
Large-scale genomic sequencing of extraintestinal pathogenic Escherichia coli strains
- Stephen J. Salipante1,4,
- David J. Roach2,4,
- Jacob O. Kitzman2,
- Matthew W. Snyder2,
- Bethany Stackhouse2,
- Susan M. Butler-Wu1,
- Choli Lee2,
- Brad T. Cookson1,3 and
- Jay Shendure2
- 1Department of Laboratory Medicine,
- 2Department of Genome Sciences,
- 3Department of Microbiology, University of Washington, Seattle, Washington 98195, USA
- Corresponding authors: stevesal{at}uw.edu, shendure{at}uw.edu
-
↵4 These authors contributed equally to this work.
Abstract
Large-scale bacterial genome sequencing efforts to date have provided limited information on the most prevalent category of disease: sporadically acquired infections caused by common pathogenic bacteria. Here, we performed whole-genome sequencing and de novo assembly of 312 blood- or urine-derived isolates of extraintestinal pathogenic (ExPEC) Escherichia coli, a common agent of sepsis and community-acquired urinary tract infections, obtained during the course of routine clinical care at a single institution. We find that ExPEC E. coli are highly genomically heterogeneous, consistent with pan-genome analyses encompassing the larger species. Investigation of differential virulence factor content and antibiotic resistance phenotypes reveals markedly different profiles among lineages and among strains infecting different body sites. We use high-resolution molecular epidemiology to explore the dynamics of infections at the level of individual patients, including identification of possible person-to-person transmission. Notably, a limited number of discrete lineages caused the majority of bloodstream infections, including one subclone (ST131-H30) responsible for 28% of bacteremic E. coli infections over a 3-yr period. We additionally use a microbial genome-wide-association study (GWAS) approach to identify individual genes responsible for antibiotic resistance, successfully recovering known genes but notably not identifying any novel factors. We anticipate that in the near future, whole-genome sequencing of microorganisms associated with clinical disease will become routine. Our study reveals what kind of information can be obtained from sequencing clinical isolates on a large scale, even well-characterized organisms such as E. coli, and provides insight into how this information might be utilized in a healthcare setting.
With the advent of high-throughput DNA sequencing technologies, it is becoming increasingly tractable to generate whole-genome sequence data from large numbers of clinically relevant bacterial isolates. However, most comparative genome sequencing efforts to date have focused on the biology and molecular epidemiology of organisms involved in disease outbreaks (Chin et al. 2011; Lieberman et al. 2011; Koser et al. 2012; Snitkin et al. 2012; Sanjar et al. 2014). Although illuminating, these studies have shed little light on the agents of bacterial disease that infect an overwhelming majority of patients: commonplace pathogens causing sporadically acquired infections. Outbreaks represent the transmission of a single bacterial clone over a short period of time (Kennedy et al. 2010), providing a necessarily biased sampling that does not encompass the general properties of disease-causing organisms within a larger species. Relatedly, genomic studies of most bacteria are consistent with the distributed genome hypothesis, which proposes that the genetic content of a species is much larger than that of any single strain (Tettelin et al. 2005), necessitating sequencing of large numbers of unrelated clones in order to accurately catalog genetic variation (Rasko et al. 2008).
Escherichia coli is among the commonest clinical pathogens and is capable of causing a spectrum of disease both within the intestinal tract (intestinal pathogenic strains) and outside of it (extraintestinal pathogenic E. coli, or ExPEC). The most potentially destructive of these illnesses is bacterial invasion of the bloodstream: E. coli is the most common Gram-negative agent of sepsis, causing ∼30% of all bacteremias and representing the tenth most common cause of death in industrialized nations (Martin et al. 2003; Jaureguy et al. 2008). Far more prevalent are E. coli urinary tract infections, which encompass ∼95% of all community-acquired cases (Lau et al. 2008; Manges et al. 2008). E. coli infections of either type incur significant morbidity and healthcare costs (Sannes et al. 2004; Lau et al. 2008; Ron 2010; Telli et al. 2010); regardless, only a handful of strains causing these diseases have been sequenced, and knowledge of ExPEC E. coli remains incomplete.
Here we performed large-scale whole-genome sequencing and analysis of clinical isolates of extraintestinal pathogenic E. coli, obtained from routine diagnostic culture of peripheral blood or urine from patients within a single hospital system. These data enable a robust pan-genome analysis of ExPEC E. coli, high-resolution molecular epidemiological analysis, and genome-wide association studies for identifying antibiotic resistance genes.
Results
Isolates
A total of 380 isolates were collected by the clinical Microbiology Laboratory of the University of Washington Medical Center. Two hundred eighty-eight uropathogenic E. coli (UPEC) were isolated from 277 patients (237 female, 36 male, 4 data not available) over a period of 5 mo (Supplemental Data Set 1). Patients had an average age of 47 yr (range, newborn to 94 yr), and two died during the healthcare encounter in which the isolate was obtained. Because of the lower incidence of E. coli bacteremia, 92 strains were isolated from blood culture of 47 patients (25 female, 22 male) over 3 yr, representing all blood-borne E. coli isolates from our hospital over that period. Patients averaged 56 yr of age (range, newborn to 85), and 10 patients died during the healthcare encounter in which the isolate was obtained.
Pan-genome and core genome analysis
We initially explored the pan-genome composition of our strain collection. The “pan-genome” of a species refers to the full range of nonorthologous genes that can be present in an organism, whereas the “core genome” comprises genes present in all representatives (Tettelin et al. 2005). E. coli is believed to have an “open” pan-genome marked by ongoing gene acquisition (Rasko et al. 2008); however, existing approximations are based on more limited numbers of previously sequenced strains (Rasko et al. 2008; Touchon et al. 2009; Kaas et al. 2012).
The pan-genome for 283 ExPEC strains passing quality control requirements for this analysis was estimated at 16,236 genes, or 14,877 genes after removing prophage and insertion sequence elements (Supplemental Fig. S1). Substantial numbers of additional nonorthologous genes were identified with each strain sequenced, confirming an open species pan-genome even when analysis is confined to ExPEC strains. We predict a core genome size of 3079 genes, or 3039 genes after removal of prophages and insertion sequences (Supplemental Fig. S1). These values compare favorably with previous pan-genome and core genome estimations (Kaas et al. 2012), although they are somewhat lower and somewhat higher, respectively, than expected from a collection of this size, likely reflecting population structure among ExPEC E. coli. Comparatively, the core genome was most highly enriched for factors involved in basic cellular functions including DNA replication, cell wall synthesis, transcription, translation, and assorted amino acid metabolic and biosynthetic processes, while the pan-genome contained more genes related to metal-ion binding and virulence (predominantly flagellar proteins, capsular pathways, and secretion systems). All gene models from ExPEC E. coli isolates that were present in all, or nearly all, strains were also represented at a high frequency in commensals, suggesting that no specific gene is essential for ExPEC pathogenesis.
Phylogenomic analysis
We next investigated genomic and epidemiological relationships among isolates. After quality filtering, 312 E. coli isolates (221 UPEC and 91 bacteremia isolates) remained in this analysis. Phylogenomic reconstruction (Delsuc et al. 2005; Kumar et al. 2012) of genomic data was robust (Fig. 1), with likelihood values approaching one for most nodes (Supplemental Fig. S2). Classification of strains to phylogenetic subgroups (Escobar-Paramo et al. 2006) and multilocus sequence types (MLST) (Tartof et al. 2005), current mainstays for E. coli classification, was performed through in silico analysis (Fig.1; Supplemental Data Set 2).
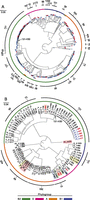
Whole-genome phylogenetic tree of ExPEC E. coli isolates. (A) Approximate maximum likelihood phylogeny showing the population structure of ExPEC E. coli. Isolates cultured from blood are represented as red terminal nodes and those cultured from urine are shown in blue. Colored ring denotes annotation of major E. coli phylogroups. Seven isolates assigned to phylogroups that are inconsistent with their phylogenomic placement are indicated with colored bars internal to this ring. The outermost ring (black) indicates groups of MLST sequence types. Sequence types with at least two representatives are numbered. The group corresponding to subclone S131-H30 is indicated. (B) Approximate maximum likelihood phylogeny of blood isolates only. Isolates are labeled according to the patient of origin and the relative day of collection (in red, ranging from day 0 for patient 43 to day 1184 for patient isolate 3_2). In instances where multiple isolates were obtained from the same patient, the order in which specimens were recovered is indicated by an underscore and a number. Patients for which multiple, genomically distinct strains were identified are highlighted. Isolates from patients 1 and 29 are indicated by blue text. The group corresponding to subclone ST131-H30 is indicated. Colored ring as in A. Scale bars are expressed in changes per site for both panels.
All four of the common ExPEC phylogroups (Carlos et al. 2010) (A, B1, B2, and D) were represented and comprised 8.3%, 8.7%, 65.7%, and 16.3% of the population, respectively, similar to reported distributions in other hospital systems (Clermont et al. 2000). Our analysis offers a more precise description of relationships among E. coli phylogroups (Fig. 1A; Supplemental Fig. S3), previously investigated through limited genetic information or using more qualitative methods (Lecointre et al. 1998; Johnson et al. 2006). Broadly, our data support the classification of phylogroups A and B1 as sister clades (Lecointre et al. 1998). Phylogroup B2, thought to be ancestral (Lecointre et al. 1998), forms a distinct clade that is divergent from the others. All phylogroups exhibited considerable genetic heterogeneity, and especially group D (Supplemental Table S1). Urine- and blood-derived isolates were extensively interleaved within the phylogeny, without significant enrichment for either infection type within any phylogroup (all comparisons P ≥ 0.05, Fisher’s exact test) and indicating that no clear phylogenomic division exists between ExPEC strains infecting these different sites.
Isolates from the same phylogroup, as determined by standard, combinatorial examination of a three-marker system (Escobar-Paramo et al. 2006), were closely related within the genomic phylogeny, with a few notable exceptions (Fig. 1A). Most strikingly, four strains classified as phylogroup D were most genomically similar to representatives of phylogroup B2 and formed a distinct clade, suggesting a common ancestry. As E. coli phylogroups reflect functional and evolutionary differences (Carlos et al. 2010), these outliers likely expose recent acquisition or loss of genetic material relevant to the phylogrouping marker system.
We looked for a possible correlation between the geographical distance separating E. coli isolates (as calculated between the centroids of patient home zip codes, average 441 km, range 0–7951 km) and the number of genomic differences among them. This analysis did not demonstrate a meaningful general correlation between genomic and geographical distance (not shown), suggesting that E. coli lineages are distributed relatively uniformly among members of our patient population.
We observed substantial clonal architecture. Two hundred eighty-five isolates were distributed among 71 known MLST types (Supplemental Data Set 2), while 13 isolates demonstrated novel sequence types bearing undescribed MLST alleles or previously unreported allelic combinations (Supplemental Table S2). Six established sequence types accounted for just over half (51%) of the population: ST131, ST95, ST127, ST73, ST69, and ST393 (Adams-Sapper et al. 2013; Toval et al. 2014). Of these, ST131 (Price et al. 2013) and ST95 (Gibreel et al. 2012; Adams-Sapper et al. 2013) were most prevalent, comprising 16.1% (50/312) and 10.8% (34/312) of the overall population, respectively.
We also observed substantial population structure within individual MLST groups (Fig. 1A). Of note, we identified two different clades of ST131 isolates (Fig. 2): one small clade restricted to urinary infections and marked by fluoroquinolone sensitivity (7/7 isolates), the other isolated from both urine and blood and marked by an increased frequency of fluoroquinolone resistance (35/47 isolates). Focused investigation revealed that the more prevalent, fluoroquinolone-resistant group corresponded to subclone H30, a recently emerged and highly pathogenic substrain (Colpan et al. 2013). A fraction of isolates from each H30 clade displayed extended-spectrum β-lactamase (ESBL) activity (Fig. 2): The most ancestral strains of the H30 subgroup exhibited concordant ESBL activity and fluoroquinolone resistance, whereas other scattered H30 isolates lacked one or more resistance phenotypes. This finding is consistent with the ST131-H30 multidrug resistance phenotype arising primarily through expansion of a single clone (Price et al. 2013) but also indicates that the phenotype has been lost by some descendants with measurable frequency (10 of the 47 isolates lacking ESBL activity, one isolate lacking fluoroquinolone resistance, and 10 having lost both).
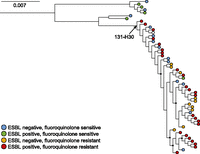
Whole-genome phylogenetic tree of ST131 isolates. The ST131-H30 subgroup is indicated and is marked by a high prevalence of fluoroquinolone resistance and extended-spectrum β-lactamase (ESBL) activity. Node color indicates the relevant drug resistance phenotype (white circle indicates missing data). Nodes supported by log likelihood values below 0.8 are marked with a black circle. Scale bar is expressed in changes per site.
Patient-level molecular epidemiology
We next examined the dynamics of E. coli infection within and among patients. One hundred isolates resulted from longitudinal sampling of the same patients, with independent collections separated by several hours or up to 2 yr. Eighteen urine-derived isolates were obtained from nine patients (two per patient), and 82 were isolated from the blood of 35 patients (90.1% of all blood-derived isolates).
We sought out subsets of strains that were virtually identical in terms of their genome sequences, which would suggest identity by descent. To first assess the magnitude of sequence artifacts introduced through sequencing, alignment, and variant calling, technical replicates of four isolates were taken through library construction and sequencing in tandem. Replicates proved highly concordant, averaging 0.25 ± 0.433 (average ± standard deviation) pairwise differences (compared to 51,801 ± 29,207 differences in an all-by-all isolate comparison) (Supplemental Data Set 3). Based on this level of uncertainty, we considered isolates genomically identical if they evidenced zero or one differentiating variant.
Seven of nine pairs of UPEC isolates were collected within 3 d of one another, and six of these pairs were genomically distinct (range of 502 to 69,681 pairwise differences). These findings are supported by different antibiotic resistance and hemolysis phenotypes in four cases and indicate a high rate of polyclonal infections. Two pairs of UPEC isolates were obtained more than a month apart: one pair was genomically identical, supporting urinary tract colonization, while the other was not (66,059 pairwise differences), suggesting independent infections. We did not find instances of genomically identical UPEC isolates obtained from different patients.
In longitudinal samples from bacteremic patients, pairs of isolates obtained from an individual within 21 d universally comprised genomically identical strains, most likely evidencing cases of ongoing infection. The mean time between all paired samplings recovering the same clone was 12.7 ± 16.3 d (mean ± standard deviation) (Supplemental Fig. S4). Intriguingly, genomically identical strains could also be recovered over substantially longer periods of time: five independent samplings from patient 29 were performed over a 5-mo period and yielded genomically identical multidrug resistant ST131-H30 isolates (Fig. 1B). Persistent E. coli bacteremia is rare but may reflect repeated translocation of a pathogenic clone from the gut (Samet et al. 2013) or other colonized organs (Alsterlund et al. 2012; Gupta et al. 2013). In contrast, we found that pairs of isolates obtained over longer periods of time (mean 251 d, standard deviation 261 d) (Supplemental Fig. S4) tended to represent genomically distinct isolates (Fig. 1B).
Of the 4190 reported E. coli MLST groups, only 23 were recovered from blood infections. Moreover, only eight sequence types were cultured from multiple patients (Fig. 1B): Combined, those lineages accounted for 76.6% (36/47) of blood infections in all patients over the study period. Included was the ST131-H30 lineage (21 independent isolates), collected from 28% (12/47) of bacteremic individuals. All 20 ST131-H30 blood isolates which underwent antibiotic resistance profiling, but only 54% (13/24) of those from urine, were fluoroquinolone-resistant, supporting a distinct phenotypic profile associated with bacteremia (Fig. 2). These results indicate that only a limited subset of closely related E. coli isolates was responsible for the majority of bacteremia cases.
The high prevalence of near-identical isolates in cases of bacteremia could reflect infection from high-prevalence endemic strains (Manges et al. 2004, 2008), nosocomial transmission of E. coli, or some combination, warranting more detailed investigation of the blood-derived isolates (Fig. 1B). There was clinical evidence consistent with nosocomial transmission in one patient (patient 1) (Fig. 1B). This immunocompromised individual became septic in the setting of graft versus host disease. Two isolates from phylogroup A, which we found to be genomically identical, were independently cultured from his blood over a period of 11 d, and the patient was transferred to an intensive care unit. Resolution of the infection was achieved and confirmed by 26 serially negative blood cultures. Forty days after transfer, the patient again developed sepsis, and cultures recovered a multi-drug resistant E. coli strain we identified as ST131-H30 (phylogroup B2). Attempts at treatment were unsuccessful and the patient died. At the time of this second infection and in the same hospital ward, patient 29 was receiving treatment for E. coli bacteremia with an identical antibiotic resistance profile and which we similarly found to be an ST131-H30 strain. This evidence would be consistent with nosocomial transmission from patient 29 to patient 1. Regardless, the ST131-H30 isolate from patient 1 differed from that of patient 29 by 355 genomic variants and harbored 11 additional virulence factors. These genomic data unambiguously demonstrate independent sources of infection, rather than nosocomial transmission.
Nevertheless, a strain genomically identical to the ST131-H30 isolate from patient 1, and with the same antibiotic resistance profile, was recovered from patient 6 over 1 yr later in a different hospital ward (Fig. 1B), convincingly supported by robust sequence coverage of both isolates (> 45× read depth). Although the epidemiological link in this instance, if any, is unknown, sharing of E. coli strains among close contacts is documented (Foxman et al. 1997; Johnson et al. 2008).
Although this strategy identifies true bacterial clones, which are by definition genomically identical, some degree of clonal diversification may occur within patients (Walker et al. 2013) or during the course of an outbreak (Lindsay 2014). We, therefore, expanded our search for potential transmission events to include pairs of strains harboring ≤ 15 genomic differences (Supplemental Data Set 3; Supplemental Fig. S5), an amount of divergence expected to accumulate more than ∼6750–15,000 bacterial generations (Barrick et al. 2009; Lee et al. 2012), and evaluated isolates with respect to temporal association. Five pairs of strains qualified under this definition. Two pairs of those isolates were distinguishable by distinct antibiotic resistance phenotypes and thus were unlikely to represent direct transmissions. However, the remaining paired comparisons comprised a trio of UPEC strains (upec-61, -106, and -249) collected within 3 mo of each other from a geographically constrained area (Supplemental Table S3) and exhibiting the same pan-antibiotic-sensitive phenotype. Given robust sequence coverage of all three isolates (> 49× read depth), we speculate that the three strains are epidemiologically linked, although contact among these patients cannot be known to us.
Distribution of virulence factors and antibiotic resistance phenotypes
Virulence factors (VFs) play an important role in conferring selective advantages to, and defining pathogenicity profiles of, E. coli strains (Nowrouzian et al. 2006; Ramos et al. 2010). Accordingly, disease-associated phylogroups of E. coli have a higher prevalence of VFs and antibiotic resistance than commensals (Picard et al. 1999; Price et al. 2013), and some subgroups are enriched for distinct subsets of VFs (Nowrouzian et al. 2006). To more fully explore the distribution of factors among ExPEC phylogroups, blood- and urine-derived isolates, and MLST groups which are under clinical selective pressure as human pathogens, we cataloged the prevalence of known VFs and antibiotic resistance phenotypes within groups of E. coli defined at these population levels (Fig. 3; Supplemental Data Sets 4, 5) and explored statistically significant differences in their enrichment or depletion. We also considered differences among these groups after accounting for population structure (Fig. 3; Supplemental Data Sets 4, 5; Price et al. 2006) in order to identify factors which may have been acquired or lost independently within lineages multiple times, rather than inherited from a single common ancestor.

Proportion and relative enrichment of virulence factors and antibiotic resistance phenotypes carried by isolates in distinct groups. Rows correspond to individual VFs (top) or antibiotic resistance phenotypes (Abx, bottom). VFs are grouped by class. (Tox) toxin, (Prot) protectin, (Iron) iron metabolism, (Tr) transporter, (Cap) capsule, (Inv) invasion, (Adh) adhesion, (Mi) miscellaneous. Columns correspond to categories of isolates grouped according to different classification schemes. Prevalence of factors within each category is shown at left for each panel (blue heatmap). Raw P-values from all possible pairwise comparisons of factor prevalence between them is shown at right for each panel (green heatmap), with the specific pairwise comparison indicated above each column. P-values were obtained after correcting for inferred population structure (left, labeled in red with “Corrected” or “C”) or without such correction (right, labeled in red “Uncorrected” or “U”). (A) Comparison of E. coli phylogroups. (B) Comparison of isolates obtained from blood (B) and urine (U). (C) Comparison of the six most prevalent MLST groups (sequence type numbers are indicated) and a seventh category encompassing all other MLST groups (“O”).
As expected, isolates from phylogroup B2 demonstrated a high frequency of carriage for the greatest number of VFs (Johnson et al. 1991; Picard et al. 1999), while phylogroup A strains displayed the lowest prevalence of VFs (Fig. 3A). Adhesins, particularly of the ecp and fim gene families, were the most prevalent VF across all phylogroups. After correcting for population structure, few differences in VF content between populations remained statistically significant, suggesting that most differences among phylogroups reflect patterns of descent. The major exceptions were several iron utilization genes (iuc and ybt families), toxin tsh, and protectin traT, which were significant after accounting for strain relatedness and implies ongoing acquisition of these genes in phylogroups B2 and D. Interestingly, differences between the related B1 and B2 phylogroups for several of these factors were only significant after accounting for population structure, possibly reflecting convergent gene acquisition.
The prevalence of virulence factor genes observed in isolates from blood or urine was similar overall (Fig. 3B), consistent with our observations about phylogenetic lineages being able to infect both bodily sites. After the population structure correction, most statistically significant differences between these groups represented only minor dissimilarities in overall VF prevalence; however, a handful of genes differed in prevalence by at least a factor of 1.5. Five such genes were enriched in blood-derived strains: invasin traJ, toxins sat and tosA, capsule papG, and adhesion papA. Notably, papA has a known role in urinary colonization (Lindberg et al. 1987; Denich et al. 1991; Johnson et al. 2000), but its preferential enrichment in blood isolates implies importance of this VF outside of uncomplicated uropathogenesis (Johnson et al. 2000). As expected, urinary-derived isolates were enriched for members of the auf adhesion family (Buckles et al. 2004; Kaper et al. 2004), and toxin vat (Spurbeck et al. 2012). Differences in the prevalence of resistance to 10/23 antibiotics were also significant independently of population structure, in all cases at higher prevalence in blood-derived isolates.
MLST groups that were recovered most frequently from our patients evidenced high prevalence for a large number of VFs compared to nondominant MLST groups, especially pronounced for group ST127 (Fig. 3C). Much like our analysis at the phylogroup level, almost no statistically significant differences among these groups were evident after correcting for population structure, indicating that the innate virulence gene repertoire of individual groups almost entirely reflects heredity from a common ancestor.
De novo identification of antibiotic resistance factors
It is now appreciated that large numbers of microbial genome sequences can be used for robust genome-wide association studies (GWAS), enabling the detection of genetic factors underlying phenotypic variation (Falush and Bowden 2006; Farhat et al. 2013; Sheppard et al. 2013; Alam et al. 2014; Laabei et al. 2014). Here, in light of the open nature of the E. coli pan-genome, we observed a significant number of novel sequences present neither in reference genomes nor previous isolates with each additional strain that was sequenced. It was consequently not possible to comprehensively or effectively perform GWAS at single-nucleotide resolution, nor in any way that relied on the use of a reference genome. As an alternative, we elected to examine associations at the level of discrete coding sequences that were identified in de novo assemblies.
We took this approach to identifying transmissible antibiotic resistance determinants within our study cohort—i.e., single gene factors conveying a phenotype that could be spread through a population via plasmids or other mobile elements. For each isolate, we determined the presence or absence of predicted genes found across the collection, then assessed the statistical significance of differences in the overall frequency that each gene was found in antibiotic-resistant and susceptible populations. As before, we performed a principal components analysis correction to account for population structure (Price et al. 2006).
With the exception of drugs from the fluoroquinolone class (which predominantly arise from chromosomal point mutation [Morgan-Linnell et al. 2009]), known resistance factors were strongly associated with a resistant phenotype for each antibiotic (P-values of 10−2.05 to 10−12.2, mean of 10−5.1) (Supplemental Table S4). Transposases, conjugation factors, transcription factors, and plasmid maintenance factors were also highly associated with antibiotic resistance, consistent with physical linkage to mobile elements. After correcting for correlation with known resistance genes, we examined the most significant genes identified for each drug and carried 17 genes forward for functional characterization in a laboratory strain. Bacteria transformed with known resistance factors (5/5) exhibited expected gains in antibiotic resistance; however, none of the potentially novel factors (0/17) conferred any detectable influence on resistance levels (Supplemental Table S5).
Discussion
Given sustained decreases in the cost of high-throughput sequencing, we are approaching a time when it will be possible for clinical laboratories to sequence all clinical bacterial isolates, even as a routine standard of care (Didelot et al. 2012; Schatz and Phillippy 2012). Here, we have attempted to provide an early glimpse as to how this kind of data can be utilized in a healthcare setting and to demonstrate what kinds of information can be readily obtained from performing bacterial sequencing of clinical isolates on a large scale.
With respect to population structure, we found no evidence of a phylogenomic division between strains infecting either the blood or urinary tracts of our patients (Fig. 1A). This contrasts with studies of human-derived and environmental E. coli (Luo et al. 2011) and suggests that specific ExPEC E. coli lineages are, in general, not restricted to invasion of one of these bodily sites or the other. The distribution of isolates across E. coli phylogroups was consistent with earlier reports; however, we identified several instances of strains being misclassified to the incorrect phylogroup based on a standard, three-marker classification system (Escobar-Paramo et al. 2006), presumably due to unexpected loss or gain of relevant genetic material. Although apparently rare, the potential for such events to mislead phylogrouping analysis should be acknowledged and argues for a comprehensive, genomic approach to phylogroup determination. Whole-genome sequencing also offers substantially higher resolution than conventional strain typing approaches, revealing population structure underlying MLST groups including the dominant ST131 clade (Fig. 1A). Interestingly, phylogenomic analysis suggests that multidrug resistance in subclone ST131-H30 is unstable and can be lost by some descendants over time (Fig. 2).
Most of the statistically significant differences in VF content and antibiotic resistance phenotypes which distinguish population-level groups of E. coli do not persist after taking population structure into account (Fig. 3), arguing that most reflect inheritance by descent from an ancestral strain and are a product of population structure alone. Nevertheless, a small subset of factors do remain significant after correction for population structure, suggesting that at least some genes have undergone multiple instances of independent acquisition or loss within lineages and potentially identifying them as important to specific lineage groups or routes of infection.
Patient-to-patient transmission of E. coli appears to be infrequent among the set of patients we examined. Anecdotal exploration of one case of suspected nosocomial transmission was ruled out in light of genomic data. Nevertheless, we found evidence for a pair of genomically indistinguishable isolates and a group of three closely related strains that were shared across multiple patients. Although the trio of related isolates was obtained within several weeks of one another, collection of the paired isolates representing a true genomic clone occurred more than a year apart and, in the absence of a clear epidemiological link, perhaps indicating an environmental reservoir or a dominant strain within the larger community (Manges et al. 2008). Indeed, rates of nosocomial transmission of lineages as assayed by conventional, lower-resolution typing technologies (Hilty et al. 2012) may have led to an overestimation of the frequency of such events, as has been observed for other bacteria (Miller et al. 2014; SenGupta et al. 2014). However, given our finding of multiple polyclonal E. coli infections, it should be noted that our conclusions may be influenced by incomplete clinical sampling of the multiple strains present in some infections (Lindsay 2014), reflecting a limitation of current clinical microbiological procedures. However, that we were able to detect potential transmission events without detailed biogeographic information or patient histories argues for the power of large-scale and unbiased sampling of clinical isolates as a means to monitor bacterial transmission. This genomic approach may provide unexpected advantages over existing molecular epidemiological techniques with lower throughput and resolution, potentially revealing outbreaks with unconventional properties (such as those spreading slowly and indolently), in addition to detailed population-level trends and direct transmission events.
Lastly, that our GWAS-type analysis was able to identify known antibiotic resistance factors, but in experimental assays failed to identify novel antibiotic resistance determinants, suggests the possibility that, in this well-studied organism, all single-gene antibiotic resistance factors present at reasonable prevalence have been previously identified. Polygenic causes of resistance may exist, and the factors we have identified in this study may contribute to multifactorial modes of resistance; however, dissecting such potentially complex pathways is outside the scope of our current work. Regardless, a GWAS-type approach based exclusively on genomic data and strain phenotypes robustly identified known antibiotic resistance genes, validating the general strategy as a means to catalog genes underlying other traits of interest and in other microbial organisms.
The ability to perform whole-genome surveys of bacteria without bias and at the scale of entire health care networks has the potential to provide in-depth information about many aspects of bacterial pathogens. As this study has demonstrated, single data sets of this nature enable more comprehensive and multifactorial examination of even well-characterized pathogens like E. coli, both in a clinical context and from a more basic science perspective.
Methods
Samples and functional strain characterization
All isolates were identified and typed by the Microbiology Laboratory at the University of Washington Medical Center (Seattle, WA), using routine clinical practices. Antibiotic resistance was assessed using a combination of Kirby-Bauer antibiotic testing and automated MIC drug testing (Sensititre system, TREK Diagnostic Systems). Use of specimens was approved by the University of Washington Human Subjects Review Committee.
Library preparation and sequencing
DNA was extracted using the Wizard Genomic DNA Purification kit (Promega). Shotgun sequencing libraries were prepared using the Nextera system V1 (Epicentre), PCR-amplified using FailSafe E PCR Mix (Epicentre), monitored by real-time PCR, and removed when exponential growth of the product was first observed. Libraries were purified using Agencourt AMPure XP (Beckman Coulter). Pools of 96 uniquely indexed samples were sequenced using an Illumina HiSeq 2000 with 101-bp paired-end chemistries.
Core and pan-genome analysis
Reads were adaptor-trimmed and subjected to de novo genome assembly using ABySS (version 1.3.5) (Simpson et al. 2009), using k-mer values (range 20–48) empirically determined to maximize contiguity on a per-sample basis (Supplemental Data Set 7). Contigs < 500 bp in length were discarded as likely misassemblies. The mean N50 statistic for all genomes was 183.6 ± 97.9 Kb (Supplemental Fig. S6). Gene predictions were made using Glimmer3.02b (Delcher et al. 2007). A “meta-reference” was next constructed to represent all unique coding sequences (CDSs) in all strains. CDSs were extracted from 53 fully sequenced E. coli reference genomes (Supplemental Data Set 8) and were first clustered using CD-HIT v4.6 (Li and Godzik 2006) (arguments -n 3 -c 0.8 -G 1 -aL 0.8 -aS 0.8 -B 1) to de-duplicate sequences ≥ 80% identical. Experimental gene predictions were compared to the de-duplicated reference CDS using BLASTP (Altschul et al. 1990), and sequences with ≥ 90% identity and ≥ 33% coverage to a reference CDS were discarded. Remaining gene predictions were de-duplicated as before and merged with the reference CDS to form the final meta-reference (Supplemental Data Set 6). Meta-reference sequences were functionally annotated using DAVID v6.7 (Huang et al. 2009), gene classes found in the pan-genome and core genome were tabulated, and classes with the greatest-fold enrichment in comparing the two sets were evaluated. BLASTX was used to search de novo assemblies against the meta-reference, and a CDS was considered present in a strain if ≥ 80% of the CDS was covered by an alignment and protein-level identity was ≥ 80%.
Pan-genome estimates were performed for sequences of ≥ 75 amino acids in length. We found that assemblies with an N50 statistic of < 5 × 104 bp did not reliably contain a full complement of essential E. coli genes (Hashimoto et al. 2005), so we limited our analysis to the 283 strains passing this cutoff. Two thousand different random input orders of genomes were performed (Touchon et al. 2009) for a subset size of 1 to 282, and quartiles were calculated for each. Estimations were performed against the complete meta-reference and after removing likely phage sequences and insertion sequences, identified by BLAST search against a prophage database as described (Zhou et al. 2011). For each gene, the highest number of strains for which the gene was present in ≥ 95% of isolates (Kaas et al. 2012) was calculated in over 2000 different random input orders. Individual genes were counted as part of the core genome for all numbers of strains up to and including this number of strains. For comparative studies of pathogenic and commensal strains, the meta-reference was subjected to BLASTX analysis against commensal reference genomes (Hall et al. 2013) as above, and sequences shared between paired queries were flagged.
Molecular epidemiology
Adaptor-trimmed sequence reads were aligned to E. coli K12 MG1665 (GenBank ID: 556503834) using BWA (v0.6.2) (Li and Durbin 2009) and SAMtools (v0.1.19) (Li et al. 2009), yielding a mean coverage depth of 39.6 ± 25.5 (Supplemental Fig. S4). Single-nucleotide variant calling was performed using SAMtools and variants supported by fewer than 10 reads or a likelihood score of < 200 marked as “unknown” data. A total of 446,152 unique variant sites were found across all isolates. To filter out low-quality genomes, isolates with ambiguous calls at 80% or more of total variant sites were excluded from phylogenetic reconstructions. Approximately maximum-likelihood phylogenetic trees were made using FastTree 2.1 (Price et al. 2010).
Isolates were assigned to phylogenetic subgroups based on a three-genetic-marker system (Escobar-Paramo et al. 2006), using a BLAST search against de novo assemblies to register presence or absence. Classification of isolates assigned to a different phylogroup than phylogenomically related isolates was confirmed by examining the depth of short reads against aligned to each of the three genetic markers. Sequence types were assigned by a BLAST search of assembled genomes to identify perfect matches against known MLST fragments from an established database (http://mlst.ucc.ie/, accessed 1/22/14). The pattern of MLST types for each locus was compared to reported sequence types. Strains for which one or more MLST loci could not be identified (14 isolates) or those bearing new locus sequences were unassigned. Assignment of strains as ST131-H30 was based on exact BLAST match to a partial fimH 30 allele (Colpan et al. 2013) (GenBank ID: 268639126).
Characterization of known virulence and antibiotic resistance factors
VF reference sequences were identified through a combination of the Virulence Factor Database (Chen et al. 2012) and primary literature review (Supplemental Data Set 9). In all strains, the presence of each VF was assessed using a BLAST search as above. VF and antibiotic resistance phenotypes were assessed within major phylogroups and sequence types containing 10 or more isolates. Technical replicates and isolates failing quality control for the pan-genome analysis were excluded. Statistical association between presence or absence of each factor (VF or resistance phenotype) and isolate classification was assessed using a logistic regression framework in R 3.1.1 (R Core Team 2014) for MLST or phylogroup membership, or for blood- or urine-derived isolates. The strength of association for factors perfectly predicted by isolate classification was assessed with a Bayesian logistic regression model (package arm) with independent Cauchy priors, mean 0, and scale 5/2 for each coefficient. Only factors meeting a significance threshold of P < 0.05 in any of the three classification models (MLST, phylogroup, or blood vs. urine source) were carried forward for post-hoc pairwise testing. Differences in factor distribution or resistance phenotype for all pairwise comparisons within a classification scheme were evaluated using Tukey’s HSD method (package lsmeans). As this method is statistically conservative, significant pairwise comparisons were identified using raw P-values. To account for possible lack of independence between isolates due to underlying population structure, the analysis was repeated using an additive logistic regression framework with three additional covariates: the first three principal components resulting from the decomposition of the matrix of presence or absence genotypes of each CDS across all isolates (described below).
Genome-wide association analysis for antibiotic resistance phenotypes
BLASTX was used to search de novo assemblies against the meta-reference, and the presence or absence every CDS ≥ 75 amino acids in length from the meta-reference was assessed using alignment and coverage metrics as before. A logistic regression model was implemented in R 3.1.1 as above. To control for population structure, the matrix of presence or absence for each CDS and for each isolate was decomposed into principal components. Q-Q plots were evaluated by manual review (Supplemental Fig. S7), and the first three principal components of the matrix were empirically determined as optimal and were retained as covariates in the model. To identify independently associated factors, known associations for each drug resistance phenotype were used as covariates. The 20 CDS with the most significant P-values were considered for each antibiotic. CDS corresponding to repetitive elements, transposons, insertion sequences, plasmid support machinery, resistance factors for other drugs, or unrelated biochemical pathways, and CDS occurring at ≥ 15% frequency in the antibiotic-sensitive population were excluded to limit spurious associations due to linkage. Genes of interest (Supplemental Table S5) were cloned into pET-9a or pET-3a expression vectors (Novagen) using the HD Infusion kit (Clontech). Transformed E. coli BL21(DE3) (NEB) were induced using 0.3 mM IPTG and subjected to antibiotic resistance testing by Etest (bioMérieux).
Data access
Sequence data generated for this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under study accession number SRP042632. Draft genomes have been submitted to NCBI GenBank (http://www.ncbi.nlm.nih.gov/genbank) under accession numbers JSFQ00000000–JSST00000000. The meta-reference sequence and other accessory data can be found in the Supplemental Material (Data sets S6, S7).
Acknowledgments
This work was supported by developmental research project grant 5U54AI057141-08REV from the National Institutes of Allergy and Infectious Diseases (NIAID)/Northwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases (NWRCE).
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.180190.114.
- Received June 19, 2014.
- Accepted November 3, 2014.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








