A zoom-in on the 1.7-Mb Chr 10 domain, with the genome coordinates of TSS on the left and red triangles indicating the Hi-C domain boundary. (B) The Hi-C interaction network between genes in the 1.7-Mb Chr 10 domain. (C) The fold enrichment of genes with the top or bottom 2.5% neural progenitor cells (NPCs) versus neuron H3K4me3/H3K27me3 ratios in each cluster (labeled C1–C6), estimated based on 100,000 permutation background (dashed line). Asterisks indicate empirical P-values by permutation (*) P < 0.05, (**) P < 0.01, (***) P < 0.001. The colors of ideogram lines, network nodes, and bar plots denote different expression clusters, respectively: orange, Cluster 1; yellow, Cluster 2; purple, Cluster 3; cyan, Cluster 4; blue, Cluster 5; gray, Cluster 6.
To further investigate chromosomal domains on 3D interaction maps, we examined the intrachromosomal interactions between curated genes using the published Hi-C data of the 8-wk mouse cortex (Dixon et al. 2012; Methods). Consistent with a spatially specific reorganization of the chromatin, there are dense interactions within the Cluster 4/5 gene segment on Chr 10, which is embedded in a Hi-C interacting domain (Fig. 3B; Methods). The Onecut3, Pip5k1c, and Atcay genes in this domain have all been reported to regulate neural differentiation and function (Bomar et al. 2003; Francius and Clotman 2010; Yu et al. 2011; Espana and Clotman 2012). Other chromosomal clustering domains and intrachromosomal interactions were also identified (Supplemental Tables S6, S7). Chromatin remodeling and chromatin interactions events centered on these domains and regulators seem to undergo an important distinctive spatial switch during neural differentiation.
To further confirm such a chromatin and epigenetic switch at Cluster 3, we investigated the relative level of histone modifications in the mouse neural progenitor cells (NPCs) (Mikkelsen et al. 2007) and cortical neurons (Kim et al. 2010) as measured by ChIP-seq. We observed a high H3K4me3 signature on Cluster 1/2 genes in NPC and Cluster 4/5 genes in neuron, and a high H3K27me3 signature on Cluster 4/5 genes in NPC and Cluster 1/2 genes in neuron (Supplemental Fig. S3; Methods). To quantify these changes, we implemented a nonparametric approach to determine the relative H3K4me3/H3K27me3 ratio in NPC versus neuron (Methods). Interestingly, although the genes with a NPC active mark and neuron inactive mark gradually drop from Cluster 1 to 5, the neuron active mark and NPC inactive mark increase sharply on Cluster 3 genes, and then slightly decrease in Cluster 4 and 5 (Fig. 3C). This not only demonstrates sharp chromatin remodeling during the phase marked by Cluster 3 but also indicates that these chromatin events activate neuronal gene expression (in Cluster 3), precede the expression of the majority of neuronal genes (in Cluster 4/5), and are likely to be causal and upstream of neuronal gene expression changes.
Using these three lines of evidence—clustering in genomic localization, chromosome interaction, and histone modification changes—we can pinpoint in vivo chromatin remodeling during neural differentiation to the spatial phase represented by Cluster 3. However, how does this chromatin remodeling event regulate the transcriptional state switch during neural differentiation? To address this question, we sought to computationally predict the regulatory circuitry.
Network analysis identifies potential key regulators of neurogenesis
Although routine motif enrichment analysis can reveal some TFs and miRNAs whose targets are enriched within each gene expression cluster (Supplemental Notes; Supplemental Tables S8, S9), it does not reveal the regulatory circuitry governing the transition between the differentiation states. We found previously that the protein-protein interactions (PPIs) occurring between transcriptionally anti-correlated genes or modules are preferentially associated with differentiation regulation (Xia et al. 2006; Xue et al. 2007). To identify such modules and the interactions between them (the interfaces between anti-correlated modules), we developed a network analysis scheme, the so-called Negative-Positive (NP) network analysis. The NP network includes all PPIs between pairs of positively and negatively transcriptionally correlated genes (Xia et al. 2006; Xue et al. 2007). According to this framework, we constructed a 423-gene NP network with 718 positive edges and 260 negative edges from the high-confidence curated PPIs from the STRING database (von Mering et al. 2005) (confidence scores > 600) (Methods), where the interacting genes are positively or negatively transcriptionally correlated across the digitized ISH profiles (PCC > 0.2 or < −0.2) (Supplemental Fig. S4A,B for threshold determination; Methods). In this NP network, 220 genes are engaged in PPIs between transcriptionally anti-correlated genes (Fig. 4A; Supplemental Table S10). To test whether the genes in the network—in particular, the PPI interfaces between the spatially transcriptionally anti-correlated modules—are significantly associated with differentiation regulation, as previously observed using temporal gene expression profiles (Xue et al. 2007), we examined their co-citation impacts (CI, the log-transformed co-cited paper count in PubMed) (see Methods) with the keyword “differentiation” using our CoCiter program (Fig. 4B,C; Qiao et al. 2013; Methods). Compared to either the curated STRING PPIs or to all of the genes curated by our image analysis pipeline, the genes in the NP network are significantly more co-cited with “differentiation” (permutation P < 1 × 10−5) (Supplemental Methods; Fig. 4B). Furthermore, the interface genes between transcriptionally anti-correlated modules are significantly more co-cited with “differentiation” than the other genes in the NP network (permutation P < 1 × 10−5) (Supplemental Methods; Fig. 4B). It is interesting to note that, although all the genes annotated are expressed in the cerebral cortex, the mere presentation at the right location and time does not necessarily mean that they participate in the differentiation process. In fact, the likelihood for them to be co-cited with differentiation is slightly lower than the STRING PPI gene background (Fig. 4B,C). This is contrary to the logic that if a gene expresses at the right place in neural tissues, it must be required in one way or another for neural differentiation. When a gene's CI with “differentiation” is plotted against the average expression PCC (AvgPCC) with all its interactors in the NP network (Han et al. 2004), those with more negative AvgPCCs exhibit a higher rate of co-citation (Fig. 4C). This indicates that the negative AvgPCCs of the genes in the ISH expression NP network are predictive for them being regulators of cell differentiation.
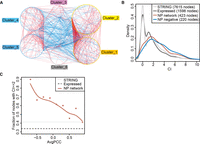
NP network module interfaces are enriched for “differentiation” functions. (A) The NP network between the six Clusters (423 nodes/genes in the network). Red edges represent PPIs between two transcriptionally correlated (PCC > 0.2) genes across the space from VZ to CP on the ISH images, while blue edges are those with PCC < −0.2. (B) Distribution of CIs with the keyword “differentiation” for the indicated gene sets. (C) Relationship between AvgPCC of genes/nodes (binned at linear intervals, at least five nodes in each bin) and the fraction of genes of at least three PubMed co-citations (CI ≥ 2) with the word “differentiation” within each bin. Gray lines indicate the average background levels.
We used Gene Set Enrichment Analysis (GSEA) (Subramanian et al. 2005) to test for pathways that are significantly associated with AvgPCC-ranked genes (normalized P < 0.01) (Methods; Table 1). Consistent with the co-citation analysis results, we found that differentiation-related pathways such as “BMP signaling pathway” and “Hedgehog signaling pathway” were overrepresented among the genes with negative AvgPCCs, and Smad5 and Gli2 have the most negative AvgPCC within these two pathways (Fig. 5A). Consistent with their regulatory role in coordinating the phase transition, BMP up-regulated genes (Methods) are significantly enriched in Cluster 1, 4, and 5, while SHH up-regulated genes (Methods) are significantly enriched in Cluster 4 and 5 (Fisher's exact test, P < 0.05 after Bonferroni correction) (Supplemental Table S5).
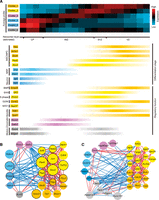
Feedback circuits identify key modules regulating differentiation. (A) The average profiles (upper panel) of the six clusters are consistent with the differentiation stages and regulatory functions, as indicated with the smoothed intensity of relevant markers (lower panel). Cortical layers (Takahashi et al. 1995a,b) can be approximately separated into four zones, the CP, IMZ, SVZ, and VZ, as indicated in the middle. (NSCs) Neural stem cells, (NPCs) neural progenitor cells, (NRPs) neuron-restricted progenitors. (B) The second largest subnetwork is enriched for regulators of neurogenesis. In this subnetwork, 22 of the 23 nodes have at least three PubMed co-citations (CI ≥ 2) with the word “differentiation.” (C) The largest subnetwork is related to mitotic spindle. In panels B and C, edge colors indicate PCC as in Figure 4A. Node colors denote the expression clusters as shown in Figure 3. TRANSFAC TFs are labeled with a black border. Node radius is proportional to CI with “differentiation.”
KEGG/GO terms enriched among the NP network genes with the most positive or negative AvgPCC
Feedback loops form the key regulatory circuitry of neurogenesis
As regulatory interactions frequently form feedback loops, to identify regulatory circuitry, we retrieved all three-step loops (or “triangles”) composed of at least one negative edge. Altogether, 617 such loops, traversing 129 nodes, were found in the NP network. These loops form 16 connected subnetworks (network graph components) (Fig. 5B,C; Supplemental Fig. S4C; Supplemental Table S11). Among them, the second largest network component is a TF-enriched subnetwork (Fisher's exact test, P = 1.8 × 10−8, and 4.49-fold enriched over the 129-node background) (Fig. 5B) that was highly co-cited with “differentiation” and recaptures several key regulators of neural differentiation, such as Pax6 and Sox2. In this subnetwork, the top six nodes with the most negative AvgPCCs—Ncor2, Hivep2, Lmo4, Satb2, Onecut3, and Rcor2, have many interactions between Cluster 1/2 (proliferative state) and Cluster 4/5 (differentiated state), suggesting that they might coordinate the transition from a proliferating cellular state to a differentiated state. Indeed, Ncor2 (Cluster 1) maintains the NSC state (Jepsen et al. 2007), Lmo4 (Cluster 4) facilitates neuronal radial migration (Asprer et al. 2011), Satb2 (Cluster 3) regulates chromatin structure during cortical development (Gyorgy et al. 2008), and Rcor2 (Cluster 3) is involved in neuronal gene chromatin regulation (Ballas et al. 2005). Interestingly, Ncor2, Rcor2, and Satb2 are all epigenetic regulators that interact with HDAC proteins (such as Hdac5 in Cluster 4) in the REST/Co-REST complexes (Ballas et al. 2005; Gyorgy et al. 2008), which is the most important epigenetic regulator of neural differentiation (Coskun et al. 2012).
For the other two nodes with negative AvgPCCs, Hivep2 (Cluster 5) has been reported to be regionally expressed during mouse brain development (Campbell and Levitt 2003); however, there is little knowledge of its molecular function in the brain, and Onecut3 (Cluster 5) was recently found to function in the development of several CNS cells/regions, such as spinal motor neurons (Francius and Clotman 2010), the locus coeruleus, and the mesencephalic trigeminal nucleus (Espana and Clotman 2012), but no function in the cerebral cortex has been reported. Their dense connections with each other and with key neural differentiation regulators (e.g., the aforementioned epigenetic regulator Rcor2) through feedback interactions, suggest that they may form regulatory circuits to coordinate neuronal fate transition through epigenetic regulations. Further supporting the regulatory roles of nodes with very negative AvgPCCs in the network (Supplemental Fig. S4C), several nodes in the remaining subnetworks have also been identified to function in neuronal development, such as Nes (Lendahl et al. 1990) and Gli2 (Takanaga et al. 2009). In general, we observed a significant anti-correlation between AvgPCC of the nodes in these feedback loops and their CI with “differentiation” (PCC = −0.21, P = 0.018).
The role of mitotic checkpoints in neuronal fate commitment
It is reassuring to have recovered the most important epigenetic regulations from the computationally predicted regulatory circuitries for the in vivo neural differentiation process. However, to our surprise, the largest network component is related to the mitotic spindle (Fig. 5C; Supplemental Table S4).
As our ISH-based gene expression profile is at the single-cell level, we further examined whether there is a mitotic checkpoint implemented at the transition stage, by examining the enrichment of cell cycle markers of different phases or checkpoints in each of the six gene expression clusters (Methods). Interestingly, while cell cycle genes at the G1/S checkpoint and G2 phases are significantly enriched in both Cluster 1 and 2, S phase genes are significantly enriched in Cluster 2 instead of Cluster 1, and G2/M checkpoint genes are enriched in Cluster 1 rather than Cluster 2 (Fig. 5A; Supplemental Table S5). This is consistent with interkinetic nuclear migration, which refers to the nuclei shuttling between the apical and basal side of the germinal zone during the cell cycle (Sun and Hevner 2014). Meanwhile, M/G1 checkpoint genes are marginally enriched in Cluster 2 (Fisher's exact test, P = 0.11 and fold enrichment = 2.2) (Fig. 5A), suggesting a potential mitotic exit checkpoint before the transition stage where chromatin remodeling occurs. This spatial difference of cell cycle status can be further supported by principal component analysis (PCA) of the cell cycle marker gene expression profiles—markers of S phase and G2/M checkpoint have separated territories, with other intermediary stage markers scattered in between (Supplemental Fig. S5).
A few mitotic checkpoint genes in our study have been found to affect brain size and brain development. For example, Nde1 (Cluster 1) at the network interface regulates cerebral cortical size (Feng and Walsh 2004), while the human homologs of Kif2a (Cluster 5) and Dync1h1 (Cluster 6), also at the network interface, cause malformation of the cortex if mutated (Poirier et al. 2013). The results of our single-cell-level analysis therefore identify the general involvement of mitotic checkpoints in cell fate transition during cerebral cortex development.
Discussion
Automated sample preparation and microscopy image acquisition have enabled generation of ISH in a high-throughput manner. To effectively analyze these data in a similar high-throughput manner remains a big challenge. To this end, the Allen Institute for Brain Science has developed an automated image-processing pipeline to provide a global view of the transcriptome. However, the pipeline depends on crude pattern recognition, which results in much lower resolutions (100-μm grid for E13.5 and 120 μm for E15.5) than the actual data. Moreover, for prenatal brains with small sizes, as the automated analysis is not precise, they were forced to perform expert-guided manual annotation of brain structures instead. Therefore, a practical and effective approach to digitizing the high-resolution ISH images remains lacking.
In this study, we developed a semi-automated method to digitize ISH images of E14.5 mouse embryos at single-cell resolution. To our knowledge, it is still technically difficult to separate intact single cells for gene expression profiling of the developing mouse cerebral cortex. Two recent studies that have come closest have used LMD (Ayoub et al. 2011) or FACS (Aprea et al. 2013) for gene expression profiles, both at much lower resolution, which use millions of cells either from three major known layers or with three marked types (Fig. 2C,D). Especially for exquisite tissues like prenatal brain, until dissecting intact single cells becomes feasible for gene expression profiling, high-resolution ISH image data-derived spatial gene expression profiling with our method remains a good alternative and approximate for in vivo single-cell-level spatial gene expression profiles.
The digitally transformed expression profiles across the cortical layers were used to finely map gene expression clusters to known and unknown transition zones or cell layers at this stage (Fig. 5A). Importantly, we found that the PPIs connecting transcriptionally anti-correlated genes, or modules, are predictive of them being regulators of differentiation/transitions across the zones. Based on this insight, we further predicted the regulatory circuits that control the fate transition from NSC to CP neuron, including the well-known REST/Co-REST epigenetic regulatory complex and mitotic checkpoint control in cell fate transition during cortical development. These regulatory circuits, together with the distinctive chromatin remodeling genes’ spatial expression at the transition zone, strongly suggest a synchronized switching of epigenetic states when cells exit the cell cycle to migrate through the IMZ, and that chromatin interaction domains serve as higher-order chromatin organization centers during neural differentiation. The regulatory function of chromatin remodelers during neurogenesis has recently attracted intense investigation (Kishi et al. 2012; Egan et al. 2013; Ronan et al. 2013; Tuoc et al. 2013). Our study thus provides an unbiased analysis that uncovers their critical role from a computational and high-resolution perspective.
While our method mainly digitized the radial organization across the cerebral wall, the IMZ may also contain a small fraction of neuron precursors that migrate temporarily orthogonally rather than strictly radially (O'Rourke et al. 1992). This kind of heterogeneity, though difficult to exclude with ISH data at only one static time point, does not affect our conclusion that the IMZ (radially spanned by Cluster 3) (Figs. 1E, 2) is a layer dedicated to chromatin remodeling for neuron progenitors during VZ-CP migration, since the small fraction of orthogonally migrating cells will eventually still contribute to the neurons in the CP.
Cell-cycle states have been recently shown to not only associate with stem cell differentiation but may play an important role in determining differentiation propensity (Pauklin and Vallier 2013). Here, we found at the single-cell level that mitotic checkpoints might be a general key regulatory event in neural differentiation.
In particular, it has been recently suggested that in the outer SVZ of humans, there is a class of progenitors named outer radial glia (oRG) cells, which arise from or undergo asymmetric division with nonvertical mitotic spindle orientation and may strongly affect mammalian cortical expansion (Shitamukai and Matsuzaki 2012; LaMonica et al. 2013). oRG-like cells are also found in the superficial SVZ and IMZ of mouse, although at much lower abundance (<10% of mitotic progenitors) (Wang et al. 2011). In our study, the observed enrichment of mitotic checkpoint M/G1 genes in Cluster 2 may signify the presence of this progenitor subtype. Although we did not detect enrichment of cell-cycle genes in the IMZ (Cluster 3), the dedicated chromatin remodeling function of the IMZ may still provide a specific environment for oRG cells. It would be interesting to further test this in prenatal human brain, where oRGs are more abundant.
Altogether, our analyses provide a novel approach of analyzing ISH image data to directly in situ capture and visualize at the single-cell level the spatial modularity, dynamic trajectory, and transient states of gene expression during embryonic neural differentiation and to infer regulatory events. Such an approach will be useful and applicable in many different systems for understanding the dynamic or spatial processes in vivo and at high resolution.
Methods
Image curation
Selected raw images and their annotations (as of January 9, 2012) were downloaded from the website http://www.eurexpress.org. After gray-scale conversion and cropping of the head region, the images were sequentially and randomly displayed for visual examination of their quality. Only the images with a clear ventricle boundary were selected and carefully cropped, using radial lines across the cortex, for downstream analysis. After background correction, the average intensities of nine neighboring pixels along the lines were binned to 20 bins each line and log2-transformed. For each of the 1816 images, a 60-bin profile (three repeat × 20 bin) was extracted to represent the pattern of a probe across the cortex, and z-score normalized before Super k-means clustering. The z-score was calculated with (X − μ)/σ, where X is bin intensity, and μ and σ are the average and standard deviation of X, respectively.
Determining the best k for Super k-means clustering
To obtain the proper number of groups for clustering, Super k-means clustering was run iteratively with k from three to 10. Then, their intra-/inter-SSDs were calculated and compared. The heuristic “elbow” method (Thorndike 1953) was used to identify the k with optimum modularity. An adaptive BIC method (Zhang et al. 2013) was also utilized.
Neighbor-joining tree analysis of gene expression profiles
The neighbor-joining tree was generated with MEGA5 (version 5.05, with analysis option “Phylogeny Reconstruction” and statistical method “Neighbor-joining”) (Tamura et al. 2011). The Euclidean distances calculated between the average profiles of the six clusters were used as the input.
RNA-seq and microarray data set processing
RNA-seq data from Ayoub et al. (2011), Belgard et al. (2011), and Aprea et al. (2013) were downloaded from the NCBI GEO database (GSE30765, GSE27243, and GSE51606), mapped to the mouse genome with TopHat v1.4.1, and summarized to expression levels with Cufflinks v.1.3.0 (Trapnell et al. 2012), based on the UCSC mm9 annotation. A normalized microarray profile of human 16pcw prenatal microdissected samples from Miller et al. (2014) was downloaded from http://brainspan.org on April 6, 2014. The up- and down-regulated genes of BMP and SHH pathways were derived from microarrays GSE48092 and GSE42565, respectively, and processed with the “affy” package in R (Gautier et al. 2004). Differentially expressed genes were determined using RankProd (Hong et al. 2006) with a false discovery rate < 0.01.
Hi-C data processing
The normalized intrachromosomal interaction matrices (40-kb bin, mm9) of Hi-C data for the 8-wk mouse cortex were downloaded from http://chromosome.sdsc.edu/mouse/hi-c/download.html (version on April 6, 2012). For each pair of genes (based on the UCSC mm9 annotation), we calculated a gene-wise interaction score—the average of normalized bin-wise interaction scores between their gene bodies, to evaluate the strength of interaction. We built the interaction network based on the combined Hi-C data, with a global gene-wise score cutoff defined as median(cutoff1, cutoff2, …, cutoff19, cutoffX), where cutoffi equals the bin-wise interaction score cutoff at the highest 1% boundary for Chromosome i.
ChIP-seq data processing and calculation of H3K4me3/H3K27me3 ratio in NPCs versus neuron
ChIP-seq data were downloaded from the NCBI GEO database (GSE12241 and GSE21161) (Mikkelsen et al. 2007; Kim et al. 2010) and mapped to the mouse genome with Bowtie v0.12.8 (Langmead et al. 2009), based on the UCSC mm9 annotation. For each RefSeq gene, fragment count (with fragment length ∼200 base pairs [bp]) was
summarized for −2000 to +2000 bp surrounding a TSS and corrected by the input (Supplemental Fig. S3). To determine the H3K4me3/H3K27me3
ratio for each gene, we first calculated the average input-corrected intensities around the TSS, replaced them with ranks
in each sample, and then defined a nonparametric rank score by
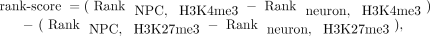 which measures the relative ratio of H3K4me3/H3K27me3 in NPCs versus neuron.
which measures the relative ratio of H3K4me3/H3K27me3 in NPCs versus neuron.
NP network analysis
The STRING PPI data (version 9.0) were downloaded on June 13, 2011. The integrated confidence scores were recalculated according to the original publication (von Mering et al. 2005) based only on “fusion,” “experimental,” and “database” scores. Only interactions with scores > 600 were used for further analysis. The NP network was constructed in a similar way to that described in Xia et al. (2006) and Xue et al. (2007), except that we required |PCC| > 0.2.
Co-citation analysis
The NCBI curated “gene2pubmed” table and PubMed abstracts from 16 common organisms were downloaded on October 11, 2011. For a given coding gene-term pair, a PubMed reference was marked as a co-citation paper if (1) it was indexed in the “gene2pubmed” table with either the mouse gene or its homologs in human, fly, and worm (determined with NCBI “homologene.data” downloaded on July 24, 2011), and (2) its abstract contained the term. For a given miRNA-term pair, a PubMed reference was marked as a co-citation paper if (1) its abstract contained the miRNA name (case-insensitive) without species prefix, e.g., “mir-124,” and (2) its abstract contained the term. CI = log2 (N + 1), where N is the co-cited paper count, was used to measure the relevance of a gene to a certain term. “Omic” papers citing more than 200 genes were ignored. The algorithm is available online (http://www.picb.ac.cn/hanlab/cociter) (Qiao et al. 2013).
Gene sets used for GSEA
The mouse GO/KEGG annotations for GSEA were downloaded from the NCBI “gene2go” table and KEGG database API (Kanehisa et al. 2012) on July 7, 2012. Only terms with a gene set overlapping with 3∼200 of the 423 NP network genes/nodes were considered.
Cell cycle marker genes
Marker genes from different phases and checkpoints of the cell cycle were derived from the mouse homologs of those identified in HeLa cells (Whitfield et al. 2002).
Acknowledgments
We thank Zhiheng Xu, Richard S. Nowakowski, Weimin Zhong, Xiaoqun Wang, Axel Mosig, Peter Stevenson, Rolf Sprengel, Zhiqi Xiong, and Feng Zhang for invaluable discussions and suggestions; and Mehmet Somel, Weiyang Chen, Yi Liu, Shengbao Suo, Wenxuan Gong, Wei Zhang, Zhijun Han, and Tiangen Chang for technical assistance. This work was supported by grants from the stem cell leading project XDA01010303, Chinese Ministry of Science and Technology (Grant #2011CB504206 and 2015CB964803), Chinese Academy of Sciences instrument development project (Grant #YZ201243), and the National Natural Science Foundation of China (Grant #91019019, 91329302, and 31210103916) to J.D.J.H. J.D.B-K. holds a Chinese Academy of Sciences Fellowship for Young International Scientists (#2011Y1SB05) and acknowledges the support of the National Natural Science Foundation of China research fund for Young International Scientists (Grant #31250110576 and 31350110530).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.181966.114.
- Received September 29, 2014.
- Accepted January 8, 2015.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








