
Origin and domestication of papaya Yh chromosome
- Robert VanBuren1,2,9,
- Fanchang Zeng2,9,
- Cuixia Chen2,9,
- Jisen Zhang1,9,
- Ching Man Wai2,
- Jennifer Han2,
- Rishi Aryal2,
- Andrea R. Gschwend2,
- Jianping Wang2,
- Jong-Kuk Na2,
- Lixian Huang1,
- Lingmao Zhang1,
- Wenjing Miao1,
- Jiqing Gou3,
- Jie Arro2,
- Romain Guyot4,
- Richard C. Moore5,
- Ming-Li Wang6,
- Francis Zee7,
- Deborah Charlesworth8,
- Paul H. Moore6,
- Qingyi Yu3 and
- Ray Ming1,2
- 1FAFU and UIUC-SIB Joint Center for Genomics and Biotechnology, Fujian Agriculture and Forestry University, Fuzhou, Fujian, 350002, China;
- 2Department of Plant Biology, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA;
- 3Texas A&M AgriLife Research, Department of Plant Pathology and Microbiology, Texas A&M University System, Dallas, Texas 75252, USA;
- 4IRD, UMR DIADE, EVODYN, BP 64501, 34394 Montpellier Cedex 5, France;
- 5Department of Botany, Miami University, Oxford, Ohio 45056, USA;
- 6Hawaii Agriculture Research Center, Kunia, Hawaii 96759, USA;
- 7USDA-ARS, Pacific Basin Agricultural Research Center, Hilo, Hawaii 96720, USA;
- 8Institute of Evolutionary Biology, University of Edinburgh, Edinburgh EH9 3JT, United Kingdom
- Corresponding author: rming{at}life.uiuc.edu
-
↵9 These authors contributed equally to this work.
Abstract
Sex in papaya is controlled by a pair of nascent sex chromosomes. Females are XX, and two slightly different Y chromosomes distinguish males (XY) and hermaphrodites (XYh). The hermaphrodite-specific region of the Yh chromosome (HSY) and its X chromosome counterpart were sequenced and analyzed previously. We now report the sequence of the entire male-specific region of the Y (MSY). We used a BAC-by-BAC approach to sequence the MSY and resequence the Y regions of 24 wild males and the Yh regions of 12 cultivated hermaphrodites. The MSY and HSY regions have highly similar gene content and structure, and only 0.4% sequence divergence. The MSY sequences from wild males include three distinct haplotypes, associated with the populations’ geographic locations, but gene flow is detected for other genomic regions. The Yh sequence is highly similar to one Y haplotype (MSY3) found only in wild dioecious populations from the north Pacific region of Costa Rica. The low MSY3-Yh divergence supports the hypothesis that hermaphrodite papaya is a product of human domestication. We estimate that Yh arose only ∼4000 yr ago, well after crop plant domestication in Mesoamerica >6200 yr ago but coinciding with the rise of the Maya civilization. The Yh chromosome has lower nucleotide diversity than the Y, or the genome regions that are not fully sex-linked, consistent with a domestication bottleneck. The identification of the ancestral MSY3 haplotype will expedite investigation of the mutation leading to the domestication of the hermaphrodite Yh chromosome. In turn, this mutation should identify the gene that was affected by the carpel-suppressing mutation that was involved in the evolution of males.
Gender in papaya is genetically controlled by a sex-linked region that behaves like an XY sex chromosome, and maleness versus hermaphroditism is controlled by slightly different sex-specific Y chromosome regions, Yh (HSY) in hermaphrodites and Y (MSY) in males. Both the HSY and MSY are ∼8.1 Mb (∼15% of the largest papaya chromosome, Chromosome 1), and recombination with the X is suppressed, so that hermaphrodite- and male-specific regions can be defined (Liu et al. 2004; Wang et al. 2012). The corresponding region of the X is only 3.5 Mb, and both the Y and Yh have increased repeat sequence content, changed physical structure, and different gene content (Wang et al. 2012). Any combination of the Y and Yh chromosomes (YY, YYh, or YhYh) is inviable, and the embryos abort 25–50 d after pollination, suggesting that the Y chromosome types are similar and that both are missing an essential gene that is functional in the X.
Wild papaya populations are dioecious, with one-half male and one-half female plants, whereas cultivated papaya is predominantly gynodioecious, with two-thirds hermaphrodite and one-third female plants, though dioecious varieties do exist. There is no direct archaeological evidence for the center of origin of papaya, but the presence of natural populations in Mexico and Central America and the cultivation in Mexico and Belize predating the Spaniards suggest a Mesoamerican origin (Colunga-GarcíaMarín and Zizumbo-Villarreal 2004). William Storey (1976) wrote, “Since dioecism seems to be the evolutionary norm in Caricaceae, it is possible that ambisexual forms owe their continued existence to human selection.” This hypothesis was previously rejected after analysis of a pair of X- and Y-specific bacterial artificial chromosomes (BACs) from an improved (but not cultivated) dioecious variety, AU9, and their homologous BAC from the gynodioecious cultivar SunUp. The resulting molecular dating estimate suggested that the Y chromosomes of males and hermaphrodites diverged ∼73,000 yr ago (Yu et al. 2008), long before the origin of agriculture in Mesoamerica ∼6200 yr ago (Pope et al. 2001). It is worth further testing to establish the age of the HSY, because if the HSY diverged very recently from the MSY, papaya could offer the opportunity to identify the gene or genes responsible for the gender difference. Such genes would be candidates for the female suppressor involved in the early stages of sex chromosome evolution in this species. The sex chromosomes in other organisms, such as mammals, are ancient (Veyrunes et al. 2008; Bellott et al. 2014; Cortez et al. 2014), and the genes involved in their initial evolution cannot be identified, because many subsequent changes, including gene gains and losses, have occurred (Hughes et al. 2010, 2012; Zhou et al. 2014). The younger sex chromosomes of some species of plants, fish, and insects may provide insights into the mechanisms involved in the early stages of the evolution of separate sexes and of sex chromosomes (Delph et al. 2010; Zhou and Bachtrog 2012).
To understand the origin and accurately estimate the divergence time of the HSY and MSY, sequencing of HSY and MSY sequences is needed. Here, we describe complete sequencing of the AU9 MSY. Moreover, sequencing multiple individuals of both males and hermaphrodites is necessary, because the origin or origins of hermaphrodites are unknown, and the AU9 MSY might not be closely related to the ancestor of the HSY, as we indeed show to be the case. Moreover, sequences from multiple individuals are needed, because the HSY and MSY of any single varieties may include mutations in genes that are not responsible for the phenotypic difference in their gender; only fixed differences between the Yh and Y are candidates for causing the functional difference (though variants in individual varieties can help exclude candidate genes because the female-suppressor is dominant, causing maleness or hermaphroditism in the heterozygous XY or XYh state, respectively). The objectives of this study were to (1) sequence the MSY as a necessary step toward identifying the genes distinguishing the Y and Yh chromosome; (2) determine which Y chromosome, the Y or Yh, is ancestral in papaya and identify the origin of the derived MSY or HSY by resequencing male and hermaphrodite genomes sampled from wild and domesticated populations; (3) use the sequences from wild populations to estimate diversity and thus test the domestication hypothesis independently of the molecular dating based on MSY–HSY divergence; and (4) use the sequences from wild populations to discover genes with fixed differences between the sets of Y-specific sequences of males and hermaphrodites, where the differences may affect gene functions, to generate candidate genes for the Y-linked carpel suppressor. The Yh chromosome of hermaphrodites differs from the Y of males by lacking a female suppressor. The finding that the papaya Yh and Y diverged very recently is therefore important, because it suggests that the number of fixed differences between the two chromosomes may be small. Sequences from papaya males from natural populations therefore offer the opportunity to identify the gene or genes responsible for the gender difference.
Results
Sequencing and analysis of a papaya MSY
The papaya HSY has been sequenced previously, using BAC libraries from the gynodioecious cultivar “SunUp,” a popular commercial transgenic cultivar from Hawaii (Wang et al. 2012). To study the MSY, the dioecious “AU9” cultivar from the USDA germplasm repository in Hilo was selected, because it was one of the few dioecious accessions available with a desirable fruit appearance. AU9 was collected from a papaya breeding program at Griffith University in Queensland, Australia, which was traced back to a papaya breeding program in Israel with plants of unknown origin. Because of the pericentric location and high repeat content of the Y-linked region, we sequenced the papaya MSY using a reiterative BAC-by-BAC approach. A physical map of the papaya MSY was constructed from the AU9 male BAC library, using probes from BACs in the HSY physical map (Gschwend et al. 2011). Candidate BACs were confirmed by a combination of PCR and BAC end sequencing, and gaps in the physical map were filled using chromosome walking (see Methods). The MSY physical map consists of a minimum tiling path of 99 BACs (Supplemental Fig. 1) with a combined nonoverlapping length of 8.1 Mb. The MSY BACs were sequenced using 454 sequencing technology (Roche) to an average depth of 50× for each BAC. Assembled contigs were anchored using the HSY as a reference, and some gaps were filled with Illumina whole-genome shotgun reads of AU9.
The HSY and MSY sequences were aligned and their nucleotide divergence was estimated to be 0.4%, with 32,517 sequence differences in total. The MSY and HSY sequences are collinear, except for an 8398-bp insertion in the MSY corresponding to a newly integrated Ty3-gypsy retrotransposon; this transposon insertion is the cause of the earlier overestimation of the 1.4% sequence divergence between MSY and HSY, which was based on only one pair of homologous BACs (Yu et al. 2008). There may be other small-scale differences between the HSY and MSY because of small sequence gaps in both assemblies. Most BACs are complete, but some highly repetitive BACs (>90% repeats) remain in multiple, ordered contigs. Overall, the gene content, exon structure, and gene order are conserved between the entire HSY and MSY regions (Fig. 1). A SVP-like gene has a copia-like retrotransposon insertion in the first intron of the HSY allele, resulting in a transcript with a partial sequence of the second intron (Ueno et al. 2014). Ninety-four transcription units were annotated on both Y chromosomes. Of these, 66 are complete, intact protein coding genes with annotated start and stop codons, and 28 (30%) are pseudogenes (truncated gene fragments, or sequences whose coding regions contain premature stop codons) (Supplemental Tables 1, 2). In HSY and MSY sequences, 39 (59%) of the intact genes have identical sequences in the coding region. The coding regions of seven genes (11%) include only synonymous differences, and 20 genes (30%) have only nonsynonymous differences (Supplemental Table 3). Below, we describe HSY and MSY sequences from more individuals, which exclude most of these genes with differences as candidates for the hermaphrodite-male difference.
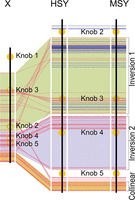
Sequence conservation between the HSY and MSY. Order of genes in the papaya MSY, HSY, and X. Red lines denote genes found in the X, Yh, and Y, and blue lines denote Y chromosome–specific genes. The heterochromatic knob structures are in yellow.
The previously sequenced HSY and X revealed two major inversions, one of which (inversion 1) was inferred to have caused the initial recombination suppression between the X and Y chromosomes (Ming et al. 2011; Wang et al. 2012). The sequence of the AU9 MSY also has these inversions, which therefore distinguish both Y chromosomes from the X. In both the MSY and the HSY, inversion 1 includes 30 of the 66 intact Y-linked genes, including all 16 that have no X-linked counterparts, as well as 21 pseudogene sequences (Supplemental Table 1). Inversion 2 includes 18 genes that appear to correspond to complete transcripts, all of them with X-linked counterparts, plus three pseudogenes. Finally the MSY and HSY share a “collinear region,” adjacent to border B of inversion 2, which includes 18 genes, all with X and MSY (and HSY) copies, and two pseudogenes.
Repetitive sequences make up ∼79.2% of the MSY pseudomolecule, similar to the HSY, which has 79.3% repetitive element content. As with the HSY, LTR elements dominate the MSY repetitive element content, making up 42.7% of these sequences from the MSY (Supplemental Table 4). In contrast, the corresponding X region has a total repetitive element content of 67.2%, lower than either Y chromosome though higher than the papaya genome-wide average of 51.9% (Ming et al. 2008; Wang et al. 2012; Na et al. 2014).
Y chromosome sequence variation, population structure, and fixed Y–Yh differences
The high sequence conservation between the MSY and HSY led us to assess their diversity in wild and cultivated papaya populations, as this can help identify both the origin of the Yh chromosome and the differences found between all such chromosomes and the Y of males. We selected hermaphrodites from 12 gynodioceous commercial cultivars with diverse origins and agronomic traits (Supplemental Fig. 2) and males from 24 dioecious wild papaya accessions collected from 10 populations across Costa Rica for whole-genome resequencing (Supplemental Table 5). The wild papaya accessions were chosen based on the previous finding, based on four pairs of X/Y genes, that three distinct populations of Y chromosomes can be identified: MSY1, MSY2, and MSY3 (Weingartner and Moore 2012). We used individuals from the same populations.
After filtering and aligning the reads to the papaya SunUp draft genome and the HSY sequence (see Methods) (Ming et al. 2008), the average sequencing depth for each individual was 15.6× for autosomal sequences and 7.8× for the haploid HSY and X regions (Supplemental Table 4). In the complete data set, we identified a total of 3,301,017 polymorphisms (SNPs and small indels) genome wide and 66,579 Y-specific variants (shared by all MSY and HSY sequences and not found among the 36 X-linked ones) (see Supplemental Table 6). The vast majority of the Y polymorphisms are in intergenic regions, and only 3885 are in coding regions (3685 in introns and 180 in exons). One hundred nine of the 180 exon variants (61%) polymorphisms are nonsynonymous; 69 (38%) are synonymous; and two (1%) cause frame shifts in coding genes. Most of the Y-linked variants are private to single (or a few) individuals. For example, one of the two frame-shift variants is in CpXY26, a phospholipid-transporting ATPase that has a premature stop codon in the cultivated variety HCAR314, and the other is a premature stop codon in gene CpXY10 in the wild male Cp112; this gene is a homolog of FLOWING LOCUS T. Neither of these is a candidate for the difference between males and hermaphrodites, as they are not fixed between the sets of plants of these two sex types. We discuss the few fixed differences below.
The SNPs in Y-specific regions of the MSY were used to investigate the population structure of the Y chromosomes. Model-based analysis, maximum likelihood phylogenetic reconstruction, and principal component analysis (PCA) all confirm the previous finding of three distinct populations of Y chromosomes (Fig. 2B–D; Weingartner and Moore 2012). The MSY1 and MSY2 Y haplotypes were found in wild populations from two geographic regions on the two opposite coasts of Costa Rica (with nine and six males from six and five subpopulations, respectively), and three of these subpopulations (operational populations 8, 10, and 2 in Supplemental Table 4) include plants with both these groups. The MSY3 haplotype group was found in a total of nine wild males, all from four subpopulation samples from the north Pacific region of Costa Rica.
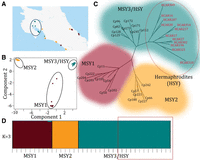
Population structure of the papaya Y chromosomes. (A) Geographic distribution of the wild papaya populations sampled in Costa Rica. Populations with the MSY1 haplotype are shown in brown; those with MSY2 in orange. Populations with MSY3 haplotypes (from which the HSY haplotype evolved) are in blue and circled. All these wild accessions have GPS coordinates, but some individuals were close together and have overlapping GPS coordinates, so that the figure has fewer sites than sampled individuals. (B) Principal component analysis (PCA) based on all 58,000 Y-specific SNPs and indels. The MSY3/HSY and MSY2 clusters each corresponds to narrow geographic distributions, while populations with MSY1 are more widely distributed. Circles signify statistically different clusters. (C) Maximum likelihood phylogenetic tree based on the 58,000 Y chromosome SNPs. Wild accessions are labeled Cp, and the domesticated varieties are labeled HCAR and are circled in red. (D) Population structure analysis using STRUCTURE (Falush et al. 2003). Each accession is represented by a vertical bar, and the lengths of the colored segments represent the contribution of each subgroup. The STRUCTURE statistics are listed in Supplemental Table 7. Domesticated varieties are outlined in red.
All the HSY haplotypes from cultivated hermaphrodites cluster with MSY3 haplotypes, strongly suggesting that the hermaphrodite Yh chromosome evolved from this region. When the Y-specific SNPs from the same genomic regions are compared in all cases, the set of HSY sequences differ from those of the nine MSY3s by an average of 2729 SNPs, an order of magnitude fewer differences than from MSY1 (31,781) or MSY2 (22,777). To quantify the differences, we compared pairwise FST values, which measure variance in allele frequencies between populations in terms of the proportion of diversity between subpopulations relative to the total genetic diversity, as opposed to within them (Charlesworth 1998). High FST values indicate population structure, and this can arise either (1) because the populations have been isolated for long enough evolutionary time to have diverged in sequence (e.g., races or subspecies) or (2) because the sequences within populations are very similar in comparison with those from different populations, which is predicted for Y chromosomes, because nonrecombining regions have reduced effective population size (Ne) (Charlesworth 2009). As shown in Figure 3, FST for the Y region is low between HSY and MSY3 (FST = 0.02) and much higher for MSY1 (FST = 0.491) or MSY2 (FST = 0.297).

Pairwise FST between the published HSY and the three other Y haplotype subgroups: MSY1 (green), MSY2 subgroup (brown), and males from the MSY3/HSY subgroup (dark gray).
The MSY1, MSY2, and MSY3 populations are not, however, highly isolated, but rather the observed high FST values are likely to be a property of the Y chromosomes, which are expected to show strong population structure for reason 2 above. Y-linked sequences are expected to have low within-population diversity, due to genetic hitchhiking processes that occur in nonrecombining regions. In Silene latifolia, for example, Y chromosome population structure is also strong (Ironside and Filatov 2005). This interpretation for the minimal differentiation (FST = 0.02) between the HSY sequences from hermaphrodites and the MSY3 sequences is supported by estimates of diversity of Y sequences within populations. Of the total of 66,579 polymorphisms found throughout all the Y chromosomes sequenced (see above), the combined MSY3/HSY set includes only 14,058; 2729 polymorphisms were found only in the wild males, and even fewer (1097) only in the cultivated hermaphrodites. As discussed further below, many fewer variants are completely associated with the difference in sex type; four variants specific to the HSY are found in all hermaphrodites; and only two variants are fixed in the MSY sequences of all males.
We also examined diversity for genes in the recombining regions of Chromosome 1 (the same chromosome whose fully sex-linked region carries the sex-determining genes). Under our interpretation, these recombining regions, the so-called pseudoautosomal region (PAR), should not show strong population structure. Population structure was indeed much less in the PAR. First, FST values for PAR sequences are very similar between the HSY sequences and MSY sequences from populations with predominantly MSY1, MSY2, and MSY3 haplotypes (FST = 0.11, 0.12, and 0.10, respectively) (Fig. 3), consistent with little population structure for this genome region. The FST values for the PAR are statistically indistinguishable between the wild populations (FST MSY1 vs. MSY2 = 0.08, MSY1 vs. MSY3 = 0.09, MSY2 vs. MSY3 = 0.09) (Supplemental Table 8), contrasting with the distinct structure observed for the fully Y-linked region. Finally, STRUCTURE, phylogenetic analysis, and PCA analysis of the PAR gene sequences are consistent with the FST analysis and show no evidence of population structure correlated with that of the Y-linked region (Fig. 4A–C). Instead, all the analyses of PAR sequences show one set of populations containing all the sequences from males and separate from the hermaphrodites, suggesting that gene flow occurs between the wild papaya populations.
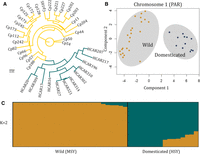
Population structure of the papaya PAR. (A) Maximum likelihood phylogenetic tree based on the 193,621 Chromosome 1 SNPs. Domesticated (hermaphrodite) varieties are shown in blue and wild (male) accessions are shown in yellow. (B) PCA based on all Chromosome 1 (PAR) SNPs and indels. The two distinct clusters, of sequences from males and hermaphrodites, are shaded in gray. (C) Population structure analysis using STRUCTURE (Falush et al. 2003). Each accession is represented by a vertical bar, and the length of each colored segment represents the contribution of each subgroup.
Gene flow is supported by the observation mentioned above of multiple Y sequences within some wild populations. Coexistence of very different Ys is not unexpected. First, even if gene flow brings different Y chromosome haplotypes into the same population, they will remain distinct because recombination between them is suppressed. Second, fixation of one Y haplotype across the species is unlikely given the large geographic region in which wild papaya populations occur, which means that mutations arising in one region will not reach distant populations for many generations.
The HSY is a product of human domestication
The scarcity of fixed differences between MSY3 and HSY sequences suggests very recent divergence. In contrast, the fully sequenced AU9 MSY does not cluster with any of the Costa Rican Y populations and is highly diverged from the other Y-linked sequences (Fig. 5). This divergence led to the previous conclusion that the hermaphrodite–male divergence time is long and greatly predates domestication. With our new sequence results, this is clearly incorrect. Rather, this accession is unusual and is not closely related to the ancestor of cultivated hermaphrodite papayas.
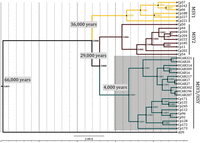
Bayesian analysis of Y chromosome divergence times. The MSY1 haplotype subgroup is shown in yellow, MSY2 in brown, and MSY3/HSY in blue. The “AU9” MSY forms an outgroup (in black) to the three populations from Costa Rica. The MYS3/HSY subgroup split is highlighted in gray. Node lengths represent estimated synonymous substitutions per site, which were used with a corrected molecular clock (see Methods) to estimate the divergence times.
We estimated the MSY3–HSY divergence time using a Bayesian approach in BEAST (Heled and Drummond 2010), using all 105,953 synonymous sites in coding sequences of the HSY and MSY regions. We used a molecular clock of 7 × 10−9 substitutions per site per year estimated for Arabidopsis, corrected by a factor of 0.672 to take account of the slower molecular evolution in papaya (see Methods). The estimated synonymous site divergence between the HSY and MSY3 yields a time of ∼4000 yr ago, with a 95% highest posterior density (HPD) of 1400–6700 yr ago. This is within the time of origin of agriculture in Mesoamerica (Fig. 5; Pope et al. 2001). The estimate for the HSY from the MSY1 group is 29,000 yr (95% HPD = 24,600–34,510) and from MSY2 is 36,000 yr (95% HPD = 27,000–42,840), both well before the origin of agriculture even in the Fertile Crescent in the Middle East ∼10,000 yr ago (Gupta 2004). We also estimated the divergence time between HSY and the MSY from AU9 using the same molecular clock. This yielded 66,000 yr (95% HPD = 54,600–78,540), similar to the 73,000 yr reported previously (Yu et al. 2008).
Patterns of divergence and diversification between the Y chromosomes and the PAR
If hermaphrodites indeed resulted from human domestication, the HSY should (unlike the wild MSY and the PAR) show evidence for a bottleneck, either due to (1) an initial selection of just one or a few hermaphrodite individuals or to (2) subsequent selection for desirable characteristics. We therefore estimated nucleotide diversity (π). Our new estimates for both Y-linked and other regions are similar to previously published estimates based on Sanger sequencing, indicating that the high-throughput sequencing approach (see Methods) yields reliable estimates (Weingartner and Moore 2012). The π value estimated for our pseudoautosomal sequences is slightly lower in cultivated than wild papaya (0.0017 vs. 0.0020), perhaps due to an initial bottleneck, but the difference is not significant (P = 0.15, based on a Wilcoxon test using all the values estimated for all genes sequenced). In contrast, nucleotide diversity is much lower among the HSY sequences (0.0004) than the wild MSY ones (0.0021, similar to the value for the PAR diversity estimated from the wild populations); the HSY–MSY difference is significant and is also significantly lower than the PAR value (in both cases Wilcoxon test, P < 1 × 10−5, Fig. 6A). The low diversity specific to the HSY suggests a reduction in diversity during domestication.
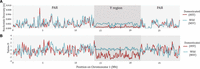
Contrasting levels of divergence and diversification between the Y chromosomes and the PAR. All analyses are plotted in intervals of 100 kb and a sliding window of 25 kb. (A) Nucleotide diversity (π) within the wild male (MSY) and cultivated hermaphrodite (HSY) groups. (B) Tajima's D-values within the males and hermaphrodites. Values near zero are consistent with a population at equilibrium under neutral evolution (i.e., without a recent bottleneck or expansion, and without strong selection).
Finally, we used Tajima's D-test to assess whether the Y chromosomes show evidence for a severe bottleneck, which should lead to negative Tajima's D-values (Tajima 1989). The HSY has the expected strongly negative mean Tajima's D (mean −1.26, P < 0.05, Wilcoxon test), consistent with a bottleneck or selective sweep during domestication. In contrast, the mean Tajima's D-values across the PAR region of Chromosome 1 are slightly negative, but close to zero in both males and hermaphrodites (−0.017 and −0.154, respectively), and that for the MSY is 0.13, also not significantly different from zero (Fig. 6B).
Discussion
We cannot, of course, identify the exact geographic location of papaya domestication, though our analyses show that the AU9 haplotype (whose origin is currently unknown) is not a potential ancestor of the HSY of the currently cultivated hermaphrodite strains. Additional populations of MSY may exist in Central America, and additional sampling of MSY wild accessions from the Yucatan peninsula in Mexico to Nicaragua bordering Costa Rica could reveal haplotypes like those in the AU9 MSY and could also reveal other populations with the MSY3 haplotype that do appear to be the progenitor of cultivated hermaphrodites. We have shown that gene flow occurs between natural papaya populations and that Y chromosomes can migrate, sometimes leading to populations with two different Y haplotypes. Discovery of the full natural range, and more surveys of Y haplotypes natural populations may reveal that the region in Costa Rica identified as the source of the hermaphrodites’ Yh haplotype is not the only one where the MSY3 haplotype occurs.
Our analyses do, however, strongly suggest that divergence of this haplotype occurred recently from the MSY3 male Y haplotype, based on the remarkable sequence similarity with the HSY of hermaphrodites, despite the gender difference. Our analyses date the divergence of HSY and the MSY3 sequence to ∼4000 yr, well after the domestication of crop plants in Mesoamerica >6200 yr ago (Pope et al. 2001) and coinciding with the rise of Maya civilization ∼4000 yr ago (Pohl et al. 1996; Colunga-GarcíaMarín and Zizumbo-Villarreal 2004). Given that no hermaphrodite papayas have been found in wild populations in Central America, this strongly suggests that the HSY resulted from papaya domestication by the Mayans or other indigenous cultures, supporting Storey's hypothesis (Storey 1976).
Now that the haplotype from which the HSY evolved has been identified, identification of the sex determination gene controlling carpel abortion that defines the hermaphrodite Yh chromosome should become possible. The evolution of separate sexes in plants requires two mutations, one resulting in male sterility and a second in female sterility (for review, see Ming et al. 2011; Bachtrog et al. 2014). The appearance of the first mutation results in sexual polymorphism, either androdioecy (males and hermaphrodites) or gynodioecy (females and hermaphrodites), until a second mutation arises at a linked site and converts the hermaphrodites into females or males (or partially female or male phenotypes). Intermediates to complete dioecy are found in many plants, such as spinach, which has mostly males and females but occasionally hermaphrodites (Bemis and Wilson 1953), and strawberry, where males, females, hermaphrodites, and neuters are present because linkage between the factors is incomplete (Spigler et al. 2008). The hypothesis that two (or more) genes are involved in plant sex determination predicts that reversion through recombinants in the region could occur. Our results indicate that gynodioecy in domesticated papaya populations is a reversion from dioecy rather than an intermediate state in the evolution of dioecy. The extreme similarity between the HSY and MSY sequences suggests that recombination within a sex-determining region was not involved; had a recombination event occurred, the HSY should include regions of sequence similar to X chromosomes from the ancestral population.
The carpel-suppressing gene in males presumably evolved from a gain-of-function mutation in the Y chromosome. Reversion to hermaphroditism could have involved either an autosomal mutation that prevents expression of the Y-linked gene controlling carpel abortion, or a loss of the carpel-suppressor function, while the sex determination gene promoting stamen development was retained by the hermaphrodites’ Yh chromosome. Unlike the hermaphrodites that have been observed in Silene latifolia (Lardon et al. 1999; Bergero et al. 2008; Fujita et al. 2012), our sequencing and assembly of the papaya HSY show clearly that deletions of parts of the Y appear not to be involved. Therefore, a small-scale, Y-linked mutation seems likely. Moreover, a Y-linked mutation predicts a strong selective sweep in the Yh, given the brief evolutionary time since the event, so that the Yh chromosome should have almost the same sequence as the ancestral Y chromosome, as is observed. We also observe the expected much more negative Tajima's D-values than for the MSY or PAR sequences analyzed. This is consistent with the population size of the HSY having been reduced to a single haplotype in a domestication event when a rare hermaphrodite was selected that carried a sex reversal mutation in the Y chromosome. If multiple hermaphrodites had been selected from a population with MSY diversity like that we estimate, this would lead to positive Tajima's D-values, because the most common haplotypes would have been selected, leading to a deficiency of rare variants after the event.
Having excluded either recombination with the X chromosome or a deletion of part of the Y chromosome, we therefore conclude that comparison of our multiple HSY and MSY sequence differences should yield candidates for a mutational difference. The gene responsible in papaya can potentially be identified by comparing the sequences of the sets of Y and Yh chromosomes. These chromosomes are extremely similar, and therefore the numbers of candidates are not large; so identification does not rely on being able to generate recombinant Y chromosomes to exclude multiple candidate differences that might be responsible.
Our study clarifies the choice of strain to be used to attempt identification of the gene controlling carpel abortion. As the AU9 variety is a distant outgroup from the MSY3 population from which the HSY evolved, it is clearly not suitable, but instead the MSY3 haplotype should be used. The information that the AU9 cultivar's MSY is distant from the HSY does, however, allow us to conclude that the many nonsynonymous differences identified between the AU9 MSY and the HSY cannot be involved in sex determination, as none of these are found between MSY3 and HSY. Interestingly, none of the fixed SNPs distinguishing the HSY or hermaphrodites from the MSY3 of males are in coding regions, suggesting that hermaphroditism may be controlled by changes in gene regulation, involving upstream or downstream changes in enhancers, trans-acting factors, small RNAs, or epigenetic effects, making the identification of the sex determination gene suppressing carpel development a challenging task.
Methods
AU9 MSY BAC library screening, minimum tiling path construction, and DNA isolation
We initiated the MSY physical map construction by screening the papaya male AU9 BAC library with probes designed from HSY-specific BAC sequences. The AU9 BAC library was screened, following the DIG high primer DNA labeling and detection starter kit II (Roche) protocol, and positive BACs were confirmed using PCR and BAC end sequencing. New probes were designed from these positive BACs to screen the BAC library. Chromosome walking continued until a minimum tiling path of MSY BACs spanned the entire sex determining region. Mini-prep BAC DNA isolation was performed to check the insert size of each BAC via CHEF gel electrophoresis. The BACs were grown overnight at 37°C using glycerol stock from a single colony, and BAC DNA was isolated using the BACMAX DNA purification kit from Epicentre (catalog no. BMAX044).
AU9 MSY sequencing and assembly
Each MSY BAC clone in the minimum tiling path of the physical map was fully sequenced utilizing 454 sequencing technology (Roche) at the Keck Center at University of Illinois Urbana-Champaign. After quality trimming, reads (with an average length of 580 bp) were assembled into contiguous sequences using gsAssembler (Roche) with the default settings. The assembled contigs (N50 35 kb) were anchored to the HSY reference sequence, and gaps were filled using a reference guided assembly approach with whole-genome shotgun reads from the AU9 cultivar in the CLC Genomics Workbench version 5.1 under default settings (CLC Bio). Genomic DNA from AU9 was extracted from young leaf tissues using the DNeasy plant mini kit (Qiagen). Paired-end DNA-seq libraries with an average insert size of 400 bp were made using the Illumina TruSeq DNA LT kit (ID: FC-121-2001) according to the manufacturer's instructions (Illumina). The AU9 MSY was annotated based on homology to genes in the HSY and de novo gene prediction and RT-PCR was used to verify the annotations.
Gene annotation in the MSY
We identified transcripts in the MSY using complementary approaches. First, we searched the papaya MSY for homologs of all the known papaya HSY and X transcripts previously reported to identify possible X and Y transcripts that had been lost in the MSY but retained in the HSY. Second, we searched for additional transcripts in the MSY. When applicable, we inversely searched the HSY for the additional genes predicted on the MSY to identify their HSY orthologs. We validated putative genes by RT-PCR using RNA from mixed tissues and through homology to orthologs in public databases. Each predicted transcript was manually annotated and translated in six frames to distinguish the protein-coding genes and pseudogenes. We classified transcripts with premature stop codons, frame-shift mutations, or truncated proteins as pseudogenes. Potential functions for each transcript were predicted using conserved domains and homologous gene functions. The CpGAT, A Comprehensive Pipeline for Genome Annotation on PlantGDB webserver (http://www.plantgdb.org), was used for additional MSY gene annotation. The repeat-masked MSY sequences were blasted (TBLASTX) to the papaya transcriptome ESTs, gene model databases, and NCBI public nr protein data sets, including protein sequences from species such as Arabidopsis thaliana, Populus trichocarpa, Vitis vinifera, Oryza sativa, and Medicago truncatula, for transcription unit identification. Gene prediction programs FGENESH (Softberry, Inc.), AUGUSTUS (Stanke et al. 2004), and GeneMark (Besemer and Borodovsky 2005) were also used to predict additional genes that may have been missed.
MSY and HSY alignments and synteny
MSY and HSY synteny analysis and dot plot comparison were performed using the SyMAP program (Soderlund et al. 2011) with the default settings. Global chromosome similarity alignments were performed using the genome alignment tool Mauve (Darling et al. 2004) with the default settings.
Whole-genome resequencing sample preparation
Samples from Costa Rica were dried on silica gel in the field and stored at −80°C, and leaf tissue from cultivated varieties was collected from greenhouse grown plants and stored at −80°C. Genomic DNA was extracted from young leaf tissues using the DNeasy plant mini kit (Qiagen). Paired-end DNA-seq libraries with an average insert size of 400 bp were made using the Illumina TruSeq DNA LT kit (ID: FC-121-2001) according to the manufacturer's instructions (Illumina).
Whole-genome resequencing, alignment, and SNP calling
Twenty-four wild male papaya plants from 10 natural populations across Costa Rica (see Results section) and 12 hermaphrodites from gynodioecious cultivars from the USDA tropical plant germplasm collection in Hilo, Hawaii were sequenced. The libraries from each of the 36 individuals were sequenced on an Illumina HiSeq 2500, generating 2.8 billion 100-bp Illumina reads, representing an average of 15.6× coverage for pseudoautosomal loci and 7.8× coverage for each of the X and Y chromosomes. The reads were aligned to the SunUp papaya draft genome sequence (Ming et al. 2008) and its HSY pseudomolecule (Wang et al. 2012) using the Burrows-Wheeler Aligner with strict parameters of maximum edit distance (-n) of 0.02 and a high mismatch penalty (-M) of five (Li and Durbin 2009).
Differentiating the two haplotypes of the X- and Y-linked regions is generally simple, as the intergenic regions are highly diverged and largely unalignable, and the genic regions have an average of 3%–7% sequence divergence across all site types (and higher for synonymous or noncoding sites). The collinear region, however, has the same gene content and order in the X and Y regions, with at most 2% sequence divergence, rising to complete identity with physical distance from the sex-linked region, and X and Y haplotypes cannot be clearly distinguished. Indeed, DNA sequence divergence cannot determine with certainty whether this region is fully sex-linked or is within PAR, because the closely linked boundary region on the PAR side is expected to be slightly differentiated, just as regions very closely linked to any site maintained polymorphic by balancing selection will exhibit sequence variants associated with the functionally different alleles (Charlesworth 2006; Kirkpatrick et al. 2010). The presumptive border region between the PAR and the collinear region (∼300 kb) was therefore excluded from our analyses of SNP density, diversity, FST, and Tajima's D. To phase reads in the rest of the (presumptively fully sex-linked) collinear region, we used strict alignment parameters (allowing two mismatches per read) and a high mismatch penalty. Collinear region SNPs were screened for previously reported polymorphisms between the X and Y sequences (Wang et al. 2012) to ensure correct haplotype phasing. The alignments were manually inspected during parameter optimization, using Tablet (Milne et al. 2010), to select for the correct stringency without excluding true polymorphisms. SNPs found in both the X- and Y-linked haplotypes were also removed from the analysis, as these may have represented alignments including both X and Y reads or repetitive regions, and we are interested largely in site differences fixed among all Y sequences and in distinguishing them from homologous X-linked sequences.
SNPs and indels were called using the SAMtools package (Li et al. 2009). A raw file of unfiltered SNPs and indels was generated using mpileup under the default parameters. Such polymorphisms were called using all individuals simultaneously to provide accuracy for low-frequency or low-coverage variants. Low-coverage and repetitive polymorphisms with coverage of less than four and more than 20–60× (representing 3× the expected genome coverage of each accession) were removed, with the upper threshold depending on the mean coverage for each individual. SNPs/indels with collective root mean square (RMS) and mapping qualities (Phred scores) less than 25 were also removed from the analysis. To test the accuracy of this method for calling SNPs, we compared SNPs generated by the SAMtools pipeline from the AU9 MSY using whole-genome shotgun reads with the BAC-by-BAC–assembled “AU9” pseudomolecule. Most SNPs (93%) were found by both analyses.
Population analysis
Maximum likelihood phylogenetic trees were constructed using SNPhylo (Ruden and Lu 2012) applied to the 66,579 Y-linked and 217,446 PAR high-quality variants. SNPhylo is able to process a large number of SNPs. The high-quality Y SNPs were concatenated and aligned by MUSCLE (Edgar 2004) and were used to construct the ML tree using DNAML in a highly automated pipeline.
To infer the population structure of the Y chromosomes and PAR genes, we used the program STRUCTURE (Falush et al. 2003). The number of clusters (K) was determined using the methods outlined by Evanno et al. (2005). K values from two to 10 were run 10 times through STRUCTURE, and the ΔK was calculated to assess the most likely number of populations. For the Y chromosome and the recombining regions of Chromosome 1, the ΔK statistic clearly supports three and two subpopulations, respectively. Ten thousand iterations were used to determine the subgroup membership of each wild and cultivated accession. The principal component analysis was performed using the PCO software (https://www.stat.auckland.ac.nz/~mja/Programs.htm).
Dating HSY divergence
The divergence time of the HSY was estimated in the Bayesian analysis program BEAST (v1.8.0) (Heled and Drummond 2010) using a modification (see below) of the molecular clock estimated for Arabidopsis (Ossowski et al. 2010). A total of 105,953 coding sites across the Y chromosome were used. The species tree was constructed using a Yule prior, which assumes that Y lineages split at a constant rate.
Fifty million Markov chain Monte Carlo (MCMC) chains were run for the BEAST analysis and, after discarding the burn-in, convergence was verified using the program TRACER (http://tree.bio.ed.ac.uk/software/tracer/). The tree with the highest clade posterior probabilities was chosen for divergence time estimates using the TreeAnnotator program from BEAST. A strict molecular clock model was used with a rate suitable for papaya of 0.0168 substitutions/synonymous site/million years, which was obtained as follows. The closest plant taxon for which a molecular clock has been estimated is the closely related family Brassicaceae. The synonymous substitution rate has been estimated for Arabidopsis relatives to be 4 × 10−9 substitutions per synonymous site per year (Beilstein et al. 2010). Assuming one generation per year for Arabidopsis thaliana, this estimate is similar to the synonymous site mutation rate estimate for the genus Arabidopsis of 7 × 10−9/generation (Ossowski et al. 2010). To take account of the slower molecular evolutionary rate with fewer generations in papaya because of its perennial nature, we reduced the latter value by a factor of 0.672 based on differences in Ks values between duplicate genes that formed in the gamma whole-genome duplication event in the core eudicots, which occurred before the split between papaya and the Brassicaceae, allowing one to estimate a factor corresponding to the relative molecular evolutionary rates of different species (Bowers et al. 2003).
Population genetic analyses
Y-specific SNPs were classified as noncoding, synonymous, or nonsynonymous based on the Y gene annotations reported by Wang et al. (2012) using the program SNPeff (Ruden and Lu 2012). Nucleotide diversity (π) and Tajima's D were calculated within the wild papaya and cultivated papaya samples in sliding windows of 100 kb with a 25-kb overlap, using a suite of programs in VCFtools, including --window-pi-step and --TajimaD, respectively (Li et al. 2009). Wilcoxon tests were implemented in R (R Core Team 2014). FST was estimated using pairwise comparisons of the HSY against the MSY sequences from the MSY3, MSY1, and MSY2 subgroups, using Weir and Cockerham's method (Weir and Cockerham 1984) implemented in VCFtools (using the --weir-fst-pop option).
Data access
Trimmed and quality-filtered Illumina reads for the 36 resequenced papaya genomes with alignment files and MSY sequence have been deposited in the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA271489 and GenBank (http://www.ncbi.nlm.nih.gov/genbank) under accession number CP010988.
Acknowledgments
This work was supported by National Science Foundation (NSF) Plant Genome Research Program Awards DBI0553417 and DBI-0922545 (to R.M., Q.Y., R.C.M., and P.H.M.) and startup funds from Fujian Agriculture and Forestry University to R.M.
Author contributions: R.M., R.V., Q.Y., and P.H.M. conceived the study. R.M., R.V., Q.Y., P.H.M., R.C.M., J.Z., and F.Z. designed the experiment. F.Z., C.C., J.W., J.K.N., J.H., A.R.G., R.A., J.G., Q.Y., and R.M. conducted physical mapping and sequencing of the MSY. L.H., L.Z., W.M., and J.Z. sequenced the 36 genomes. R.V., F.Z., C.C., J.Z., C.M.W., J.H., R.A., A.R.G., J.W., J.K.N., L.H., L.Z., M.W., J.A., D.C., R.G., Q.Y., and R.M. contributed to the analyses. R.V., R.M., and D.C. wrote the manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.183905.114.
- Received September 14, 2014.
- Accepted February 9, 2015.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








