
Abstract
We used comparative and population genomics to study intron evolutionary dynamics in the fungal model genus Neurospora. For our investigation, we used well-annotated genomes of N. crassa, N. discreta, and N. tetrasperma, and 92 resequenced genomes of N. tetrasperma from natural populations. By analyzing the four well-annotated genomes, we identified 9495 intron sites in 7619 orthologous genes. Our data supports nonhomologous end joining (NHEJ) and tandem duplication as mechanisms for intron gains in the genus and the RT-mRNA process as a mechanism for intron loss. We found a moderate intron gain rate (5.78–6.89 × 10−13 intron gains per nucleotide site per year) and a high intron loss rate (7.53–13.76 × 10−10 intron losses per intron sites per year) as compared to other eukaryotes. The derived intron gains and losses are skewed to high frequencies, relative to neutral SNPs, in natural populations of N. tetrasperma, suggesting that selection is involved in maintaining a high intron turnover. Furthermore, our analyses of the association between intron population-level frequency and genomic features suggest that selection is involved in shaping a 5′ intron position bias and a low intron GC content. However, intron sequence analyses suggest that the gained introns were not exposed to recent selective sweeps. Taken together, this work contributes to our understanding of the importance of mutational bias and selection in shaping the intron distribution in eukaryotic genomes.
The presence of introns in protein coding DNA was discovered in 1977 (Berget et al. 1977; Chow et al. 1977; Evans et al. 1977; Goldberg et al. 1977), and since then, introns have been identified as a typical feature of eukaryotic nuclear genomes. Yet, the functional role of introns and the factors affecting their turnover are only beginning to be understood (Chorev and Carmel 2012). The growing availability of large-scale genomic data sets constitutes an important step toward addressing these fundamental issues in genome biology. For example, recent studies have shown that the genome of the last common ancestor among all eukaryotes is likely to have been intron-rich (Csuros et al. 2011) and that intron losses have dominated in many eukaryotic species; however, a few bursts of substantial intron gains have taken place, for example, during the origin of metazoan and land plants (Csuros et al. 2011; Rogozin et al. 2012). Among all the eukaryotic branches, the fungal group Ascomycota shows a novel pattern, with a relatively high level of intron losses and gains (Carmel et al. 2007). Currently, there is a shortage of scientific data about intron turnover within the Ascomycota, including population-level intron changes and the molecular mechanisms and selective forces underlying intron gains and losses.
Fink (1987) first proposed that intron loss is mediated by reverse-transcribed mRNA (RT-mRNA). In this classical model, mRNA is first reverse-transcribed into the intronless cDNA and then converted back into its original genomic location by homologous recombination (Bernstein et al. 1983; Lewin 1983; Boeke et al. 1985). Since reverse transcription is processed from 3′ to 5′, and often terminates prematurely, intron losses are expected to occur more frequently in the 3′ end of the genes (Fink 1987; Mourier and Jeffares 2003). Nonetheless, subsequent analyses revealed that the highest intron loss rate is found in the internal part of a gene for many species (Nielsen et al. 2004; Sharpton et al. 2008; Zhang et al. 2010), and other mechanisms for intron gain and loss have been proposed. The DNA repair mechanism, nonhomologous end joining (NHEJ) (Johnson and Jasin 2000; Rebuzzini et al. 2005; Preston et al. 2006; Mao et al. 2008; Shimizu et al. 2010), has also been suggested to mediate intron gain and loss in eukaryotes (Rebuzzini et al. 2005; Preston et al. 2006; Li et al. 2009; Farlow et al. 2010, 2011; Shimizu et al. 2010; Zhang et al. 2010; Yenerall et al. 2011). Additional proposed mechanisms for intron gains include intron transposition, transposon insertion, tandem genomic duplication, intronization, and multiplication of introner-like elements (ILE) (van der Burgt et al. 2012; Yenerall and Zhou 2012), all of which needs further investigation.
Population-level analyses are keys to fully understanding intron evolution, since intra-species dynamics ultimately determine whether introns become fixed or lost in the genome. Until now, however, few studies have used a population genetic approach to study intron evolution. Population data from intron polymorphic sites in Drosophila and Zymoseptoria tritici have shown the action of positive selection on some intron variants (Llopart et al. 2002; Brunner et al. 2014), whereas in Daphnia, the majority of intron gains are deleterious (Li et al. 2009). The recent emergence of population level genomic data sets has opened up the possibility to study intron evolutionary dynamics on a genomic scale (Li et al. 2009; Torriani et al. 2011; Croll and McDonald 2012).
In the present study, we assessed rates, patterns, and mechanisms of intron gain and loss at both the inter- and intra-species level in the fungal model genus Neurospora (phylum Ascomycota). Neurospora is an intron-sparse group (∼1.7 introns/gene) with a 5′ positional biased intron distribution in the genome (Galagan et al. 2003; Jeffares et al. 2006). While most species forming the terminal clade in the genus are self-sterile (heterothallic) and thereby largely outcrossing (Ellison et al. 2011a; Nygren et al. 2011), one member of the genus, N. tetrasperma, has evolved pseudohomothallism, a mating system dependent on heterokaryosis for mating-type and intratetrad selfing (Dodge 1927; Corcoran et al. 2014). Pseudohomothallism in N. tetrasperma is associated with a large region of suppressed recombination (∼8 Mbp) on the mating-type chromosomes (Menkis et al. 2008; Ellison et al. 2011b; Sun et al. 2012), while other regions are typically freely recombining. This mating system has resulted in a high degree of homoallelism across recombining regions of the genome, as compared to the recombinationally suppressed segment on the two different mating-type chromosomes (Sun et al. 2012). Taken together, these features in Neurospora allowed us to study several key questions associated with intron turnover, such as the cause of the 5′ positional biased intron pattern and intron dynamics within various recombination backgrounds. For the analyses, we conducted comparative analyses across four well-annotated Neurospora genomes to identify patterns of intron gains and losses in this genus. Second, using genomic data from populations of N. tetrasperma, we assessed the selective forces acting on intron gains and losses at the intraspecific level and propose a model of the factors giving rise to the current intron distribution in the genomes of this model genus.
Results
Four well-annotated genomes were included in the study, that of N. crassa, N. discreta, and two genomes of N. tetrasperma. The two genomes of N. tetrasperma originate from a single heterokaryotic wild-type strain (i.e., they shared a common cytoplasm in the mycelium) and are of different mating type (mat A and mat a); for simplicity, we refer to them as N. tetrasperma A and N. tetrasperma a in this study. We identified 7619 orthologous genes from ∼10,000 genes of the investigated genomes (9907 genes in N. crassa, 10,380 in N. tetrasperma A, 11,192 in N. tetrasperma a, and 9948 in N. discreta [Galagan et al. 2003; Ellison et al. 2011b]). From the orthologous gene alignments, we identified a total of 9495 sites in the genome that contained an intron in at least one species (hereafter referred to as intron sites). In order to investigate the impact of recombination on the intron gains and losses patterns, we divided the intron sites into two categories based on the genomic location: the region of suppressed recombination (SR) in N. tetrasperma, i.e., the central part of the mating-type chromosome, and the recombining (R) regions, i.e., the remaining genome, as defined in Ellison et al. (2011b). After filtering, we found 2107 intron sites (1.30 introns/gene on average) in the SR region, and 7388 intron sites (1.21 introns/gene) in the R regions, a difference not significantly different (χ2 test, P > 0.05).
Interspecies analyses of intron gains and losses
Intron gains and losses over evolutionary time in Neurospora
Based on previous studies, we assume that the phylogenetic relationship of the investigated strains is {N. discreta (N. crassa [N. tetrasperma A, N. tetrasperma a])} (Fig. 1A; Dettman et al. 2003; Menkis et al. 2009). By using this phylogeny in combination with Dollo parsimony, which assumes that introns are unlikely to gain in the exact position twice in different phylogenetic lineages, whereas independent losses of introns are allowed (Rogozin et al. 2003), we identified 15 distinct patterns of intron gains and losses in N. crassa and N. tetrasperma (Fig. 1B). Among the 9495 intron sites, we found 60 sites for which we could trace intron gains in N. crassa or N. tetrasperma, 66 sites with intron losses, 9286 sites with intron presence in all four genomes, and 83 sites with uncertainty about the ancestral state. The fact that the investigated species are closely related (genome-wide dS of 0.12 between N. discreta and either of N. crassa and N. tetrasperma, and 0.049 between N. crassa and N. tetrasperma as estimated from the orthologous genes) minimizes the risk of investigating multiple gains and losses at the same intron site. We found intron gains and losses in both the SR and R regions, as shown in Figure 1B.
Pattern of intron gains and losses in Neurospora. (A) Phylogenetic relationship of the four Neurospora genomes in this study. (B) Classification of intron presence (1) and absence (0) for the 9495 intron sites. The number of intron sites for each pattern is indicated at the bottom of the figure. SR indicates that the intron position is located within the region of suppressed recombination of the mating-type chromosomes in N. tetrasperma, and R in the normal recombination region of the genome.
For interspecies comparisons of intron gains and losses among Neurospora species, we selected one genome per species (N. discreta, N. crassa, N. tetrasperma a) and then mapped all intron gain and loss events on the Neurospora phylogenetic tree (Fig. 2). Using this approach, we found that after the split from the common ancestor with N. tetrasperma, the number of intron gains and losses for N. crassa are 24 and 23, and for N. tetrasperma, 29 and 42 (Fig. 2). We did not find significant differences for intron gain and loss numbers between N. crassa and N. tetrasperma (Pearson χ2 test, P > 0.11), and for N. tetrasperma, between SR and R region (P > 0.20).
Neurospora phylogenetic tree with numbers of intron gains and losses found at the branches delineating N. tetrasperma and N. crassa. Plus (+) indicates intron gain events, and minus (−) indicates losses. Numbers in parentheses indicate the number of the introns that are located within the region of suppressed recombination of the mating-type chromosome of N. tetrasperma (SR), and in the normal recombination region of the genome (R), as (SR/R). Note that in N. crassa, recombination is not suppressed in the SR region, but numbers are given for comparative purposes.

Based on the genomic dS of 0.0486 between N. crassa and N. tetrasperma a, estimated from the orthologous gene alignments, and the synonymous substitution rate of 7.969 × 10−9 per site per year obtained from the study by Kasuga et al. (2002), we calculated the divergence time for N. crassa and N. tetrasperma a as 3.05 × 106 years. The intron gain rate (given as introns per nucleotide site per year) was estimated to be 5.78 × 10−13 for N. crassa, and the loss rate (given as introns per intron site per year) to be 7.53 × 10−10. For N. tetrasperma a, the intron gain rate was 6.89 × 10−13 introns per nucleotide site per year, and the loss rate 13.76 × 10−10 introns per intron site per year.
Positional bias in intron location
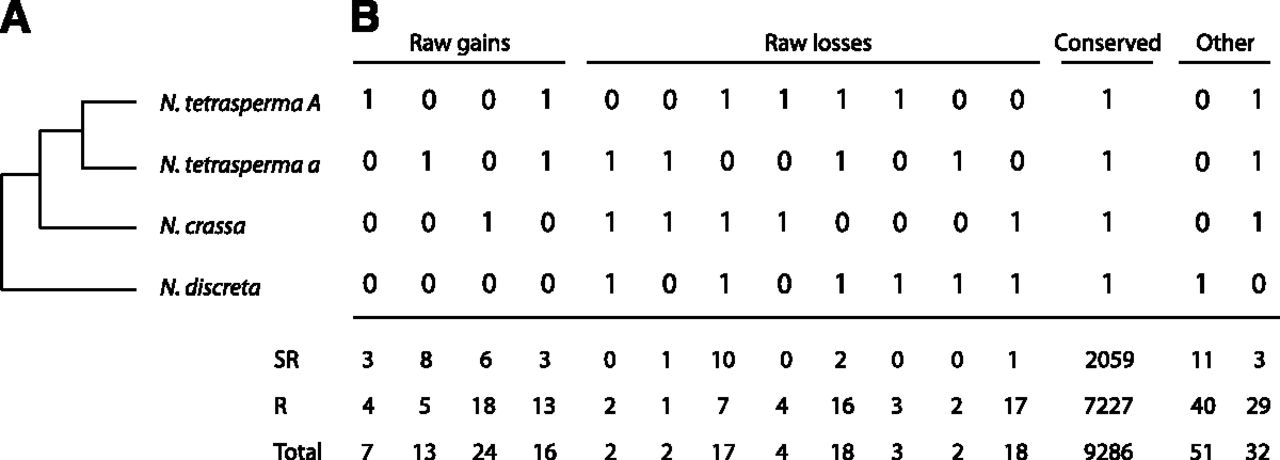
We divided all introns into three categories (5′, internal, and 3′), based on their relative position within a gene (calculated as [no. of bases in the coding sequence upstream of the current intron]/[total no. of bases in the coding sequence]). For both N. crassa and N. tetrasperma a, we found a statistically significantly higher number of introns located in the 5′ end of a gene than in the internal and 3′ regions (χ2 test, P < 1 × 10−4) (Fig. 3A), consistent with previous observations in N. crassa on a smaller data set (Nielsen et al. 2004). Within the N. tetrasperma a genome, this pattern was found both for the SR and the R regions (P < 1 × 10−4) (Fig. 3A). Figure 3, B through E shows the number of intron gains and losses since the last common ancestor of N. crassa and N. tetrasperma a, and the intron gain and loss rates; for these estimates, the data set is too small for meaningful statistical tests. Nevertheless, we see trends in the data that are worth noting. First, in both species, the number of gained and lost introns showed the pattern of 5′ > internal > 3′ (Fig. 3B). For the intron gain rate, we found a 5′ elevation in the N. crassa and the N. tetrasperma a combined data sets, a trend found in the SR region of N. tetrasperma a but lacking in the N. tetrasperma a R data set (Fig. 3C). The intron loss rates were highest at the internal position, followed by the 5′ or 3′ in N. crassa, N. tetrasperma a, and N. tetrasperma a R, but not in N. tetrasperma a SR (Fig. 3D). For both species, the intron gain/loss rate ratio showed a consistently biased pattern, with a higher gain/loss ratio in 5′ than internal and 3′ positions (Fig. 3E). We found a higher intron gain/loss rate ratio in the SR region compared to the R region (5′ SR > 5′ R, internal SR > internal R, 3′ SR > 3′ R) (Fig. 3E), but the overall pattern of 5′ positional bias was not affected (5′ > internal > 3′ in both regions). It is worth pointing out that the trends observed in Figure 3, B through E are not statistically significant, and thus, we cannot exclude the possibility that they are caused by chance alone.
Positional biases of introns and their gains and losses in N. crassa and N. tetrasperma a. Introns are binned into three categories (5′, internal, and 3′) based on the intron position within a gene, with each category representing one-third of the coding sequence length. In each plot, the bar or point from left to right represents 5′, internal, and 3′ intron data. Data from N. crassa are presented in the left panel and N. tetrasperma is presented as all intron positions. (SR) Intron positions located within the suppressed recombination region of the mating-type chromosome, and (R) in the normal recombination region of the genome. (A) Number of introns. (B) Number of intron gains and losses since the last common ancestor of N. crassa and N. tetrasperma. Bars above the x-axis represent intron gain number, below, intron loss. (C) Intron gain rate. (D) Intron loss rate. (E) Intron gain/loss rate ratio.
Intron phases, length, and GC content
Based on the position in a codon, we categorized the introns as phase 0 (located between codons), phase 1 (located after the first nucleotide of a codon), and phase 2 (after the second nucleotide of a codon) introns. We detected a statistically significant excess of phase 0 introns, as compared to phase 1 and 2, in both N. crassa and N. tetrasperma genomes, thus rejecting the null hypothesis that introns have an equal distribution across phases (χ2 test, P < 1 × 10−4). For N. crassa, we detected a significantly higher number of gained introns in phase 1 (P = 0.04), and an excess of lost introns in phase 2 (P < 1 × 10−4), while no statistically significant differences in the phase distribution of either gained or lost introns were found in N. tetrasperma (P > 0.15).
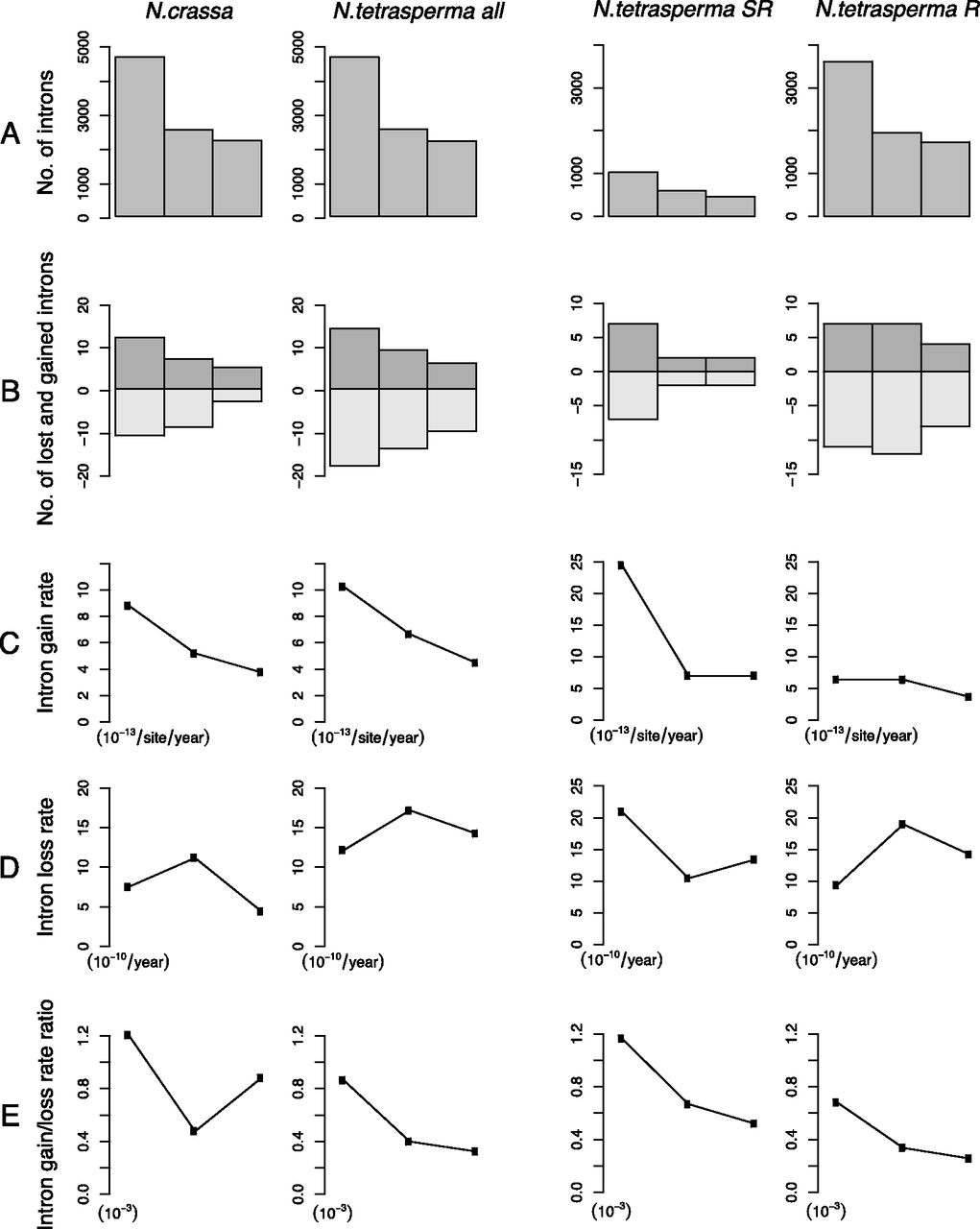
To assess whether intron length affects intron turnover, we plotted the intron length distribution for conserved, gained, and lost introns (estimated based on the existing introns [cf. Zhang et al. 2010]) in Neurospora. We found that the distribution peak is ∼60–69 base pairs (bp) for all three categories (Fig. 4), indicating that intron length does not affect intron gains or losses in Neurospora. Nevertheless, we found two long (> 1000-bp) intron gains, including the second intron of gene NCU00850T0 (name shortened to NCU00850T0-2, same rule applied for all introns hereafter) in N. tetrasperma A (2389 bp long), and NCU08524T0-2 in N. tetrasperma A and a, (6651 and 6652 bp long, respectively) but did not detect any intron losses with length > 500 bp (Fig. 4).
Intron length distribution for the conserved, gained, and lost introns. The binned intron lengths are shown on the x-axes and number of introns on the y-axes.
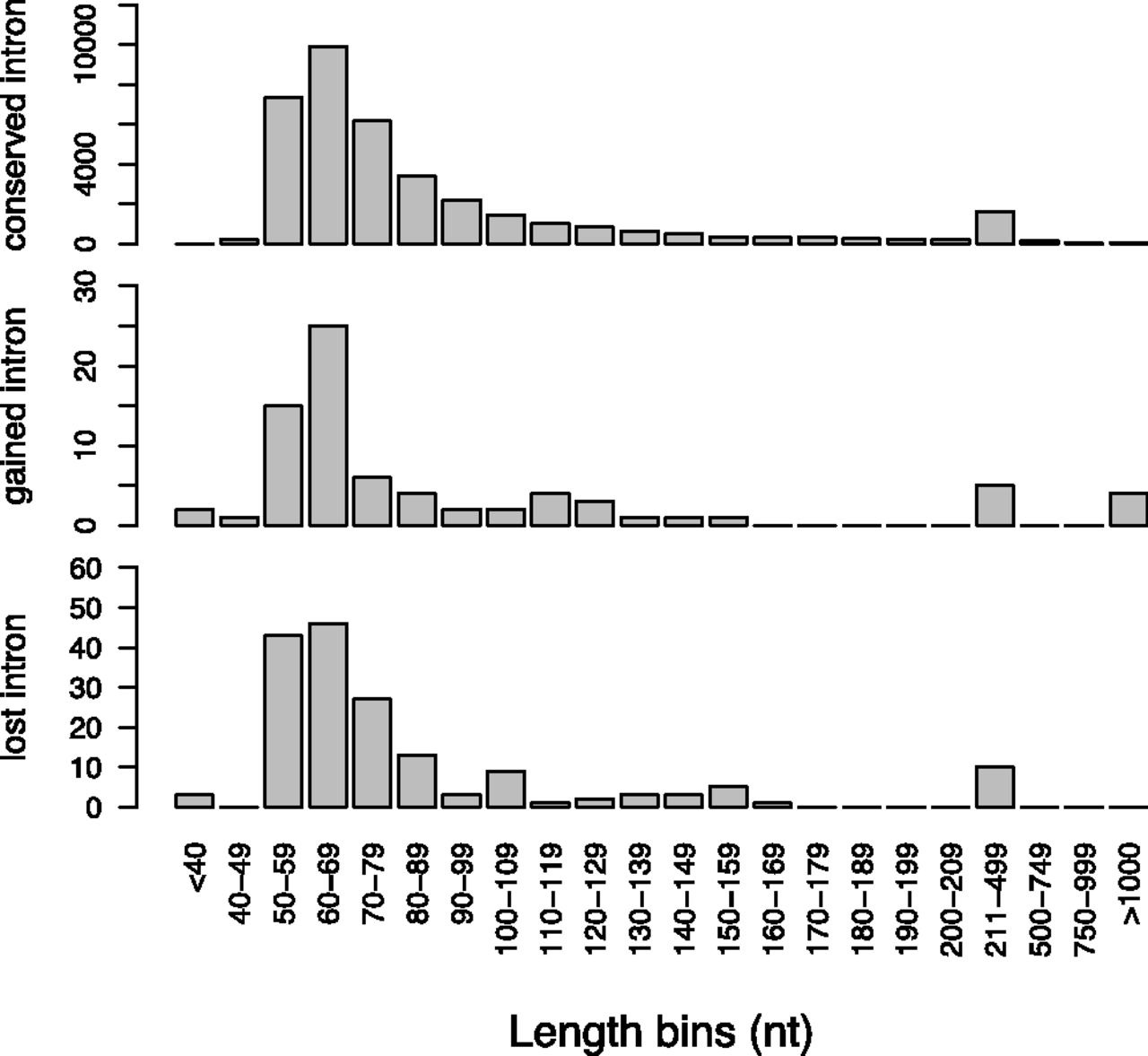
To statistically test the differences in GC content, we investigated the nuclear and mitochondrial DNA in 1-kb fragments. We found that the GC content was significantly higher in nuclear than in mitochondrial DNA (t-test, P < 1 × 10−10), a pattern reported in several previous studies (e.g., Castellana et al. 2011; Amit et al. 2012). Furthermore, we found a significantly higher GC content in CDS than intronic regions (t-test,P < 1 × 10−10), as reported previously (Whittle et al. 2011b), but similar values among conserved, gained, and lost introns (∼45%–46%) (Fig. 5). The aforementioned two long gained introns showed a much lower GC content than the genome average (NCU00850T0-2, GC% 22.9%; NCU08524T0-2, GC% 27.0%). We searched the sequence similarities between these two introns and the mitochondrial genome and found no significant hit (E-value cutoff 1 × 10−4). However, the abundance of AT repeats in these sequences (data not shown) suggests they have been mutated by a fungal-specific mechanism, the repeat-induced point mutation process (RIP), known to increase AT content of a sequence (Galagan et al. 2003).
GC content for the nuclear genome, the mitochondrial genome, all pooled coding sequences (CDS), and for conserved, gained, and lost introns, from left to right. Standard error is plotted on each bar.
Mechanisms for intron gain
Intron gain via NHEJ is expected to leave short direct repeats (∼5–12 bp) spanning both sides of intron-exon borders, a pattern that may be gradually erased by accumulated nucleotide substitutions over time (Li et al. 2009; Farlow et al. 2011). We scanned the intron-exon border for repeats in Neurospora, by extracting 20 bp of nucleotide sequences spanning intron splicing sites and scoring sequences with ≥ 5-bp identical matches on both sides of the splicing site as repeats. Using this approach, we found that the sequences spanning the border of gained introns are significantly enriched in short direct repeats, as compared to the conserved introns (χ2 test, P < 1 × 10−4; gained introns: 32 [42.1%] with repeats, 44 [57.9%] without repeats; conserved introns: 6627 [18.8%], 30,613 [81.2%]), suggesting that a NHEJ mediated process is involved in intron gain in Neurospora.
To identify the source of gained introns in N. crassa and N. tetrasperma A and a, we compared gained intron sequences against the corresponding genomes (including mitochondria) by using BLAT (Kent 2002) and a minimum length and sequence similarity of 50 bp and 90%, respectively. One intron (NCU09740T0-2, N. tetrasperma A) showed a significant similarity to the adjacent 5′ exon (614 bp, 100% sequence identity), suggesting an intron gain mediated by a tandem duplication (cf. Yenerall and Zhou 2012). For the remaining gained introns, we did not find any significant sequence similarities to the corresponding genomes, except the self-hits.
Our findings suggest that intron gains in Neurospora are not mediated by transposon insertions, intron transpositions, or the multiplication of introner-like elements (repeats within introns that are known TE characters [van der Burgt et al. 2012]). Specifically, we searched for sequence similarities of the gained introns in several repeat databases at the RepeatMasker web server (Smit et al. 1996–2010), and found no hits with retroelements or DNA transposons. Furthermore, we searched for sequence similarities between the gained introns and conserved/lost intron sequences in the Neurospora genomes and found no sequences with significant similarity, allowing us to reject intron transposition as a mechanism for intron gain (cf. Torriani et al. 2011; Yenerall et al. 2011). Finally, we did not find any polypyrimidine tracts or increased length of gained introns in comparison to conserved and lost introns (Fig. 4)—hallmarks of the multiplication of ILEs (van der Burgt et al. 2012).
Mechanisms for intron loss
In genes where multiple introns have been lost, the RT-mRNA process is expected to leave a signature of loss of adjacent introns, while NHEJ is expected to delete one intron at a time by microhomologous pairing between the 5′ and 3′ end of the intron (Roy and Gilbert 2006; Farlow et al. 2011). We found that among the 60 genes that lost introns in Neurospora, 57 had lost only one intron (Supplemental Table S1) and are thereby not informative for using this criterion to differentiate between the two mechanisms. However, three of the genes had lost two or more introns, and all of them lost introns adjacently (Supplemental Table S1), consistent with the RT-mRNA as an intron loss mechanism for these particular genes.
No statistically significant differences were detected in the number of short direct repeats for the lost introns (inferred by the sequence from the existing introns at these sites in the other Neurospora species) as compared to the conserved introns (χ2 test, P = 0.43; lost introns, 34 [20.1%] with repeats, 135 [79.9%] without repeats; conserved introns, 6627 [18.8%], 30,613 [81.2%]), suggesting that NHEJ is not the major mechanism for intron loss in Neurospora. Since an intron loss mediated by the NHEJ mechanism could result in an imperfect loss of an intron, we extended this analysis to include introns called with relaxed filtering parameters for intron positions (cf. Roy and Gilbert 2006) and obtained the same result.
Population level analyses of intron gains and losses
Intron polymorphisms in N. tetrasperma
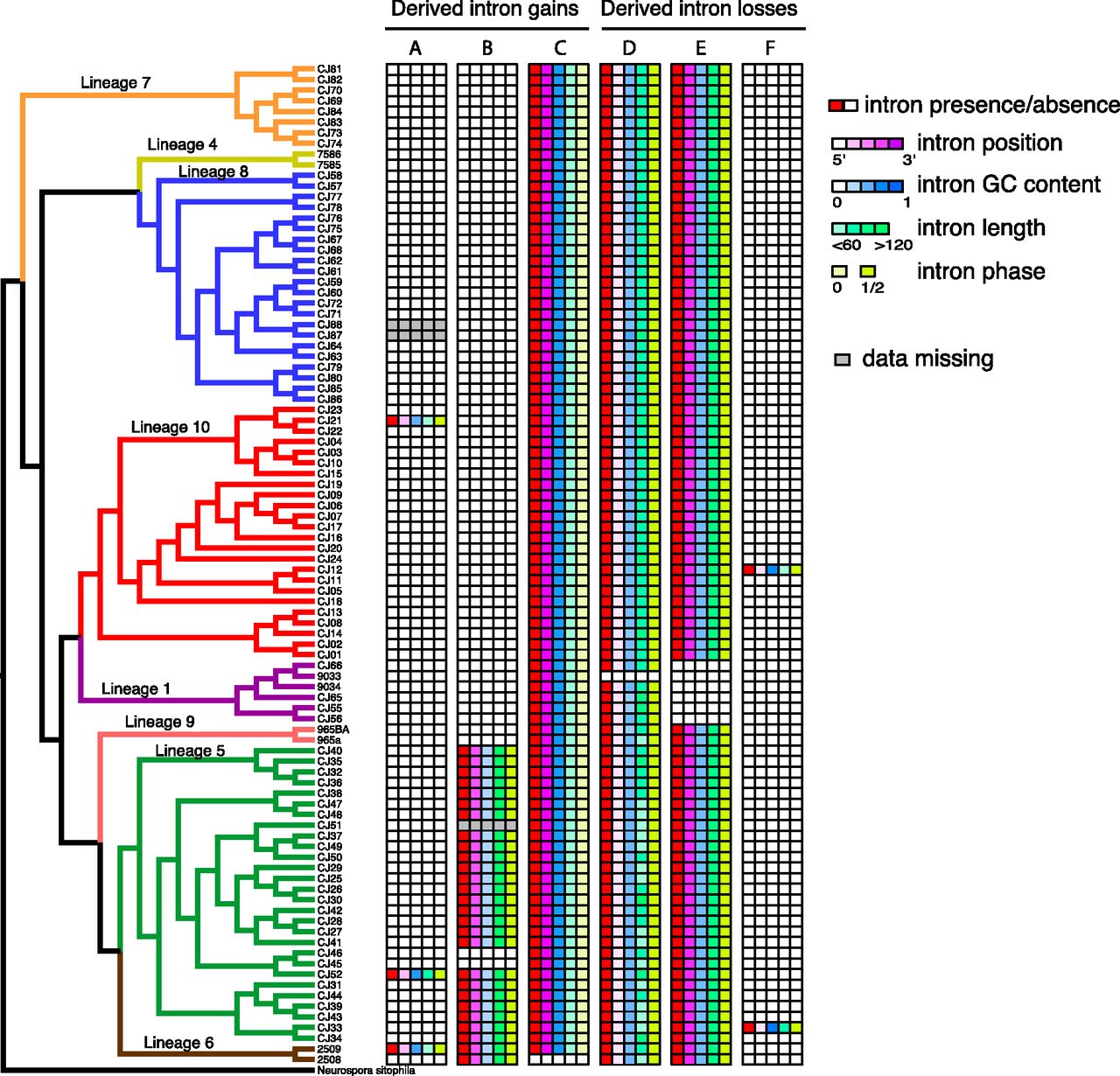
Based on the intron presence/absence pattern for the 9495 intron sites (identified by using the well-annotated genomes), we identified 40 polymorphic sites in the de novo genome assemblies of 92 strains of N. tetrasperma. With the assumptions of Dollo parsimony (Rogozin et al. 2003) and no ancestral intron-polymorphisms shared between species, we divided them into 14 derived polymorphic intron-gains and 26 derived polymorphic intron-losses in N. tetrasperma. To visualize the distribution of intron polymorphisms in N. tetrasperma populations, we plotted the presence, absence, and genetic features for all 14 polymorphic intron gains and 26 polymorphic intron losses, across the phylogeny of the investigated strains (Supplemental Figs. S1, S2). We found variable population frequencies for different intron polymorphisms; Figure 6 shows illustrative examples. First, the intron NCU02668T0-1 (Fig. 6A) exhibits an example of a low-frequency derived intron gain polymorphism, while intron NCU08524T0-3 (Fig. 6B) illustrates an example of an intron gain of intermediate frequency and population bias in lineage 5 and 6, and the intron NCU12060T0-9 (Fig. 6C) is close to fixation in all lineages of N. tetrasperma. For derived intron losses, the intron NCU02539T0-1 (Fig. 6D) was lost in only one strain and represents an example of a low-frequency derived intron loss, the intron NCU01095T0-3 (Fig. 6E) was lost in lineage 1 but not other lineages, illustrating a population structure bias in intron loss, while the intron NCU08145T0-2 (Fig. 6F) is an example of a high-frequency intron loss.
Examples illustrating intron polymorphic data in N. tetrasperma populations. Phylogenetic tree for N. tetrasperma populations is presented in the left panel, and branches for different lineages are shown with different colors. Six intron polymorphisms are chosen to represent (A) intron gain with low frequency, (B) intron gain within lineages, (C) intron gain with high frequency, (D) intron loss with low frequency, (E) intron loss within lineages, and (F) intron loss with high frequency. For each intron data set, the first column indicates intron present (red) or absent (white), and the second to fifth columns indicate intron position (magenta), intron GC content (blue), intron length (green), and intron phase (yellow). Gray indicates data missing.
Frequency spectrum of derived intron gains and losses
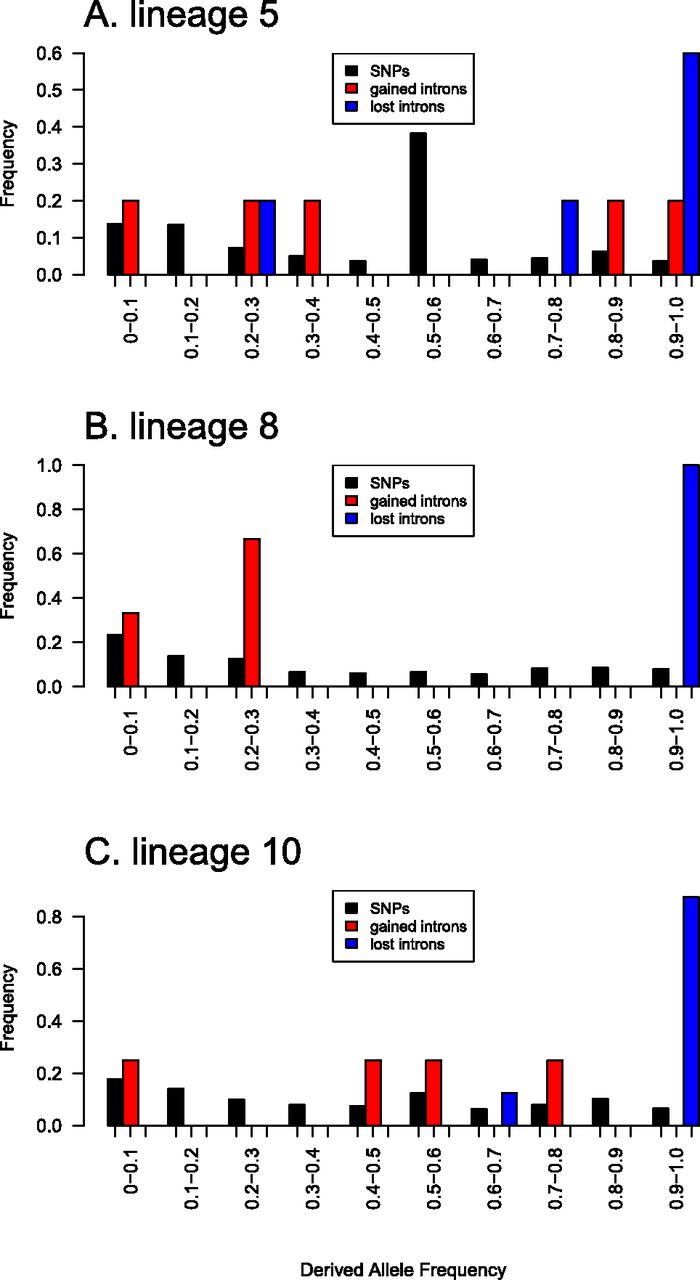
To estimate if selection is involved in shaping intron frequency, we compared the frequency spectrum for the gained and lost intron polymorphisms with a neutral reference. For the latter, we measured the frequency of derived single nucleotide polymorphisms (SNPs) at polymorphic sites, inferred from the outgroup by parsimony. The SNPs were biallelic and fourfold degenerate. Previous studies have reported that the codon usage in Neurospora evolves under selective pressure (Whittle et al. 2011a,b, 2012), and therefore we used only SNPs causing changes between nonpreferred codons in the analyses (cf. Akashi and Schaeffer 1997; Przeworski et al. 1999; Haddrill et al. 2008) and refer to them herein as neutral SNPs. Furthermore, previous studies have shown that N. tetrasperma comprises multiple phylogenetically and reproductively isolated lineages (Menkis et al. 2009; Corcoran et al. 2014). Thus, we conducted separate frequency spectrum analyses for neutral SNPs, intron gains and intron losses polymorphic in each of three lineages represented with N > 10 in the resequenced N. tetrasperma samples (Supplemental Table S2): lineage 5, lineage 8, and lineage 10. See Supplemental Figure S3 for the frequency spectra of neutral SNPs in the R and SR regions of the genomes. For these three lineages, the mean Kst (Hudson et al. 1992) over nine noncoding nuclear loci ranges from 0.10 to 0.37 (Corcoran et al. 2014).
When comparing the frequency distribution of SNPs and intron polymorphisms, we found that intron losses were significantly skewed toward high frequencies, as compared to neutral SNPs, in all three lineages (Wilcoxon-Mann-Whitney test, P < 0.01), while for gained introns, the frequencies were not skewed (P > 0.2) (Fig. 7). To investigate if the pattern was affected by the fixation of gained introns in separate lineages, we included information from intron gains and SNPs that are polymorphic in N. tetrasperma but fixed in separate lineages. In lineages 5 and 10, we found a higher proportion of fixed gained introns than fixed neutral SNPs (Supplemental Fig. S4), but in none of the three lineages were these distributions significantly different (P > 0.1). When merging data from all samples of N. tetrasperma (Supplemental Table S2), both the derived gained and lost introns were statistically significantly skewed toward higher frequencies as compared to the derived neutral SNPs (given as mean ± standard error: gained introns 53.8 ± 11.7%, lost introns 64.9 ± 9.0%, SNPs 23.2 ± 0.1%; P < 0.016), while no difference was found between the frequency distributions of gained and lost introns (P = 0.78) (Supplemental Fig. S5). To investigate if recombination affects selection of derived introns, we conducted frequency spectrum analyses separately for the R and SR regions in N. tetrasperma (Supplemental Fig. S6): Intron losses were significantly skewed toward higher frequencies as compared to neutral SNPs in both R and SR regions; however, intron gains were only significantly skewed in the R region (P < 0.05; R region, gained intron 62.0 ± 12.5%, lost intron 66.5.0 ± 9.9%, SNPs 26.3 ± 0.1%; SR region, gained intron 38.9 ± 21.3%, lost intron 58.0 ± 23.3%, SNPs 14.4 ± 0.2%) (Supplemental Fig. S6). Finally, we excluded the possibility that the analysis of intron gains was affected by a putative sample bias toward gains of high frequency (resulting from the fact that we did not call gains de novo in the population data set and thus are restricted to gains found in at least one of the two N. tetrasperma well-annotated genomes) by verifying the results for the gains outlined above using SNPs with the same sample bias (data not shown).
Frequency spectrum of SNPs and gained and lost intron polymorphisms in three lineages of N. tetrasperma: (A) lineage 5, (B) lineage 8, and (C) lineage 10.
Intron frequency and genetic parameters
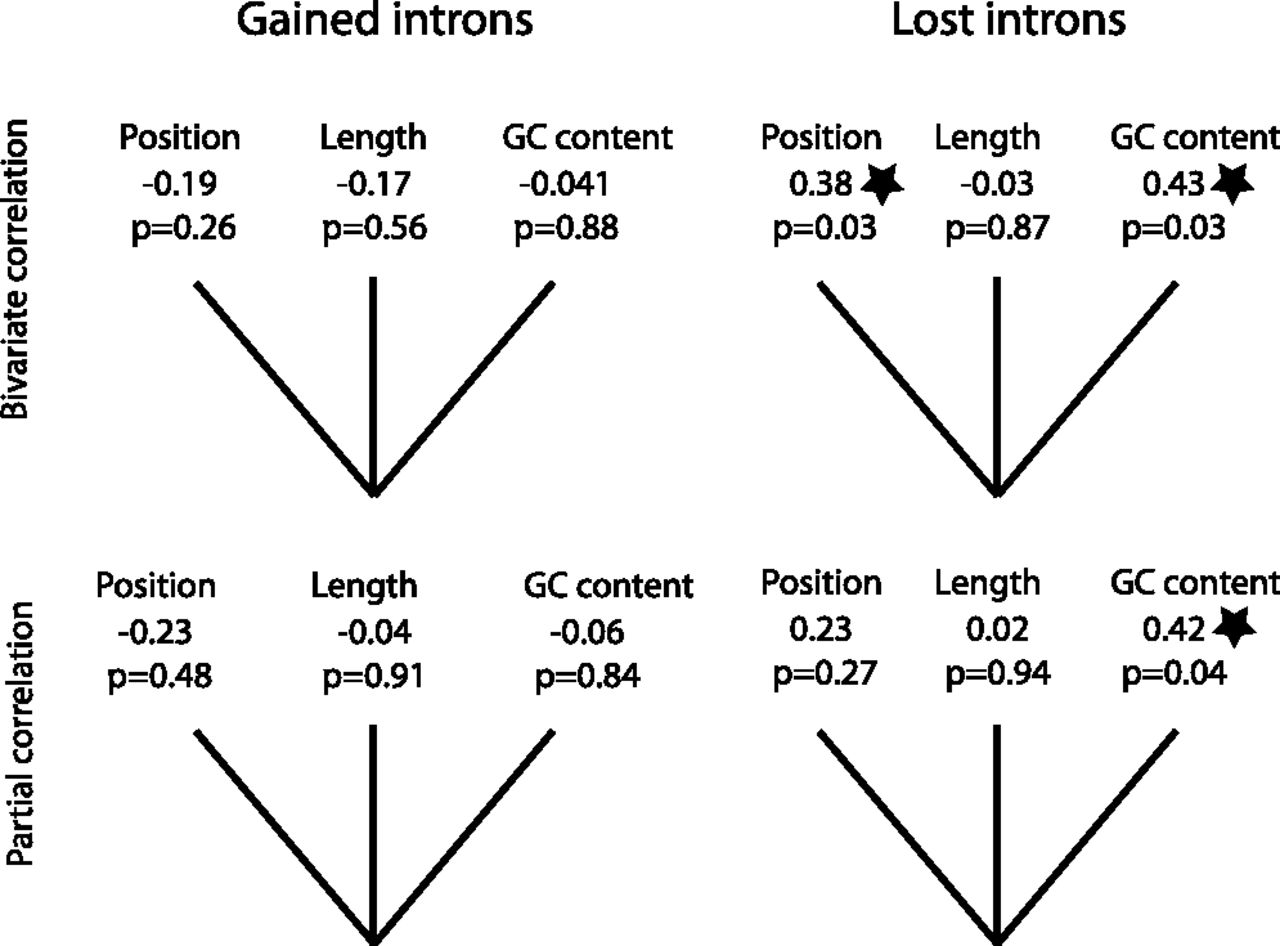
To test if the frequency of derived intron gains and losses in N. tetrasperma populations is associated with genomic parameters, such as the intron position in the gene, intron length, and GC content, we conducted bivariate and partial correlation tests using the 14 derived gained and 26 derived lost intron sites (Fig. 8). In these tests we found a negative, albeit not significant, correlation between gained intron frequency and intron position (Spearman test, r = −0.19, P = 0.26; r = −0.23, P = 0.48) (Fig. 8) and a positive correlation between the frequency of lost introns and intron position (a correlation that was significant in the bivariate but not in the partial correlation test: r = 0.38, P = 0.03; r = 0.23, P = 0.27) (Fig. 8). This result indicates that intron losses in the 5′ end of a gene are less likely to go to fixation than intron losses in the 3′ end. In contrast to intron position, intron length did not show any correlation with intron frequency (r = −0.17, r = 0.04; r = −0.03, r = 0.02; P > 0.56) (Fig. 8), indicating that intron length is not an important factor shaping the intron gain or loss. The GC content is not correlated with the frequency of gained introns (r = −0.04, P = 0.88; r = −0.06, P = 0.85) but shows a significant positive correlation with lost intron frequency (r = 0.43, P = 0.03; r = 0.42, P = 0.04), indicating introns with high GC content are more likely to be lost in N. tetrasperma populations (Fig. 8).
Bivariate and partial correlation test between intron population frequency and intron position, length, and GC content for gained and lost introns. The correlation coefficient and P-value are listed below each category. Tests with statistically significant results are marked with stars.
Furthermore, to test if intron frequency is associated with intron phase, we conducted a χ2 test with the gained and lost introns. We found no correlation between intron phase and intron frequency distribution (P > 0.3), suggesting intron phase is not a driving factor shaping intron turnover in N. tetrasperma populations.
Sequence analyses of gained introns
Recently gained introns are expected to show reduced nucleotide diversity within populations when compared to older introns, due to the shorter time available to accumulate mutations. Additionally, introns gained in a species that have recently increased in frequency due to a selective sweep, as opposed to genetic drift, are expected to show reduced nucleotide diversity and an excess of low frequency variants compared to the genomic background of introns conserved between species. We did not find a pattern of lower nucleotide diversity in the gained introns in N. tetrasperma in comparison to introns shared by all strains (Supplemental Fig. S7), suggesting that the gained introns are not of recent origin. Furthermore, we found no evidence supporting a recent history of selective sweep on the gained introns in N. tetrasperma, with the gained introns not showing more negative Tajima’s D values when compared with conserved introns of similar size in the genome (data not shown).
Discussion
Moderate intron gain and high intron loss rate in Neurospora
Our results provide novel insights into intron evolution in eukaryotes, and particularly for the fungal group Ascomycota. First, we report an exceptionally high rate of intron loss in Neurospora (7.53–13.76 × 10−10 per intron sites per year). This rate is more than 70-fold higher than in the fungal clade Cryptococcus (0.1 × 10−10) (Sharpton et al. 2008) and close to the highest levels reported to date in Saccharomyces pombe (1.3–2.0 × 10−9) and Caenorhabditis elegans (1.6–2.2 × 10−9) (The C. elegans Sequencing Consortium 1998; Wood et al. 2002; Roy and Gilbert 2005; Carmel et al. 2007). The intron gain rate in Neurospora (5.78–6.89 × 10−13 per nucleotide site per year) is in a similar range to certain eukaryotes, such as Drosophila melanogaster (0.7.0–0.9 × 10−12), Anopheles gambiae (0.8.0–0.9 × 10−12) and S. pombe (0.6.0–0.8 × 10−12) (Roy and Gilbert 2005). It is lower than C. elegans (3.4–4.8 × 10−12), Arabidopsis thaliana (2.2–2.9 × 10−12) (Roy and Gilbert 2005), and Daphnia (1.2 × 10−11) (Li et al. 2009), but much higher than the human, mouse, and dog lineages, which have no evidence for intron gains within the last 80 million years (Roy et al. 2003; Coulombe-Huntington and Majewski 2007a).
Overall, we conclude that Neurospora exhibits a high intron loss rate and a moderate gain rate as compared to other eukaryotes. The derived intron gains and losses are skewed to a high frequency, relative to neutral SNPs, in populations of N. tetrasperma (Fig. 7; Supplemental Figs. S4–S6), suggesting that positive selection is involved in maintaining a high intron turnover. This pattern is particularly clear for intron losses, while we found only weak support for selection acting on intron gains. Furthermore, the lack of evidence for lower genetic variation within the gained introns in N. tetrasperma in comparison to conserved introns (Supplemental Fig. S7) indicates that the gained introns are not of recent origin. Thus, our data do not support a history of selective sweeps driving the increase in frequency of the gained introns in N. tetrasperma. This finding is in contrast to the recent report of positive selection and genetic hitchhiking driving the rapid gain of introns in the pathogenic fungus Zymoseptoria tritici (Brunner et al. 2014).
Based on our present results and previous studies of ascomycetes (Carmel et al. 2007), we propose that the high intron loss rate has been maintained throughout the evolutionary history of the Ascomycota, including the recently diverged branches, such as Neurospora. For intron gains, we hypothesize that the ancestor of Ascomycota experienced a burst of intron gains, but the high intron gain rate was not maintained over evolutionary time and therefore is not found in recently diverged branches. This concept is consistent with the punctuated intron gain model, which suggests the dynamics of intron turnover are mainly dominated by losses, with a few episodes of major gains (Carmel et al. 2007; Csuros et al. 2011; Rogozin et al. 2012; Roy and Irimia 2012).
Factors underlying positional biases for intron gains and losses in Neurospora
Our data indicate that, in Neurospora, the 5′ positional biased intron distribution is influenced by multiple factors, including mutational bias and selection. For intron gains, our data suggest that NHEJ is involved as a mechanism, but this process is unlikely to be biased by position in the gene. Nevertheless, our data suggest a possible link between the intron gain rate and gene position; although not statistically significant, we found an elevated intron gain rate at the 5′ end of genes in N. crassa and N. tetrasperma (Figs. 3, 8) in both species-level and population-level analyses. Furthermore, in our merged population data set, we found that the intron gains are skewed toward high frequencies as compared to neutral SNPs (Supplemental Fig. S5), and the frequency is negatively correlated (−0.19/−0.23) with intron position within a gene (Fig. 8), suggesting that the 5′ biased intron gains are favored by natural selection in Neurospora population. This finding is in accordance with previous studies indicating that introns located in the 5′ end of the gene have many important functions. For example, U12 introns are associated with speed of splicing and are concentrated in the 5′ end of the genes in animals and plants, and in Drosophila, the 5′ introns harbor abundant cis-regulatory elements (Marais et al. 2005; Basu et al. 2008). Thus, it is reasonable to hypothesize that an intron gain in the 5′ end of a gene might be driven to fixation by being advantageous for transcription and gene regulation.
In contrast to the pattern of a 5′ gain rate in Neurospora, trends observed in our data suggest an internal-biased intron loss pattern in interspecific comparison (Fig. 3), which has also been found in several other fungal species (Nielsen et al. 2004; Zhang et al. 2010), and a 3′ biased pattern for intron losses in intraspecific comparisons (Fig. 8). We propose that this specific intron-loss pattern is mainly shaped by two factors: mutational bias from RT-mRNA and selection. Specifically, we found RT-mRNA to be a potential mechanism for intron loss in Neurospora. The classic RT-mRNA model predicts a 3′ biased intron loss pattern by this mechanism, due to premature termination in the reverse transcription process (Mourier and Jeffares 2003), and the modified RT-mRNA model suggests internally biased intron losses, due to a high recombination rate in the middle of gene during the reverse transcription process (Zhang et al. 2010). Thus, a mutational bias for RT-mRNA is expected to shape the internal or 3′ biased intron loss pattern, as we found indicated in this study (Figs. 3, 8). Moreover, our data suggest that selection may be driving 3′ losses of introns; in our population level analyses, we found that derived intron losses are significantly skewed toward a high frequency as compared to neutral SNPs (Fig. 7; Supplemental Figs. S4–S6), and the frequency is significantly positively correlated with intron position (Fig. 8), suggesting selection increases the intron loss variants in the 3′ end of a gene. It has been suggested that most introns have a small transcriptional and mutational cost to their host genes (Jackson et al. 2000; Lynch 2002), and selection is strong enough to drive intron loss-variants to high frequency in species with large effective population size (Ne ∼ 106–108) but not for species with a small effective population size (Ne ∼ 104–105) (Lynch and Conery 2003; Charlesworth 2009; Rogozin et al. 2012). The Ne for N. crassa is ∼106 (Ellison et al. 2011a), which is one or two orders of magnitude higher than many vertebrates (Lynch and Conery 2003), and thus we find it reasonable to argue that selection is involved in shaping the intron loss pattern in Neurospora.
Taken together, these findings lead us to hypothesize that the 5′ biased intron gains and internal/3′ biased intron losses have resulted in the accumulation of introns in the 5′ end of genes in the long term, as shown in Figure 3A.
The role of intron length, phase, and GC content for intron turnover
Our analyses of introns in Neurospora have illuminated the association between intron turnover and intron length, intron phase, and GC content. First, the data from this study indicate that intron length is not a major factor determining intron dynamics in Neurospora (Figs. 4, 8), which is in contrast to other studies (of humans and Drosophila) in which selection is involved in shaping intron length distribution (Coulombe-Huntington and Majewski 2007b; Wang and Yu 2011; Leushkin et al. 2013). Furthermore, as for the majority of other organisms that have been analyzed to date (Denoeud et al. 2010; Rogozin et al. 2012), we found a strong phase 0 bias of intron position in Neurospora. Introns in phase 1 and 2 are suggested be of negative influence on organism fitness, as compared to phase 0 introns, due to intron sliding and false splicing, and are thus less likely to go to fixation within a population (Li et al. 2009). For the intron phase 0 bias, two alternative hypotheses exist: the “intron-early hypothesis” suggests the pattern of excess phase 0 introns were inherited from prokaryotic ancestors, whereas the “intron-late hypothesis” suggests phase 0 introns continuously emerged throughout eukaryotic evolution (Gilbert 1987; Logsdon 1998). By showing ongoing fixation of phase 0 introns in a recently diverged eukaryotic branch Neurospora (Supplemental Table S1), our results support the intron-late hypothesis.
Until now, the relationship between base composition and intron turnover has not been investigated thoroughly in microorganisms. It has been reported that in human populations, selection favors high GC content for short introns and low GC content for long introns (Wang and Yu 2011). The study of 11 representative eukaryotic species suggested that differential exon-intron GC content is favored by selection, as it “marks” the exon region from the intron and facilitates spliceosomal recognition (Amit et al. 2012). In this study, we found a statistically significant positive correlation between intron GC content and intron loss frequency (Fig. 8), suggesting that selection favors removal of high GC introns in N. tetrasperma populations. Previously, it was found that codon usage in Neurospora is under weak selection, and preferred codons are more likely to end with either G or C (Whittle et al. 2011b). Thus, we propose that the pattern observed in Figure 5, i.e., a significantly lower GC content for introns as compared to surrounding CDS in Neurospora, is shaped both by selection for enhanced GC content in CDS regions and selection for reduced GC content in introns. We speculate that the pattern of differential GC content in CDS and introns could resemble the splicing signal recognition system in mammals.
Mechanisms for intron gain and loss in Neurospora
Many theories and empirical studies have been put forth with respect to intron gain mechanisms (Yenerall and Zhou 2012), and it appears certain organisms invoke more than one process (Denoeud et al. 2010; Torriani et al. 2011; Yenerall et al. 2011; Roy and Irimia 2012; Collemare et al. 2013). Similarly, our results indicate that intron gains in Neurospora are mediated by several mechanisms: mainly by NHEJ, and likely also by tandem genomic duplication. In contrast, intron losses in N. tetrasperma appear to be medicated by a single process, RT-mRNA, as has been reported for various eukaryotes (Derr and Strathern 1993; Coulombe-Huntington and Majewski 2007b; Sharpton et al. 2008; Zhang et al. 2010; Yenerall et al. 2011), but other causes cannot be fully excluded (e.g., Douglas et al. 2001; Katinka et al. 2001; Mourier and Jeffares 2003).
Conclusions
In this study, we used publicly available Neurospora genomic data, combined with 92 resequenced N. tetrasperma genomes, to study intron evolution in the genus. By the interspecific analyses, we were able to contribute to our understanding of the mechanisms causing gains and losses of introns in this model system of filamentous fungi. Furthermore, even though the numbers of polymorphic gains and losses were small in the investigated populations of N. tetrasperma, and thereby our analyses are influenced by chance alone, we were in this study able to use population-level analyses to shed light on the evolutionary dynamics of these ubiquitous features of the eukaryote genomes. Specifically, our data indicate that positive selection and mutational bias for intron gain and loss variants are important factors driving a high intron turnover. In addition, the data indicated that selection is involved in shaping the 5′ bias in intron position; thus we propose that selection could explain similar biases reported in many unicellular and fungal systems. Furthermore, we showed that intron frequency distributions are associated with genetic parameters, such as intron GC content, in N. tetrasperma populations. Overall, this study is pioneering in showing the importance of natural selection and mutational bias in shaping intron evolution in natural populations. Future population level intron studies in other eukaryotes species will be needed to understand the generality of our findings.
Methods
Genome sequences and annotation of four well-annotated Neurospora genomes
The genomic DNA sequence, amino acid sequence, coding sequence (CDS), and whole genome annotation files were acquired from publicly available data sets, as N. crassa (FGSC 2489, mating type A, finished v 10.0) from the Broad Institute (http://www.broadinstitute.org/), N. discreta (FGSC 8579, mating type A, v 1.0), and N. tetrasperma (FGSC 2508, mating type A, v 2.0; FGSC 2509, mating type a, v 1.0) from the DOE Joint Genome Institute (JGI) (http://genome.jgi-psf.org/). All of these genomes originate from haploid and homokaryotic tissue. Gene prediction and annotation of the genomes are based on sophisticated bioinformatics modeling and experimental EST-data, completed in previous studies (Galagan et al. 2003; Ellison et al. 2011b).
Resequencing, assembly, and analyses of 92 Neurospora tetrasperma genomes
The 92 strains of N. tetrasperma used for genome resequencing are presented in Supplemental Table S2. The strains are haploid and homokaryotic and were obtained from the Fungal Genetics Stock Center (FGSC), University of Missouri, Kansas City, KS. Genomic DNA was extracted from mycelial tissue using the Easy-DNA kit (Invitrogen). Illumina 500-bp paired-end libraries were prepared from each sample at BGI, and each sample was sequenced using Illumina HiSeq 2000, producing paired-end reads of 90 bp in length. The filtered reads for each strain were mapped to the N. tetrasperma 2509 reference genome using BWA (v 0.6.1) (Li and Durbin 2009), and de novo assemblies of all strains were done by SOAPdenovo (version 1.05) (Li et al. 2010). For detailed information on filtering, assembly, and SNP calling, see Supplemental Methods.
The phylogenetic relationship of the 92 resequenced N. tetrasperma strains was inferred from variable SNP sites from the autosomes (i.e., all chromosomes but the mating-type chromosome), by using RAxML v 7.3.1 (-m GRTCAT –f d –N 20) (Stamatakis 2006).
Identification of orthologous genes and introns, and estimation of the intron gain and loss rates, in the Neurospora genomes
We used the method outlined previously (Corcoran et al. 2014) to detect orthologs between N. crassa and each of the other three well-annotated genomes. Our method to identify introns in these genes followed the basic principle of Nielsen et al. (2004): The amino acid sequences for the orthologs were aligned by MAFFT (v 6.717b) with option LINSI (Katoh et al. 2002). To exclude the introns associated with annotation error and poorly aligned regions from the analyses, we filtered out introns that (1) were close (≤ 5 amino acids) to any of the alignment borders, (2) were immediately adjacent to alignment gaps, and (3) had < 50% sequence identity in the 10-amino acid residues on both sides of the intron.
The intron gain and loss rates over time in the N. crassa and N. tetrasperma lineages were calculated as in Roy and Gilbert (2005) and Lynch (2007). For detailed information on rate calculations and statistical tests, see Supplemental Methods.
Population level analyses of introns
With the aim to identify the presence/absence of the introns at each site identified by the four well-annotated genomes in the 92 de novo assembled N. tetrasperma genomes (Supplemental Table S2), we used a sequence alignment-based method. First, the two exon-sequences spanning each filtered intron identified as above were extracted from the annotated N. tetrasperma a genome, and searched for in each of the 92 N. tetrasperma genomes by LASTZ (Harris 2007). If the two exons hit on the same contig in a resequenced genome, and the distance between the two exons was equal or longer than 30 bp, we scored it as an intron presence in the corresponding genome. If the two exons hit on the same contig at a distance shorter than 30 bp, we scored it as an intron absence. For each gained intron polymorphism, the intron sequences was aligned by MAFFT with option LINSI (Katoh et al. 2002), and the alignment quality was inspected manually to confirm that the gained sequences are homologous and not the result of parallel gains of introns at the same location. See the example of an alignment of an intron of gene NCU02858 in Supplemental Figure S8.
To test whether the gained introns were young and have been the subject of a recent selective sweep, we compared the nucleotide diversity and Tajima’s D statistic (Tajima 1989) for each gained intron (fixed or polymorphic–excluding singletons) in N. tetrasperma versus all conserved introns of a similar size (±10 bp) in the genome, both within lineages (L5, L8, and L10) and over all N. tetrasperma strains. Any gained intron that had a nucleotide diversity and Tajima’s D < 95% of the conserved introns of similar size was considered as an intron that may have undergone a selective sweep.
Data access
The reads for the resequenced strains of Neurospora tetrasperma generated in this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession numbers SRP040006 and SRP040007.
Acknowledgments
We thank Siv Andersson from the Department of Molecular Evolution, Uppsala University, and three anonymous reviewers for helpful comments and suggestions. We also thank the Swedish Research Council, Helge Ax:son Johnsons stiftelse, and Nilsson-Ehle-Donationerna from Kungl. Fysiografiska Sällskapet i Lund for financial support. We also thank the BGI for their contribution in data generation.
Notes
[1] Supplementary material [Supplemental material is available for this article.]
[2] Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.175653.114.
References
- ↵Akashi H, Schaeffer SW. 1997. Natural selection and the frequency distributions of “silent” DNA polymorphism in Drosophila. Genetics 146: 295–307.
- ↵Amit M, Donyo M, Hollander D, Goren A, Kim E, Gelfman S, Lev-Maor G, Burstein D, Schwartz S, Postolsky B, . 2012. Differential GC content between exons and introns establishes distinct strategies of splice-site recognition. Cell Reports 1: 543–556.
- ↵Basu MK, Makalowski W, Rogozin IB, Koonin EV. 2008. U12 intron positions are more strongly conserved between animals and plants than U2 intron positions. Biol Direct 3: 19.
- ↵Berget SM, Moore C, Sharp PA. 1977. Spliced segments at the 5′ terminus of adenovirus 2 late mRNA. Proc Natl Acad Sci 74: 3171–3175.
- ↵Bernstein LB, Mount SM, Weiner AM. 1983. Pseudogenes for human small nuclear RNA U3 appear to arise by integration of self-primed reverse transcripts of the RNA into new chromosomal sites. Cell 32: 461–472.
- ↵Boeke JD, Garfinkel DJ, Styles CA, Fink GR. 1985. Ty elements transpose through an RNA intermediate. Cell 40: 491–500.
- ↵Brunner PC, Torriani SF, Croll D, Stukenbrock EH, McDonald BA. 2014. Hitchhiking selection is driving intron gain in a pathogenic fungus. Mol Biol Evol 31: 1741–1749.
- ↵The C. elegans Sequencing Consortium. 1998. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science 282: 2012–2018.
- ↵Carmel L, Wolf YI, Rogozin IB, Koonin EV. 2007. Three distinct modes of intron dynamics in the evolution of eukaryotes. Genome Res 17: 1034–1044.
- ↵Castellana S, Vicario S, Saccone C. 2011. Evolutionary patterns of the mitochondrial genome in Metazoa: exploring the role of mutation and selection in mitochondrial protein coding genes. Genome Biol Evol 3: 1067–1079.
- ↵Charlesworth B. 2009. Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat Rev Genet 10: 195–205.
- ↵Chorev M, Carmel L. 2012. The function of introns. Front Genet 3: 55.
- ↵Chow LT, Gelinas RE, Broker TR, Roberts RJ. 1977. An amazing sequence arrangement at the 5′ ends of adenovirus 2 messenger RNA. Cell 12: 1–8.
- ↵Collemare J, Burgt Avd, de Wit PJ. 2013. At the origin of spliceosomal introns: Is multiplication of introner-like elements the main mechanism of intron gain in fungi? Commun Integr Biol 6: e23147.
- ↵Corcoran P, Dettman J, Sun Y, Luque E, Corrochano L, Taylor J, Lascoux M, Johannesson H. 2014. A global multilocus analysis of the model fungus Neurospora reveals a single recent origin of a novel genetic system. Mol Phylogenet Evol 78: 136–147.
- ↵Coulombe-Huntington J, Majewski J. 2007a. Characterization of intron loss events in mammals. Genome Res 17: 23–32.
- ↵Coulombe-Huntington J, Majewski J. 2007b. Intron loss and gain in Drosophila. Mol Biol Evol 24: 2842–2850.
- ↵Croll D, McDonald BA. 2012. Intron gains and losses in the evolution of Fusarium and Cryptococcus fungi. Genome Biol Evol 4: 1148–1161.
- ↵Csuros M, Rogozin IB, Koonin EV. 2011. A detailed history of intron-rich eukaryotic ancestors inferred from a global survey of 100 complete genomes. PLoS Comput Biol 7: e1002150.
- ↵Denoeud F, Henriet S, Mungpakdee S, Aury JM, Da Silva C, Brinkmann H, Mikhaleva J, Olsen LC, Jubin C, Canestro C, . 2010. Plasticity of animal genome architecture unmasked by rapid evolution of a pelagic tunicate. Science 330: 1381–1385.
- ↵Derr LK, Strathern JN. 1993. A role for reverse transcripts in gene conversion. Nature 361: 170–173.
- ↵Dettman JR, Jacobson DJ, Taylor JW. 2003. A multilocus genealogical approach to phylogenetic species recognition in the model eukaryote Neurospora. Evolution 57: 2703–2720.
- ↵Dodge BO. 1927. Nuclear phenomena associated with heterothallism and homothallism in the ascomycete Neurospora. J Agric Res 35: 0289–0305.
- ↵Douglas S, Zauner S, Fraunholz M, Beaton M, Penny S, Deng LT, Wu X, Reith M, Cavalier-Smith T, Maier UG. 2001. The highly reduced genome of an enslaved algal nucleus. Nature 410: 1091–1096.
- ↵Ellison CE, Hall C, Kowbel D, Welch J, Brem RB, Glass NL, Taylor JW. 2011a. Population genomics and local adaptation in wild isolates of a model microbial eukaryote. Proc Natl Acad Sci 108: 2831–2836.
- ↵Ellison CE, Stajich JE, Jacobson DJ, Natvig DO, Lapidus A, Foster B, Aerts A, Riley R, Lindquist EA, Grigoriev IV, . 2011b. Massive changes in genome architecture accompany the transition to self-fertility in the filamentous fungus Neurospora tetrasperma. Genetics 189: 55–69.
- ↵Evans RM, Fraser N, Ziff E, Weber J, Wilson M, Darnell JE. 1977. The initiation sites for RNA transcription in Ad2 DNA. Cell 12: 733–739.
- ↵Farlow A, Meduri E, Dolezal M, Hua L, Schlotterer C. 2010. Nonsense-mediated decay enables intron gain in Drosophila. PLoS Genet 6: e1000819.
- ↵Farlow A, Meduri E, Schlotterer C. 2011. DNA double-strand break repair and the evolution of intron density. Trends Genet 27: 1–6.
- ↵Fink GR. 1987. Pseudogenes in yeast? Cell 49: 5–6.
- ↵Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, Jaffe D, FitzHugh W, Ma LJ, Smirnov S, Purcell S, . 2003. The genome sequence of the filamentous fungus Neurospora crassa. Nature 422: 859–868.
- ↵Gilbert W. 1987. The exon theory of genes. Cold Spring Harb Sym 52: 901–905.
- ↵Goldberg S, Schwartz H, Darnell JE Jr. 1977. Evidence from UV transcription mapping in HeLa cells that heterogeneous nuclear RNA is the messenger RNA precursor. Proc Natl Acad Sci 74: 4520–4523.
- ↵Haddrill PR, Bachtrog D, Andolfatto P. 2008. Positive and negative selection on noncoding DNA in Drosophila simulans. Mol Biol Evol 25: 1825–1834.
- ↵Harris RS. 2007. “Improved pairwise alignment of genomic DNA.” PhD thesis, The Pennsylvania State University, State College, PA.
- ↵Hudson RR, Boos DD, Kaplan NL. 1992. A statistical test for detecting geographic subdivision. Mol Biol Evol 9: 138–151.
- ↵Jackson DA, Pombo A, Iborra F. 2000. The balance sheet for transcription: an analysis of nuclear RNA metabolism in mammalian cells. FASEB J 14: 242–254.
- ↵Jeffares DC, Mourier T, Penny D. 2006. The biology of intron gain and loss. Trends Genet 22: 16–22.
- ↵Johnson RD, Jasin M. 2000. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J 19: 3398–3407.
- ↵Kasuga T, White TJ, Taylor JW. 2002. Estimation of nucleotide substitution rates in Eurotiomycete fungi. Mol Biol Evol 19: 2318–2324.
- ↵Katinka MD, Duprat S, Cornillot E, Metenier G, Thomarat F, Prensier G, Barbe V, Peyretaillade E, Brottier P, Wincker P, . 2001. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature 414: 450–453.
- ↵Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30: 3059–3066.
- ↵Kent WJ. 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664.
- ↵Leushkin EV, Bazykin GA, Kondrashov AS. 2013. Strong mutational bias toward deletions in the Drosophila melanogaster genome is compensated by selection. Genome Biol Evol 5: 514–524.
- ↵Lewin R. 1983. How mammalian RNA returns to its genome. Science 219: 1052–1054.
- ↵Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760.
- ↵Li W, Tucker AE, Sung W, Thomas WK, Lynch M. 2009. Extensive, recent intron gains in Daphnia populations. Science 326: 1260–1262.
- ↵Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, . 2010. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20: 265–272.
- ↵Llopart A, Comeron JM, Brunet FG, Lachaise D, Long M. 2002. Intron presence-absence polymorphism in Drosophila driven by positive Darwinian selection. Proc Natl Acad Sci 99: 8121–8126.
- ↵Logsdon JM. 1998. The recent origins of spliceosomal introns revisited. Curr Opin Genet Dev 8: 637–648.
- ↵Lynch M. 2002. Intron evolution as a population-genetic process. Proc Natl Acad Sci 99: 6118–6123.
- ↵Lynch M. 2007. The origins of genome architecture. Sinauer Associates, Sunderland, MA.
- ↵Lynch M, Conery JS. 2003. The origins of genome complexity. Science 302: 1401–1404.
- ↵Mao Z, Bozzella M, Seluanov A, Gorbunova V. 2008. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst) 7: 1765–1771.
- ↵Marais G, Nouvellet P, Keightley PD, Charlesworth B. 2005. Intron size and exon evolution in Drosophila. Genetics 170: 481–485.
- ↵Menkis A, Jacobson DJ, Gustafsson T, Johannesson H. 2008. The mating-type chromosome in the filamentous ascomycete Neurospora tetrasperma represents a model for early evolution of sex chromosomes. PLoS Genet 4: e1000030.
- ↵Menkis A, Bastiaans E, Jacobson DJ, Johannesson H. 2009. Phylogenetic and biological species diversity within the Neurospora tetrasperma complex. J Evol Biol 22: 1923–1936.
- ↵Mourier T, Jeffares DC. 2003. Eukaryotic intron loss. Science 300: 1393.
- ↵Nielsen CB, Friedman B, Birren B, Burge CB, Galagan JE. 2004. Patterns of intron gain and loss in fungi. PLoS Biol 2: e422.
- ↵Nygren K, Strandberg R, Wallberg A, Nabholz B, Gustafsson T, Garcia D, Cano J, Guarro J, Johannesson H. 2011. A comprehensive phylogeny of Neurospora reveals a link between reproductive mode and molecular evolution in fungi. Mol Phylogenet Evol 59: 649–663.
- ↵Preston CR, Flores CC, Engels WR. 2006. Differential usage of alternative pathways of double-strand break repair in Drosophila. Genetics 172: 1055–1068.
- ↵Przeworski M, Charlesworth B, Wall JD. 1999. Genealogies and weak purifying selection. Mol Biol Evol 16: 246–252.
- ↵Rebuzzini P, Khoriauli L, Azzalin CM, Magnani E, Mondello C, Giulotto E. 2005. New mammalian cellular systems to study mutations introduced at the break site by non-homologous end-joining. DNA Repair (Amst) 4: 546–555.
- ↵Rogozin IB, Wolf YI, Sorokin AV, Mirkin BG, Koonin EV. 2003. Remarkable interkingdom conservation of intron positions and massive, lineage-specific intron loss and gain in eukaryotic evolution. Curr Biol 13: 1512–1517.
- ↵Rogozin IB, Carmel L, Csuros M, Koonin EV. 2012. Origin and evolution of spliceosomal introns. Biol Direct 7: 11.
- ↵Roy SW, Gilbert W. 2005. Rates of intron loss and gain: implications for early eukaryotic evolution. Proc Natl Acad Sci 102: 5773–5778.
- ↵Roy SW, Gilbert W. 2006. The evolution of spliceosomal introns: patterns, puzzles and progress. Nat Rev Genet 7: 211–221.
- ↵Roy SW, Irimia M. 2012. Genome evolution: where do new introns come from? Curr Biol 22: R529–R531.
- ↵Roy SW, Fedorov A, Gilbert W. 2003. Large-scale comparison of intron positions in mammalian genes shows intron loss but no gain. Proc Natl Acad Sci 100: 7158–7162.
- ↵Sharpton TJ, Neafsey DE, Galagan JE, Taylor JW. 2008. Mechanisms of intron gain and loss in Cryptococcus. Genome Biol 9: R24.
- ↵Shimizu K, Li HM, Virtudazo EV, Watanabe A, Kamei K, Yamaguchi M, Kawamoto S. 2010. Deletion of CnLIG4 DNA ligase gene in the fungal pathogen Cryptococcus neoformans elevates homologous recombination efficiency. Mycoscience 51: 28–33.
- ↵Smit AFA, Hubley R, Green P. 1996–2010. RepeatMasker Open-3.0. http://www.repeatmasker.org.
- ↵Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22: 2688–2690.
- ↵Sun Y, Corcoran P, Menkis A, Whittle CA, Andersson SG, Johannesson H. 2012. Large-scale introgression shapes the evolution of the mating-type chromosomes of the filamentous ascomycete Neurospora tetrasperma. PLoS Genet 8: e1002820.
- ↵Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
- ↵Torriani SF, Stukenbrock EH, Brunner PC, McDonald BA, Croll D. 2011. Evidence for extensive recent intron transposition in closely related fungi. Curr Biol 21: 2017–2022.
- ↵van der Burgt A, Severing E, de Wit PJ, Collemare J. 2012. Birth of new spliceosomal introns in fungi by multiplication of introner-like elements. Curr Biol 22: 1260–1265.
- ↵Wang D, Yu J. 2011. Both size and GC-content of minimal introns are selected in human populations. PLoS ONE 6: e17945.
- ↵Whittle CA, Sun Y, Johannesson H. 2011a. Degeneration in codon usage within the young segment of suppressed recombination in the mating type chromosomes of Neurospora tetrasperma. Eukaryot Cell 10: 594–603.
- ↵Whittle CA, Sun Y, Johannesson H. 2011b. Evolution of synonymous codon usage in Neurospora tetrasperma and Neurospora discreta. Genome Biol Evol 3: 332–343.
- ↵Whittle CA, Sun Y, Johannesson H. 2012. Genome-wide selection on codon usage at the population level in the fungal model organism Neurospora crassa. Mol Biol Evol 29: 1975–1986.
- ↵Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, Sgouros J, Peat N, Hayles J, Baker S, . 2002. The genome sequence of Schizosaccharomyces pombe. Nature 415: 871–880.
- ↵Yenerall P, Zhou L. 2012. Identifying the mechanisms of intron gain: progress and trends. Biol Direct 7: 29.
- ↵Yenerall P, Krupa B, Zhou L. 2011. Mechanisms of intron gain and loss in Drosophila. BMC Evol Biol 11: 364.
- ↵Zhang LY, Yang YF, Niu DK. 2010. Evaluation of models of the mechanisms underlying intron loss and gain in Aspergillus fungi. J Mol Evol 71: 364–373.