
Novel H3K4me3 marks are enriched at human- and chimpanzee-specific cytogenetic structures
- 1Center for Integrative Genomics, University of Lausanne, 1015 Lausanne, Switzerland;
- 2Swiss Institute of Bioinformatics, 1015 Lausanne, Switzerland
- Corresponding authors: giuliana.giannuzzi{at}unil.ch, alexandre.reymond{at}unil.ch
Abstract
Human and chimpanzee genomes are 98.8% identical within comparable sequences. However, they differ structurally in nine pericentric inversions, one fusion that originated human chromosome 2, and content and localization of heterochromatin and lineage-specific segmental duplications. The possible functional consequences of these cytogenetic and structural differences are not fully understood and their possible involvement in speciation remains unclear. We show that subtelomeric regions—regions that have a species-specific organization, are more divergent in sequence, and are enriched in genes and recombination hotspots—are significantly enriched for species-specific histone modifications that decorate transcription start sites in different tissues in both human and chimpanzee. The human lineage-specific chromosome 2 fusion point and ancestral centromere locus as well as chromosome 1 and 18 pericentric inversion breakpoints showed enrichment of human-specific H3K4me3 peaks in the prefrontal cortex. Our results reveal an association between plastic regions and potential novel regulatory elements.
Chromosomes, the DNA–protein structures that carry genetic information, undergo structural rearrangements, including fusion and fission events, as well as inversions, translocations, duplications, and deletions. Human and chimpanzee karyotypes differ by one chromosomal fusion that gave rise to human chromosome 2 (HSA2) from two ancestral chromosomes coupled to the inactivation of one of the two centromeres, at least nine pericentric inversions, and in the content of constitutive heterochromatin (Yunis et al. 1980; IJdo et al. 1991; Baldini et al. 1993; Nickerson and Nelson 1998). Seven of these inversions, mapping to human chromosomes 4, 5, 9, 12, 15, 16, and 17, are specific to the chimpanzee lineage (Marzella et al. 2000; Kehrer-Sawatzki et al. 2002; Locke et al. 2003; Goidts et al. 2005; Kehrer-Sawatzki et al. 2005a,b,c; Shimada et al. 2005; Szamalek et al. 2005), while the remaining two, mapping to HSA1 and HSA18, appeared in the human lineage after separation from the chimpanzee (Yunis and Prakash 1982; McConkey 1997; Dennehey et al. 2004; Weise et al. 2005; Szamalek et al. 2006). These reorganized structures became fixed during evolution either by providing an advantage or by mere genetic drift.
Human subtelomeric regions, as well as pericentromeric ones, are hotspots of segmental duplications that were reshaped over recent evolutionary time (Horvath et al. 2000; Mefford and Trask 2002; She et al. 2004; Linardopoulou et al. 2005). Indeed, while human and chimpanzee genomes are 98.77% identical within comparable sequences, they show an increased divergence (15%) in the terminal 10 Mbp (millions of base pairs) of chromosomes (The Chimpanzee Sequencing and Analysis Consortium 2005). These highly plastic segments of the human genome show qualitative and quantitative differences in the distribution of segmental duplications when compared with the great apes, consistent with their recent origin and human-specific sequence transfers (Horvath et al. 2001; Bailey et al. 2002; Horvath et al. 2003; Linardopoulou et al. 2005; Locke et al. 2005). In addition, regions enriched in segmental duplications are more prone to both interspecies and intraspecies structural variation (Newman et al. 2005; Sharp et al. 2005), since these repeated segments may mediate nonallelic homologous recombination (NAHR) (Hastings et al. 2009).
It is still unclear whether chromosomal rearrangements and structurally different loci played a role in the human–chimpanzee speciation. Indeed the hypothesis that they affected the rate of genetic divergence between humans and chimpanzees does not have enough support (Kehrer-Sawatzki and Cooper 2007). Previous studies revealed no evidence of accelerated evolution for genes on rearranged versus colinear chromosomes (Lu et al. 2003; Navarro and Barton 2003; Vallender and Lahn 2004; Zhang et al. 2004; The Chimpanzee Sequencing and Analysis Consortium 2005; Marques-Bonet et al. 2007) and showed that chromosomal rearrangements have generally no impact on gene expression except in a few particular cases (Munoz and Sankoff 2012). However, chromosomal rearrangements appear to be associated with higher divergence in gene-expression levels in the brain (Marques-Bonet et al. 2004) and genes located on rearranged chromosomes showed reduced recombination rate compared with colinear ones (Farré et al. 2013).
In this study we analyzed the chromosomal distribution of human- and chimpanzee-specific enrichment/depletion of H3K4me3 histone modifications in the prefrontal cortex (Shulha et al. 2012) and lymphoblastoid cell lines (LCLs) (Cain et al. 2011) and tested their accumulation at genomic regions with species-specific structure. H3K4me3 is an epigenetic mark broadly associated with RNA polymerase II occupancy at transcription start sites and RNA expression levels (Wang et al. 2008; The ENCODE Project Consortium 2012; Kilpinen et al. 2013). We detected a higher density of human- and chimpanzee-specific H3K4me3 peaks in subtelomeric regions both in the prefrontal cortex and LCLs. The human prefrontal cortex similarly showed a higher density of species-specific H3K4me3 marks at other human-specific genomic structures. Our results provide evidence for a possible functional and regulatory role in human and chimpanzee evolution of recently acquired structural and chromosomal differences.
Results
A recent analysis compared the genome-wide profiles of H3K4me3 histone modifications in the prefrontal cortex of human, chimpanzee, and macaque (Shulha et al. 2012). It identified 471 loci with significant changes in histone modification rates in human (enrichment, 410; loss, 61) when compared with the two nonhuman primate species. Additionally, the investigators detected 33 human specifically enriched loci that were selectively methylated in neuronal versus nonneuronal cells (Shulha et al. 2012), among which they pinpoint DPP10 (chromosomal band 2q14.1), CNTN4, and CHL1 (both at 3p26.3), three genes conferring susceptibility to neurological disease (Sakurai et al. 2002; Fernandez et al. 2004; Marshall et al. 2008; Glessner et al. 2009; Roohi et al. 2009; Salyakina et al. 2011). They describe and further analyze the DPP10 and 16p11.2–12.2 loci, the latter among the 410 loci with human-specific enrichment in H3K4me3 modifications. They conclude that “coordinated epigenetic regulation via newly derived transcription start site chromatin could play an important role in the emergence of human-specific gene expression networks in brain” (Shulha et al. 2012). It is noteworthy that all four of the featured loci map to regions that were modified in recent human evolution. CNTN4 and CHL1 are within the subtelomeric region of the HSA3 short arm. The DPP10 locus (2q14.1) maps only 1 Mbp away from the above-mentioned fusion point of HSA2 (IJdo et al. 1991; Fan et al. 2002; The Chimpanzee Sequencing and Analysis Consortium 2005), while the 16p11.2–12.2 region has seen a rapid integration of segmental duplications in the last 15 million years of hominoid evolution that contributed to a profound modification of these chromosomal bands (Johnson et al. 2001; Antonacci et al. 2010), putting them at risk for recurrent pathogenic rearrangements (Fig. 1; Girirajan et al. 2010; Walters et al. 2010; Jacquemont et al. 2011). These observations suggest that we should consider the possibility that the differences reported in regulatory footprints (Shulha et al. 2012) might be associated with the specific genomic organization of these loci in human and/or with their localization to or closeness to highly plastic sections of the human genome.
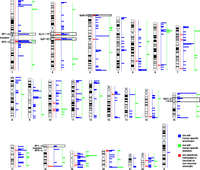
Karyotype-wide mapping of the regions with human-specific enrichment (n = 410, blue) and depletion (n = 61, green) of H3K4me3 modifications in prefrontal neurons. The localizations of the 33 human-specific loci selectively methylated in neuronal versus nonneuronal cells are similarly pinpointed in red (n = 33 regions). The human-specific pericentric inversion breakpoints (BP1 and BP2) of HSA1 and HSA18, the fusion point (FP) and ancestral centromere (AC) of HSA2, and the 16p11.2–12.2 and 3p26.3 regions mentioned in the text are indicated.
The chromosome-wide distribution of the 410 H3K4me3 peaks with human-specific enrichment in prefrontal neurons is not uniform (Fig. 1). We assessed the possible association between regions with significantly different epigenetic panorama in humans when compared with other primates (Shulha et al. 2012) and genomic segments around loci that were structurally modified during the recent evolution of the human genome, i.e., HSA2 fusion point and ancestral centromere, as well as HSA1 and HSA18 inversion breakpoints (Yunis et al. 1980; Dennehey et al. 2004; Szamalek et al. 2006), with HSA1 also encompassing pericentromeric heterochromatin that is absent in its chimpanzee homolog (Yunis et al. 1980). Additionally, we assessed highly plastic segments such as human-specific segmental duplications (Sudmant et al. 2013) together with subtelomeric and pericentromeric regions (Yunis et al. 1980; Bailey et al. 2001; Bailey et al. 2002; Horvath et al. 2003; The Chimpanzee Sequencing and Analysis Consortium 2005; Linardopoulou et al. 2005; Locke et al. 2005).
The human-specific prefrontal cortex H3K4me3-enriched sites significantly accumulate at subtelomeric (fivefold) and pericentromeric sites (threefold) in both the number of peaks (P-value = 4 × 10−73 and 3 × 10−15, respectively; permutation P-value = 0.001) and the amount of base pairs covered (P-value = 2 × 10−56 and 2 × 10−13, respectively; permutation P-value = 0.001) (Fig. 2A,B; Table 1). For example, 87% and 82% of the subtelomeric and pericentromeric regions of autosomes (34/39 and 18/22, respectively) contain at least one H3K4me3 human-enriched peak (Fig. 1). Interestingly, the density and fraction of these peaks show a sharp increase toward the chromosomal ends (Fig. 2C,D).
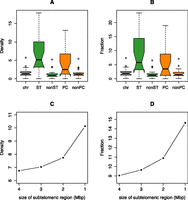
Distribution of the density (A) and fraction (B) of H3K4me3-enriched human-specific regions in all chromosome (chr), subtelomeric (ST), nonsubtelomeric (nonST), pericentromeric (PC), and nonpericentromeric (nonPC) regions. Note the significant concentration of these epigenetic decorations in the highly plastic subtelomeric and pericentromeric sections of the human genome (density, one-way χ2 test, P-values = 4 × 10−73 and 3 × 10−15, respectively; fraction, Fisher’s exact test, P-values = 2 × 10−56 and 2 × 10−13, respectively). See Table 1 for nomenclature. Comparison of density (C) and fraction (D) of human-specific H3K4me3 modifications in subtelomeric regions of different size (4, 3, 2, and 1 Mbp).
Distribution of the 410 regions with human-specific enrichment of H3K4me3 modifications in prefrontal cortexa (including and excluding segmental duplications and chromosome X from the counts)
We detected a significant eightfold increase in both the density (P-value = 2 × 10−21, permutation P-value = 0.001) and amount of base pairs covered (P-value = 7 × 10−11, permutation P-value = 0.001) of human specifically enriched histone modification marks in human-specific segmental duplications (Sudmant et al. 2013). Albeit not significant, the segmental duplication-rich 16p11.2–12.2 cytogenetic bands showed a consistent twofold enrichment of these epigenetic marks when compared with the genome average.
Despite a certain overlap of highly plastic genome sections such as subtelomeric, pericentromeric, and duplicated regions with human lineage-specific rearrangement breakpoints (e.g., HSA1 p-arm inversion breakpoint overlaps with its pericentromeric region and HSA18 p-arm inversion breakpoint overlaps with its subtelomeric region), we independently assessed possible enrichment in the latter. The fusion point (chr2:113–116 Mbp) and ancestral centromere (2q21) loci of HSA2 overlap clusters with higher density (14- and sixfold, respectively, P-value = 0.001) and sequence coverage (15- and sixfold, P-value = 0.001 and 0.002, respectively) of H3K4me3 peaks when compared with the genome-wide average (Figs. 1, 3; Table 1). The HSA1 inversion breakpoints show 21-fold enrichment in both density and sequence coverage (P-value = 0.001), while the HSA18 inversion breakpoints show 10- (P-value = 0.006) and 18-fold (P-value = 0.001) higher density and sequence coverage than the genome average, respectively (Figs. 1, 3; Table 1). As a negative control, we tested chimpanzee-specific inversion breakpoints for which the human genome organization preserves the ancestral state (i.e., the ones mapping to chromosomes 4, 5, 9, 12, 16, and 17), and found no difference in either density or sequence coverage (P-values = 1) of H3K4me3 lineage-specific peaks compared with the genome average (Table 1).
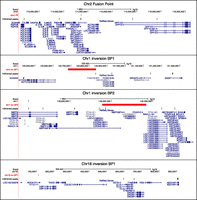
Localization of human specifically enriched H3K4me3 peaks in prefrontal cortex that map in the proximity of the HSA2 fusion point (n = 6; top panel), HSA1 inversion breakpoints (n = 4 and 7, respectively, for BP1 and BP2; center panels), and HSA18 inversion breakpoint 1 (n = 3; bottom panel). The human-specific H3K4me3 peaks indicated by black vertical ticks and the position of the break- and fusion points marked in red are shown together with the genes mapping within these regions (blue).
HSA19 is the human chromosome with the highest gene density (Grimwood et al. 2004) and the highest density of H3K4me3-specific peaks (fourfold enrichment when compared with the average results of autosomes) (Fig. 1). Although it has no large-scale structural differences when compared with its chimpanzee and macaque homologs (The Chimpanzee Sequencing and Analysis Consortium 2005; Rhesus Macaque Genome Sequencing and Analysis Consortium 2007), HSA19 is the autosome with the highest human–chimpanzee sequence divergence besides HSA21 (The Chimpanzee Sequencing and Analysis Consortium 2005) and one of the chromosomes with the highest segmental duplication density (Bailey et al. 2001; Bailey et al. 2002).
We observed similar results for the 33 H3K4me3 human-enriched regions selectively methylated in neuronal versus nonneuronal chromatin (Shulha et al. 2012) as 10 (fivefold enrichment) map to subtelomeric regions, two map to the ancestral fusion locus of HSA2 (see above), and two are at the ancestral centromere locus of the same chromosome (Fig. 1; Supplemental Table S1). Thus 51% of the methylated regions (17 out of 33) overlap at least one of the features. Conversely, the 61 human-specific prefrontal cortex H3K4me3-depleted sites show no enrichment at these regions (Fig. 1; Supplemental Table S2).
We evaluated whether enrichments of human-specific H3K4me3 sites at genome structures unique to humans were specific to the prefrontal cortex or also present in other tissues. We used H3K4me3 ChIP-seq data of human, chimpanzee, and macaque LCLs (Cain et al. 2011) to identify autosomal human specifically enriched or depleted peaks. We found significant fourfold enrichments of H3K4me3-enriched peaks at subtelomeric regions in LCLs (density P-value = 2 × 10−23, permutation P-value = 0.001; fraction P-value = 4 × 10−19, permutation P-value = 0.001), together with enrichment trends of density (sixfold) and fraction (fourfold) at the HSA2 fusion point and threefold enrichment at 16p11.2–12.2 (Fig. 4; Table 2; Supplemental Table S3). These results show that the enrichment of human-specific H3K4me3 sites at subtelomeric regions is not unique to brain structures. Similar to prefrontal cortex, the 109 autosomal regions with human-specific depletion of H3K4me3 marks in LCLs showed no enrichment at these regions (Supplemental Table S4).
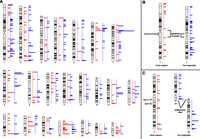
(A) Human karyotype-wide mapping of the regions with human-specific (n = 164, red) and chimpanzee-specific (n = 224, blue) enrichment of H3K4me3 modifications in LCLs. Chimpanzee-specific regions in panel A are positioned on the human karyotype; however, some chromosomes are structurally different between these species. For example, chromosome 1 differs because of a pericentric inversion and heterochromatin content (B); chromosome 2 differs because of a chromosomal fusion in human and the presence of subtelomeric heterochromatic caps (C).
Summary of density and fraction enrichments of human and chimpanzee prefrontal cortex (PC) and LCLs in enriched H3K4me3-marked sites
Next we assessed if concentrations of lineage-specific H3K4me3 sites at lineage-specific genome structures were unique to humans. Toward this goal we determined the chimpanzee-specific H3K4me3 peaks in LCLs (n = 224 enriched and n = 36 depleted) and used the published chimpanzee and nonhuman peaks of the prefrontal cortex (n = 523 enriched and n = 327 depleted) (Shulha et al. 2012). We identified significant accumulation at subtelomeric regions of H3K4me3 sites that are chimpanzee-enriched (fourfold enrichment) or chimpanzee-depleted (twofold) in the prefrontal cortex and of chimpanzee-enriched H3K4me3 sites in LCLs (twofold) (Fig. 4; Table 2; Supplemental Tables S5–S8). For example, the segment orthologous to the fusion point of chromosome 2, which has a subtelomeric location in the chimpanzee genome (Fig. 4C), showed a fivefold enrichment of density and fraction in the prefrontal cortex. The situation is less clear pericentromerically and at species’ inversion breakpoints, because we detected no enrichment at these sites in chimpanzee (Fig. 4B; Table 2; Supplemental Tables S5, S6).
As we observe a general increase of species-specific H3K4me3 peaks in segmental duplications, we then asked whether these observed accumulations at structurally different regions were possibly an indirect effect of their high duplication content. Alternatively, we considered that part of the signal might arise from the erroneous inclusion of false positive species-specific peaks due to poor annotation of multicopy sequences in some species/regions (Pickrell et al. 2011). To this end, we repeated our enrichment analyses in the prefrontal cortex excluding duplicated regions (Table 1; Supplemental Tables S1, S2, S5–S7). Barring all duplicated regions of the genome did not abrogate the reported enrichment at subtelomeric regions for the two species (human and chimpanzee) studied and at the HSA2 fusion point. Conversely, removing these regions abolished the enrichment of human-specific prefrontal cortex H3K4me3 sites at the HSA2 ancestral centromere locus.
To gain insights into the possible cause(s) of these enrichments, we then studied the features of both the affected genomic regions and the human-specific peaks they encompass after ensuring first that the observed increased densities could not be explained simply by the wide heterogeneity of the genome. We found that the distributions of the lineage-specific marks were not random (P-values ≤ 4 × 10−6), except the human depleted one (P-value = 0.4) (Supplemental Table S9; Methods). In the genomic regions we investigated the concentration of repetitive elements, genes, protein-coding genes, and recombination hotspots (Table 3). The latter were associated with testis-specific trimethylation of H3K4 in mouse (Smagulova et al. 2011), while repetitive elements were shown to create novel regulatory elements (Feschotte 2008). We observed no correlation between repetitive elements and the increases of H3K4me3 marks. Similarly, gene density alone cannot explain the totality of the observed enrichments of species-specific H3K4me3 peaks, as its increase compared with genome average is narrower than that of the chromatin marks. For example, while subtelomeric regions display a 1.61-fold inflated gene density (twofold if considering coding genes), the corresponding increase of H3K4me3 lineage-specific peaks varies from two- to fivefold depending on tissue and species. As subtelomeric regions are both gene-richer and the cradle of an abundance of recombination hotspots, it is possible that these two characteristics together with the higher divergence of these portion vis-a-vis the chimpanzee genome explain the observed enrichments. Additionally, we computed the content in repetitive elements, assessed sequence similarity with the chimpanzee and overlap with mapped recombination hotspots of human-enriched peaks at subtelomeric regions, HSA2 fusion point and HSA1 and HSA18 inversion breakpoints (the latter in prefrontal cortex only), and compared them with those of the complete set of H3K4me3 human-enriched peaks. We found that the peaks at subtelomeric regions and rearrangement break- and fusion points presented greater sequence divergence (measured both through identity percentage and aligned fraction) (Fig. 5). As these concomitantly show no enrichment/depletion in their repetitive element content, it suggests that the emergence of these lineage-specific peaks might be favored by sequence changes but that the latter are not due to lineage-specific inclusion of repeated elements. The higher percentage of specific peaks overlapping recombination hotspots at subtelomeric regions (∼1.7× higher than the genome average) is correlated to the higher density of recombination hotspots in these portions (1.6× higher) (Table 3), confirming that, similarly to gene density, recombination hotspots do not explain the observed enrichments of specific peaks at these segments entirely (Table 4).
Features of the genomic regions analyzed
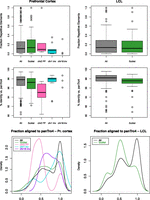
Human-enriched H3K4me3 peaks in prefrontal cortex (left panels) and LCLs (right panels). Fraction of repetitive elements (top panels), percentage of identity (center panels), and fraction aligned to the chimpanzee genome (panTro4) (bottom panels) of all human enriched peaks and peaks mapping to subtelomeric regions, HSA2 fusion points, HSA1 inversion breakpoints, and HSA18 inversion breakpoints (the rearranged points for prefrontal cortex only).
Number and percentage of H3K4me3 human-enriched peaks in PC and LCLs mapped to a recombination hotspot
Shulha and colleagues found that four of the 33 neuronal peaks (12%) were associated with novel RNA expression specific to human prefrontal cortex and 18 of the 410 human-enriched peaks (4%) with different RNA levels (Shulha et al. 2012). We similarly assessed how many human/chimpanzee enriched/depleted H3K4me3 LCL peaks corresponded to changes in expression of embedded exons. We uncovered an overlap between such exons and lineage-specific peaks ranging from 13% to 26% (Supplemental Table S10), suggesting that a noteworthy fraction of the human–chimpanzee variation in H3K4me3 peaks has an effect on RNA expression.
Discussion
We analyzed the location of human-specific and chimpanzee-specific H3K4me3 histone modification marks—a proxy for promoters/TSSs—in prefrontal neurons (Shulha et al. 2012) to assess if particular chromosomal portions and lineage-specific chromosomal rearrangements provide fertile ground for new regulatory elements. Our analyses stemmed from the observation that the DPP10 locus, featured in Shulha et al. (2012) because it is characterized by both human-specific and neuronal-specific epigenetic marks in the prefrontal cortex, maps 1 Mbp away from the HSA2 fusion site. Besides gauging enrichment of human specifically enriched marks at the HSA2 fusion point, we also evaluated the number of human-specific H3K4me3-marked sites at other human lineage-specific genomic structures such as the HSA2 ancestral centromere locus as well as the HSA1 and HSA18 pericentric inversion breakpoints. Additionally, we considered segments of the human genome known to be structurally different between apes and humans, like subtelomeric and pericentromeric intervals as well as segmental duplications.
Our results suggest that a significant fraction of the newly acquired human topological domains characterized by lineage-specific epigenetic decorations in the prefrontal cortex (Shulha et al. 2012) overlaps with domains having new cytogenetic architecture generated by evolutionary chromosomal rearrangements or with rapidly evolving sites like subtelomeric and pericentromeric regions and segmental duplications. For example, the majority of regions selectively methylated in neurons identified in Shulha et al. (2012) map to highly plastic chromosome regions and/or with a known human-specific organization (Fig. 1; Supplemental Table S1).
To understand whether this propensity exists in other tissue and species as well, we analyzed another cell type, LCLs, and another species, the chimpanzee. We found a consistent and general increase of diversity and novelty in H3K4me3 epigenetic marks at subtelomeric regions in both human and chimpanzee and both prefrontal cortex and LCLs. In human prefrontal cortex, both the density and fraction of specifically enriched H3K4me3 sites sharply increase as we approach the chromosomal ends (Fig. 2C,D), suggesting that the conjunction of an increased divergence between the human and chimpanzee genome sequences, as we progress toward the telomere (The Chimpanzee Sequencing and Analysis Consortium 2005) and distinctive contents in recombination hotspots, heterochromatin, and duplicated regions at these sites, supports novelty in regulatory elements. The novel human epigenetic marks at the chromosome 2 fusion point might also reflect its ancestral subtelomeric location. Of note, increased epigenetic diversity at subtelomeric regions was observed in two recent comparisons: the 5-hydroxymethylcytosine epigenetic mark between induced pluripotent stem cells (iPSCs) and embryonic stem cells (ESCs) in human (Wang et al. 2013) and higher order chromatin structure between human and mouse (Chambers et al. 2013). Moreover, an example of the emergence of novel promoters and expression modules through segmental duplications was described for the human core duplicon LRRC37 (Bekpen et al. 2012; Giannuzzi et al. 2013).
The loss of enrichments at pericentromeric regions, and HSA2 ancestral centromere locus when excluding human prefrontal cortex peaks mapped to duplications, suggests that the higher concentration of species-specific H3K4me3 peaks at these sites may derive from the high content in duplicated sequences rather than their cytogenetic localization. Similarly, the lack of enrichment of chimpanzee-specific peaks at chimpanzee inversion breakpoints suggests that the human concentration at these sites may not be associated with the structural change per se but rather with their subtelomeric/pericentromeric localization or presence of duplicated blocks. Of note, the correct identification of H3K4me3 peaks in duplicated regions might require separate efforts due to the misannotation of recent and highly similar segmental duplications in genomes.
While the enrichment at subtelomeric regions was consistently seen across species and tissues, at pericentromeric sites enrichment was only detected in the human prefrontal cortex, not in human LCLs or in the chimpanzee. Is this human-specific enrichment in cortex truly singular or a mere reflection of the lower quality achieved by the chimpanzee compared with the human genome, especially within pericentromeric regions? Further studies are warranted to confirm/refute these differences.
Our results support the existence of an evolutionary role for chromosomal rearrangement loci and subtelomeric regions. These segments of the genome harbor new sequences, which arose both from increased divergence and species-specific organization, and/or more recombination events. The convergence of these features possibly allows chromatin reconfiguration and thus the appearance of novel H3K4me3 sites, partly associated with modification in gene expression. Our findings suggest that evolutionary novelties and neighboring sequences should be investigated not only for gene expression differences and as fertile ground for the emergence of novel genes and transcripts, but also in the quest for lineage-specific epigenetic and regulatory changes. Our results also indicate how the duplicated regions that border copy number variants could play a role in the modification of the expression of normal copy number flanking genes (Merla et al. 2006; Reymond et al. 2007; Henrichsen et al. 2009a,b), an effect that can extend over the entire length of the affected chromosome (Ricard et al. 2010).
Methods
Genomic regions
Coordinates refer to the human reference sequence hg19/GRCh37. Subtelomeric regions were defined as the first and the last 4 Mbp of chromosome sequence (for acrocentric chromosomes only the last 4 Mbp were considered). Similarly, pericentromeric regions were the first 4 Mbp on either side (p and q chromosomal arms) of the centromere and, when present, the heterochromatin gaps, and the first 4 Mbp on the q-side for acrocentric chromosomes. Coordinates of human-specific and chimpanzee-specific (i.e., present only in human and chimpanzee, respectively) segmental duplications (both fixed duplications and expansions) (Sudmant et al. 2013) were converted from the hg18 to the hg19 release using the liftOver tool with default parameters. We retrieved coordinates of HSA1 inversion breakpoints from Szamalek et al. (2006); coordinates of HSA18, PTR4 (Pan troglodytes), PTR5, PTR12, PTR16, and PTR17 inversion breakpoints from the UCSC Genome Browser (Chiaromonte et al. 2002; Kent et al. 2002, 2003; Schwartz et al. 2003); coordinates of PTR9 inversion breakpoint on HSA9q21 from Kehrer-Sawatzki et al. (2005c); coordinates of PTR15 inversion breakpoint on HSA15q13 from Locke et al. (2003). A window spanning 1 Mbp upstream of and 1 Mbp downstream from the breakpoints was considered. We note that some of these features overlap: (1) HSA1 p-arm inversion breakpoint overlaps with its pericentromeric region; (2) HSA18 p-arm inversion breakpoint overlaps with its subtelomeric region; (3) HSA16 pericentromeric region overlaps with 16p11.2–12.2 cytogenetic bands; (4) human-specific segmental duplications overlap with all other features, i.e., human inversion breakpoints, HSA2 fusion point and ancestral centromere loci, 16p11.2–12.2, and subtelomeric and pericentromeric regions. Sequence gaps were excluded in all calculations. Chromosome Y and unplaced contigs were excluded from all analyses. The same regions with the exclusion of human and chimpanzee segmental duplications and chromosome X were considered as well. In the computation of enrichments for the chimpanzee, we redefined the coordinates of subtelomeric and pericentromeric regions considering the structural differences between human and chimpanzee chromosomes, i.e., chromosomal fusion and pericentric inversions.
Locations of recombination hotspots from Phase II HapMap data (release 21) (McVean et al. 2004; The International HapMap Consortium 2005; Winckler et al. 2005) were converted from hg17 to hg19 using liftOver (minMatch = 0.9). Repeat annotation for the GRCh37/hg19 human genome release was downloaded from the UCSC Genome Browser. Gene annotation refers to Ensembl v74 (Flicek et al. 2014).
H3K4me3 species-specific peaks
We obtained coordinates of 410 and 61 H3K4me3 peaks with human-specific enrichment/depletion, respectively, in prefrontal cortex, 33 human-specific neuronal H3K4me3 peaks, and 551 and 337 H3K4me3 peaks with chimpanzee-specific enrichment/depletion, respectively, in prefrontal cortex from Shulha et al. (2012).
We identified autosomal H3K4me3 peaks in LCLs using ChIP-seq data from Cain et al. (2011). We mapped human, chimpanzee, and macaque reads to the human genome (GRCh37/hg19) using Bowtie (version 0.12.9) (Langmead et al. 2009) and called human and chimpanzee peaks using MACS (Zhang et al. 2008). We filtered peaks with FDR < 0.1, >500 bp, mapped on autosomes, and not overlapping gaps or human/chimpanzee duplications (Sudmant et al. 2013). We defined enriched regions 500 bp around the peak summit as suggested in Bardet et al. (2012) and merged the human- and chimpanzee-enriched regions. We extended mapped reads of 100 bp and counted the coverage for the enriched regions. We identified the regions with FDR < 0.01 and at least twofold enriched or depleted in human and chimpanzee using limma (Law et al. 2014).
The coordinates of chimpanzee-specific (enriched and depleted) regions in the prefrontal cortex (Supplemental Tables S7, S8 of Shulha et al. 2012) were converted from panTro2 to GRCh37/hg19 using the liftOver tool (minMatch = 0.7). We manually checked through BLAT regions with a size difference >20% of the original size in the chimpanzee genome. This procedure allowed the conversion of 523 out of 551 and 327 out of 337 prefrontal cortex chimpanzee-enriched/depleted peaks.
We computed the intersection between peaks and genomic regions, recombination hotspots, repetitive elements, and genes using BEDTools (Quinlan and Hall 2010). To estimate the divergence of human peaks versus chimpanzee, we aligned the peak sequences to the chimpanzee genome (panTro4) using BLAT with default parameters. For each peak, we considered the best alignment and analyzed the percent identity and ratio between the alignment length and the size of the peak sequence. Boxplots and density curves were drawn using the R package (R Development Core Team 2014).
RNA-seq data analysis
We aligned LCL RNA-seq reads from Cain et al. (2011) to the human genome (hg19) using TopHat2 (Kim et al. 2013), assembled transcripts using Cufflinks (Trapnell et al. 2010), and joined human and chimpanzee transcripts using Cuffmerge. We counted reads in the transcripts using two python scripts from Anders et al. (2012) and identified differentially expressed exons (FDR < 0.01 and at least twofold change) among those that overlap with H3K4me3 marked regions using limma (Law et al. 2014).
Statistical analysis
We assessed the statistical significance of the density and fraction enrichments using the one-way χ2 and Fisher’s exact tests, respectively, and permutation tests (Davison and Hinkley 1997) by replacing peaks across the genome using shuffleBed 1000 times (Quinlan and Hall 2010). We adjusted P-values for multiple comparisons using the Bonferroni correction method. Permutation P-values were calculated by P-value = (E + 1)/(R + 1), where R is the number of permutations, equal to 1000, and E is the number of permutation test statistics that are greater than or equal to the observed test statistic.
The same enrichments were computed twice, including and excluding species-specific peaks mapped to duplicated sequences and chromosome X. To this end, human and chimpanzee duplication data (both fixed duplications and expansions) (Sudmant et al. 2013) were converted from the hg18 to the hg19 release using the liftOver tool (-minMatch = 0.5 -minBlocks = 0.5).
We divided the human genome, excluding centromeric gaps, into 675 4-Mb segments and predicted the expected number of segments with different counts of marks using the Poisson distribution. We compared these predicted values with the observed ones (H3K4me3 human/chimpanzee-enriched/depleted peaks in prefrontal cortex and the 33 neuronal peaks) using a G-test and found that the human-enriched, chimpanzee-enriched, and chimpanzee-depleted peaks were not randomly distributed. We also performed permutation tests by replacing subtelomeric, pericentromeric, HSA2 fusion point, HSA2 ancestral centromere, HSA1 and HSA18 inversion breakpoint regions as well as chimpanzee inversion breakpoint regions across the genome using shuffleBed 1000 times (Quinlan and Hall 2010). The results of both approaches similarly showed that the lineage-specific peaks—except the human-depleted ones—were not randomly distributed along the genome.
Acknowledgments
We thank Maria Nicla Loviglio for discussions. This work was supported by the Swiss National Science Foundation (SNSF) and an SNSF Sinergia grant to A.R. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The computations were performed at the Vital-IT (http://www.vital-it.ch) Center for high-performance computing of the SIB Swiss Institute of Bioinformatics.
Author contributions: G.G. designed the study and conducted the analyses. G.G. and E.M. performed the statistical tests. G.G. and A.R. wrote the manuscript. All authors read and approved the final manuscript.
Footnotes
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.167742.113.
- Received October 1, 2013.
- Accepted June 9, 2014.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








