
Discovery of recurrent structural variants in nasopharyngeal carcinoma
- Anton Valouev1,5,6,
- Ziming Weng2,3,5,
- Robert T. Sweeney2,
- Sushama Varma2,
- Quynh-Thu Le4,
- Christina Kong2,
- Arend Sidow2,3,6 and
- Robert B. West2,6
- 1Division of Bioinformatics, Department of Preventive Medicine, University of Southern California Keck School of Medicine, Los Angeles, California 90087, USA;
- 2Department of Pathology, Stanford University School of Medicine, Stanford, California 94305, USA;
- 3Department of Genetics, Stanford University School of Medicine, Stanford, California 94305, USA;
- 4Department of Radiation Oncology, Stanford University School of Medicine, Stanford, California 94305, USA
-
↵5 These authors contributed equally to this work.
Abstract
We present the discovery of genes recurrently involved in structural variation in nasopharyngeal carcinoma (NPC) and the identification of a novel type of somatic structural variant. We identified the variants with high complexity mate-pair libraries and a novel computational algorithm specifically designed for tumor-normal comparisons, SMASH. SMASH combines signals from split reads and mate-pair discordance to detect somatic structural variants. We demonstrate a >90% validation rate and a breakpoint reconstruction accuracy of 3 bp by Sanger sequencing. Our approach identified three in-frame gene fusions (YAP1-MAML2, PTPLB-RSRC1, and SP3-PTK2) that had strong levels of expression in corresponding NPC tissues. We found two cases of a novel type of structural variant, which we call “coupled inversion,” one of which produced the YAP1-MAML2 fusion. To investigate whether the identified fusion genes are recurrent, we performed fluorescent in situ hybridization (FISH) to screen 196 independent NPC cases. We observed recurrent rearrangements of MAML2 (three cases), PTK2 (six cases), and SP3 (two cases), corresponding to a combined rate of structural variation recurrence of 6% among tested NPC tissues.
Nasopharyngeal carcinoma (NPC) is a malignant neoplasm of the head and neck originating in the epithelial lining of the nasopharynx. It has a high incidence among the native people of the American Arctic and Greenland and in southern Asia (Yu and Yuan 2002). NPC is strongly linked to consumption of Cantonese salted fish (Ning et al. 1990) and infection with Epstein-Barr virus (EBV) (Raab-Traub 2002), which is almost invariably present within the cancer cells and is thought to promote oncogenic transformation (zur Hausen et al. 1970). A challenging feature of NPC genome sequencing is that significant lymphocyte infiltration (e.g., 80% of cells in a sample) (Jayasurya et al. 2000) is common, requiring special laboratory and bioinformatic approaches not necessary for higher-purity tumors (Mardis et al. 2009).
Currently, cancer genomes are analyzed by reading short sequences (usually 100 bases) from the ends of library DNA inserts 200–500 bp in length (Meyerson et al. 2010). For technical reasons, it is difficult to obtain deep genome coverage with inserts exceeding 600 bp using this approach. Much larger inserts can be produced by circularizing large DNA fragments of up to 10 kb (Fullwood et al. 2009; Hillmer et al. 2011), and subsequent isolation of a short fragment that contains both ends (mate pairs). Large-insert and fosmid mate-pair libraries offer several attractive features that make them well-suited for analysis of structural variation (Raphael et al. 2003; International Human Genome Sequencing Consortium 2004; Tuzun et al. 2005; Kidd et al. 2008; Hampton et al. 2011; Williams et al. 2012). First, mate pairs inherently capture genomic structure in that discordantly aligning mate-pair reads occur at sites of genomic rearrangements, exposing underlying lesions (Supplemental Fig. S1a). Second, large-insert mate-pair libraries deliver deep physical coverage of the genome (100–1000×), reliably revealing somatic structural variants even in specimens with low tumor content (Supplemental Fig. S1a,b). Third, variant-supporting mate-pair reads from large inserts may align up to several kilobases away from a breakpoint, beyond the repeats that often catalyze structural variants (Supplemental Fig. S1c). For these reasons, large insert mate-pair libraries have been used extensively for de novo assembly of genomes and for identification of inherited structural variants (International Human Genome Sequencing Consortium 2001). In principle, they should also be well-suited for analysis of “difficult” low-tumor purity cancer tissues such as NPCs. In a recent study, paired-end fosmid sequencing libraries with nearly 40 kb inserts were adapted to Illumina sequencing and applied to the K562 cell line (Williams et al. 2012). Due to low library complexity, of 33.9 million sequenced read pairs, only about 7 million were unique (corresponding to about 0.5× genome sequence coverage), but nonetheless facilitated structural variation detection.
Mate-pair techniques have not yet been applied to produce truly deep sequencing data sets of tumor-normal samples (30× or greater sequence coverage), presumably due to the difficulty of retaining sufficient library complexity to support deep sequencing. We have improved the efficiency of library preparation by combining two existing protocols (Supplemental Fig. S2), and so were able to generate 3.5-kb insert libraries with sufficient genomic complexity to enable deep sequencing of two NPC genomes. To take full advantage of unique features offered by large-insert libraries, such as the large footprints of breakpoint-spanning inserts (Supplemental Fig. S1c) and the correlation between the two ends of alignment coordinates of breakpoint-spanning inserts, we also developed a novel somatic structural variant caller. SMASH (Somatic Mutation Analysis by Sequencing and Homology detection) is specifically designed to accurately map somatic structural lesions, including deletions, duplications, translocations, and large duplicative insertions via direct comparison of tumor and normal data sets.
Structural variation methods, such as GASV (Sindi et al. 2009), SegSeq (Chiang et al. 2009), DELLY (Rausch et al. 2012b), HYDRA (Quinlan et al. 2010), AGE (Abyzov and Gerstein 2011), and others (Lee et al. 2008; for review, see Snyder et al. 2010; Alkan et al. 2011), generally utilize (1) read-pair (RP) discordance, (2) increase or reduction in sequence coverage, (3) split reads that span breakpoints, and (4) exact assembly of breakpoint sequences. These tools were primarily designed for variant detection from a single data set, such as a normal genome, and are suited for cataloguing structural polymorphisms in the human population (Kidd et al. 2008, 2010; Mills et al. 2011). However, specific detection of somatic structural variants in cancer using these tools typically requires additional downstream custom analysis to enable “subtraction” of germline variants from the tumor variant calls (Rausch et al. 2012a). This limits the general utility of such tools for somatic variant detection. Recently, as an increasing number of studies specifically focused on somatic mutations, dedicated somatic variant callers such as CREST (Wang et al. 2011) have been developed. CREST relies on detection of partially aligned reads (known as “soft clipping”) across a breakpoint.
SMASH adopts a hybrid approach to somatic variant detection. It relies on read-pair discordance to discover somatic breakpoints and then uses split reads to refine their coordinates. Furthermore, SMASH incorporates a number of important quality measures and filters, which are critical for minimizing the rate of false positive somatic SV calls (Supplemental Material). As we demonstrate here with both simulated and real sequence data, such a hybrid approach delivers high sensitivity of somatic SV detection due to the read-pair discordance, high accuracy of breakpoint coordinates enabled by split reads, and overall low false discovery rate due to extensive use of quality measures.
Results
We obtained fresh-frozen NPC tissue and matched normal blood from two independent NPC cases. NPC-5989 is an untreated NPC tumor, and NPC-5421 is the first recurrence after chemotherapy and radiation treatment. Using our approach to building mate-pair libraries (Supplemental Fig. S2), we sequenced 3.5-kb mate-pair libraries for NPC-5989 on the Illumina sequencing platform, producing 887 million tumor read pairs (58× sequence coverage and 752× physical coverage) and 900 million matched-normal read pairs (58× sequence and 781× physical coverage). For NPC-5421, we utilized an early SOLiD mate pair protocol and obtained 433 million read pairs (365× physical coverage) using 3.5-kb and 1.5-kb insert libraries and the SOLiD sequencing platform, as well as 837 million read pairs (664× physical coverage) from the matched normal sample using 3.4- and 1.5-kb libraries. Rough estimates from histological sections indicated that the majority of cells in NPC-5989 were abnormal, but that tumor content in NPC-5421 was only ∼20%.
Consistent with the involvement of Epstein-Barr Virus (EBV) in NPC etiology and previous estimates of 1–30 viral genome copies per cell (Nanbo et al. 2007; Liu et al. 2011), we detected large amounts of EBV genomic sequence in NPC-5989 but not in its matched normal sample (1.86 million versus 521 read pairs). We estimate that there were about 108 EBV genome copies per cell in the tumor sample (Supplemental Material). We found that a significant portion of the EBV genome existed in a circular form (7736 read pairs supported circularization of the EBV genome spanning both ends of the genome at coordinates 169 kb and 0 kb). However, we could not identify any breakpoints joining human and EBV regions, indicating that the virus was likely not integrated into the host genome.
To systematically identify somatic rearrangements or other large lesions in the NPC tumor genomes, we applied SMASH to the mapped-read data. Somatic rearrangements produce breakpoints, which are comprised of two disjoint reference regions that are fused in the tumor genome but not in the normal genome. Tumor DNA inserts spanning the breakpoints result in discordantly mapping read pairs, which do not conform to the distribution of insert lengths in the rest of the sequencing library if the lesion involves a sufficiently large region. Some variants (deletions, insertions, tandem duplications) produce a single breakpoint (Supplemental Fig. S3a–c,e), whereas others (inversions, duplications, and balanced translocations) produce two or more breakpoints (Supplemental Fig. S3d,f,g).
SMASH detects somatic variants from SAM files (Li et al. 2009) using three main steps: breakpoint detection (Fig. 1A,B), elimination of germline breakpoints (Fig. 1C), and refinement of breakpoint coordinates using split reads (Fig. 1D). Initially, SMASH calculates the empirical distribution of tumor library insert sizes, which is used to find discordant tumor read pairs. Discordantly mapping reads include (1) paired-end reads that map to different chromosomes; (2) paired-end reads for which read orientation is inconsistent with the structure of the library; and (3) paired-end reads for which the distance between coordinates of paired ends deviates significantly from the expectation. We ran SMASH such that it flagged inserts as discordant when the inferred insert size was three or more standard deviations away from the empirical mean.
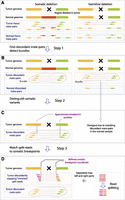
(A) SMASH workflow illustrated by comparing a hypothetical somatic structural variant (left portion of the figure) and a hypothetical germline structural variant (right portion of the figure). The region that is deleted is pointed out by a black cross. Arrows connected by dashed lines represent read pairs, in which reads of concordant pairs have the same color and those of discordant pairs have a different color. Different colors also represent different genomic regions that have been juxtaposed by a structural variant. (B) Step 1: SMASH eliminates concordant pairs and retains discordant pairs, and groups discordant read pairs from the tumor sample into bundles (contoured by gray lines) based on proximity of underlying read coordinates and consistency of orientations. (C) Step 2: Approximate coordinates of breakpoints are derived from read bundles; then each normal read pair is compared to tumor breakpoints and all breakpoints that have normal read pairs supporting them are eliminated. (D) Step 3: Sequencing reads are split and ends are mapped independently; discordant split read coordinates are used to further refine breakpoints.
The coordinates of discordant read pairs, such as those mapping to different chromosomes (Supplemental Fig. S3e), indicate potential breakpoints. Mate-pair libraries contain some ‘background’ discordant read pairs generated by circularization of two (rather than a single) genomic fragments into a single construct. Such discordant mate pairs are randomly distributed throughout the genome and not expected to cluster. In contrast, the discordant reads that result from rearrangements will form large clusters near the breakpoint positions. Thus, to minimize potential false positives resulting from presence of chimeric reads, SMASH groups discordant read pairs into discrete bundles representing grouped read pairs supporting a single breakpoint (Fig. 1B). A breakpoint is inferred to be somatic if no mate pairs support the same breakpoint in the normal sample. Based on the coordinates of bundle reads, the genomic coordinates of the somatic breakpoints (Fig. 1C) are estimated and finally refined using split reads (Fig. 1D). In this refinement step, the breakpoint is inferred from the terminal alignment positions of the discordantly mapping split reads, which pinpoint breakpoint positions with nearly base-pair level accuracy. As a final filter, SMASH performs sequence homology comparison of regions flanking the breakpoints using local alignment (Smith and Waterman 1981) and eliminates potential artifacts resulting from inaccurate read mapping due to sequence homology between the two genomic regions.
To ask whether SMASH performs correctly under idealized conditions, we applied it to 1 billion simulated tumor mate pairs and 1 billion simulated normal read pairs that were derived from a simulated genome with 35 simulated breakpoints, representing five types of structural variants (Supplemental Material). SMASH demonstrated good accuracy of inferred breakpoint coordinates without any false positive calls. Except for small insertions, SMASH consistently detected breakpoints with allelic frequencies >1.7%. To see if SMASH produces results comparable to other SV algorithms, we compared its breakpoint detection to HYDRA (Quinlan et al. 2010; Malhotra et al. 2013) on a set of simulated breakpoints (Supplemental Material; Supplemental Table T3). Overall, SMASH breakpoint detection rates were similar to HYDRA, with SMASH more accurately resolving breakpoint coordinates and HYDRA more consistently detecting short insertions. In a separate simulation, we detected breakpoints previously reported as a part of human structural variant resource (Kidd et al. 2010; Supplemental Material), demonstrating good performance of SMASH on real-world SVs; here, breakpoint detection was limited by the ability to map variant reads into nonunique regions of the genome. In general, homology of sequences in the immediate neighborhood of breakpoints represents a major challenge for accurate SV detection by variant callers that rely on split reads and read-pair discordance.
In NPC-5989, SMASH identified a total of 10 somatic breakpoints (Supplemental Table T1) representing six structural variants: one deletion of 25 kb (one breakpoint), one deletion of 590 kb (one breakpoint), one 13 kb tandem duplication (one breakpoint), one 95 Mb/18 Mb duplicative translocation (one breakpoint), and two “coupled inversions” 5.6 and 6.1 megabases in size (six breakpoints, see below).
The largest variant was a duplicative translocation (Fig. 2A), which moved the telomeric 95 Mb of chromosome 1q to the telomeric end of chromosome 8q, resulting in a concomitant loss of 18 Mb of chromosome 8q. Consistent with this event, we saw a 28% gain of sequence coverage on the telomeric portion of chromosome 1q and a 32% reduction of sequence coverage on the telomeric portion chromosome 8q. Based on these coverage changes, we estimated that 56%–64% of cells in the sample carried this duplicative translocation (Supplemental Material). To provide alternative validation of the observed copy number variants in NPC-5989, we performed array CGH (aCGH) analysis on the same sample (Fig. 2B,C). The duplication of 1q was detected by probes spanning coordinates 154.1–249.2 Mb (Supplemental Table T2). aCGH analysis also successfully detected loss of the telomeric end of Chr8 starting at the coordinate 128.8 Mbp, as well as a 596 Kbp copy number “neutral” region within the amplified telomeric end of Chr1 (starting at 155.1 Mb), which was due to a SMASH-reported deletion breakpoint. Thus, some of our calls were confirmed by aCGH; but as expected, our sequence-based analysis found more structural variants than aCGH.
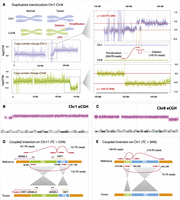
Examples of structural variants detected within NPC-5989. (A) Schematic representation of chromosomal rearrangements affecting Chr1 (blue) and Chr8 (green). Coverage plots demonstrate an increase in copy number of Chr1q and a reduction of copy number of Chr8q. Each data point represents the log ratio of tumor read counts to normal read counts in 5-kb bins across the chromosome. Red lines represent mean coverage corresponding to regions with different copy numbers. Based on the ratio levels, we estimate that tumor content is 56% (based on amplification of Chr1q) and 64% (based on deletion of Chr8q). The magnified regions contain two breakpoints representing duplicative translocation and deletion (shown as linked red arrows), supported by 204 and 145 read pairs, respectively. Coordinates of breakpoints match closely with positions of copy number changes both on Chr1 and Chr8, supporting the same rearrangement event. Deletion breakpoint coordinates also match coordinates of the region on Chr1 where coverage visibly drops. The duplicative translocation results in a region of LOH 18 Mb in size at the end of Chr8, and in the amplification of much of Chr1q. (B,C) Results of the array CGH analysis of NPC-5989 on Chr1 and Chr8. The x-axis represents genomic coordinates, whereas the y-axis represents probe saturation which is converted into copy number calls. (D) Coupled inversion involving 6-Mb and 0.1-Mb regions on Chr11, which results in the YAP1-MAML2 gene fusion product. The coupled inversion is represented by three breakpoints (linked red arrows). (E) Coupled inversion involving 0.7-Mb and 4.9-Mb regions on Chr1.
Six of the detected somatic breakpoints represented two distinct copy-number neutral structural variants of a novel type, which we call “coupled inversion” (Fig. 2D,E; Supplemental Table T1). These variants are characterized by two outer breakpoints, an inner breakpoint, and two in situ inverted fragments bordering a central breakpoint. A coupled inversion on Chr11 (Fig. 2D) inverted a 6.0-Mb and a 0.1-Mb fragment, disrupting the MAML2 and YAP1 genes and producing an in-frame YAP1-MAML2 gene fusion. The two outer breakpoints were supported by 141 and 157 mate pairs, and the inner breakpoint was supported by 155 mate pairs. A coupled inversion on Chr1 was generated by inverting a 0.7-Mb and a 4.9-Mb fragment (Fig. 2E), supported by 199 and 216 mate pairs for the outer breakpoints and 75 mate-pair reads for the inner breakpoint. By counting the number of discordant read pairs representing each breakpoint, we estimated allelic frequencies of the two coupled inversions to be 0.26 and 0.28, corresponding to an estimated tumor content of 53% and 56%. Because assessed clone frequencies exceed 50%, both the coupled inversions and the Chr1-Chr8 duplicative translocations are likely present in the same tumor clone, representing ∼54% of the cells in the tumor sample. Also present in the tumor clone is the loss of chromosome 16q, which we detected by coverage analysis. It was not detected by SMASH presumably because truncations do not generate breakpoints with discordant mappings.
For validation, we designed specific primers against all 10 breakpoints and performed PCR amplification. We observed specific PCR products in tumor but not in matched normal samples, confirming 100% of the NPC-5989 somatic structural variant calls (Fig. 3A). After Sanger sequencing of tumor-specific PCR products (see Supplemental Material), we determined precise locations of each breakpoint by aligning Sanger sequences using BLAST (Altschul et al. 1997), and were able to confirm all 10 breakpoint coordinates. We found that on average, mate-pair analysis had reported the breakpoints to within 43 bp of the actual position, and split reads improved the average accuracy to 2.5 bp (Supplemental Table T4).

Validation of somatic structural breakpoints by PCR (images were inverted for better presentation). (A) Agarose gel of PCR products amplified from genomic DNA, targeting breakpoints detected by SMASH in NPC-5989. Because the breakpoints are somatic, specific PCR bands only occur in the tumor sample (pointed out by red arrows). (T) tumor sample; (N) matched normal sample from blood; (W) no-DNA control; (L) 1 kb plus ladder. (B) Agarose gel of PCR products amplified by RT-PCR on tumor RNA corresponding to somatic gene fusions in NPC-5989 and NPC-5421. RT primers were ∼200 bp downstream from the fusion points. PCR primers were within 150 bp upstream of and downstream from the fusion points to specifically amplify across it. (T) tumor; (C) control sample (different tumor); (L) 1 kb plus ladder. Specific products within the expected size range are pointed out by red arrows.
We also analyzed these 10 breakpoints within an archival (FFPE) sample of the same NPC5989 tumor (Supplemental Fig. S4). Only 4 of 10 breakpoints were detectable within the FFPE sample, including all three breakpoints representing the coupled inversion that produced the fusion YAP1-MAML2 product, and a 25-kb deletion on Chr2 that affected two genes with unknown function (AK131224 and FLJ16124). The presence of all three breakpoints of one coupled inversion and absence of all three breakpoints of the second coupled inversion are consistent with these variants, representing two distinct structural events. These results also suggest that the FFPE sample represents an earlier state of the tumor, which did not yet acquire some of the structural variants that we observed in the sequenced sample. These observations are consistent with YAP1–MAML2 fusion representing an early-stage driver mutation.
The fusion site within the MAML2 gene (Fig. 4A) is identical to that reported previously in a CRTC1-MAML2 fusion in mucroepidermoid carcinoma, in which exons 2–5 (aa 172–1156) were fused to exon1 of CRTC1 (previously MECT1) (Tonon et al. 2003). The YAP1-MAML2 fusion of NPC5989 contains sequences encoding both the TEAD1-interaction domain and the transactivation domain of MAML2, as well as a partial WW1 domain potentially involved in protein–protein interaction (Fig. 4B).
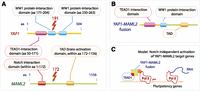
YAP1-MAML2 gene fusion protein domains. (A) Structural domains of the YAP1 gene include the TEAD1-interaction domain (amino acids 50–171) and two WW1 protein interaction domains with unknown function. MAML2 contains a Notch-interaction domain (somewhere within amino acids 1–172), which is responsible for MAML2 transactivation function in the presence of Notch signaling. Transactivation domain of MAML2 is located somewhere within amino acids 172–1156. The two genes are fused at the amino acid 191 of YAP1 gene and 172 of MAML2 gene. The resulting gene (B) contains the TEAD1-interaction domain and a truncated WW1 domain from YAP1 and the transactivation domain from MAML2. (C) Under the proposed model, the fusion protein is recruited via TEAD1 binding to target genes of TEAD1, many of which are important in embryonic stem cells (ESCs). Because the MAML2 Notch interaction domain is absent, transactivation of ESC target genes may occur constitutively, possibly leading to dedifferentiation or proliferation.
Analysis of a second NPC case (NPC5421) revealed 35 candidate somatic structural breakpoints (Supplemental Table T1), but most had weak support likely because of the low tumor content of the sample. The greatest number of mate pairs supporting a single breakpoint was 22, corresponding to an estimated allele frequency of 6%. This may indicate either a high degree of sample heterogeneity or a single low-frequency tumor clone with a highly rearranged genome. Nonetheless, the structural variants predicted three candidate in-frame gene fusions involving PTPLB-RSRC1, SP3-PTK2, and GLYAT-NLRC5, only one of which did not validate (see below). This highlights that even in low-tumor content samples, mate-pair libraries coupled with SMASH analysis allows specific detection of somatic structural variants.
To investigate whether the detected gene fusions are expressed, we carried out RT-PCR on four in-frame gene fusions (YAP1-MAML2, PTPLB-RSRC1, SP3-PTK2, GLYAT-NLRC5) and one out-of-frame fusion (ACTN4-FBX017). PCR primers were placed into the exons of the fusion pair genes, flanking the breakpoint positions (see Supplemental Material). Because no RNA was available from the matched blood, we used the two NPC RNA samples to control each other. Using this design, PCR products were expected to specifically amplify the fused portion of the transcript. Our RT-PCR analysis detected the presence of four of five fusion products (Fig. 3B) in the correct NPC samples, demonstrating that the four fusion gene pairs (YAP1-MAML2, PTPLB-RSRC1, SP3-PTK2, and ACTN4-FBX017) are specifically expressed in the corresponding NPC tissues. One of the fusions from the low-tumor content NPC-5421 (GLYAT-NLRC5) did not validate by RT-PCR or breakpoint PCR, and therefore likely represents a false positive breakpoint call. Sanger sequencing of the specific RT-PCR bands of the four fusion genes demonstrated that the fusion sequences, as expected, contained the N-terminal portion of one gene and the C-terminal portion of the other, with the two exons forming the junction. Surprisingly, we detected weak expression of the out-of-frame ACTN4-FBX017 fusion, indicating either that nonsense-mediated decay of this message is not fully efficient, or that an undetected mechanism such as alternative splicing resulted in an in-frame gene product.
Finally, we investigated whether any of the six genes involved in in-frame fusions (YAP1, MAML2, PTPLB, RSRC1, SP3, and PTK2) were also rearranged in other NPC cases. We compiled an NPC tissue microarray (Battifora 1986) using 196 independent samples and performed fluorescent in situ hybridization (FISH). We observed that of six tested genes, three were indeed recurrently rearranged: MAML2, which underwent rearrangements in three cases (Fig. 5A); PTK2, six cases (Fig 5B); and SP3, two cases (Fig. 5C). Due to the limited resolution of FISH probes and fairly large probe spacing (0.2–0.4 Mb), it is generally difficult to assess consistency of the precise breakpoints across the different NPC samples.

Recurrent rearrangements detected by fluorescent in situ hybridization of tissue microarrays. (A) Rearrangement of MAML2 in case NPC-22525. Probes were selected to flank centromeric (green probe) and telomeric (red probe) ends of the MAML2 gene. FISH images show cells with abnormal alleles. Approximate tumor cell boundaries are outlined with yellow dashed lines. Non-rearranged MAML2 alleles show colocalization of green and red probes (yellow arrows). Isolated green (or red) probes represent rearranged alleles and are marked by green (or red) arrows. (B) Two cases showing rearrangements of PTK2. (C) Two cases with a rearranged SP3 gene. NPC-22480 represents a core of a sequenced sample NPC-5421, which contains a rearranged SP3 allele, corresponding to the isolated centromeric probe (green).
Discussion
This first characterization of NPC by deep whole genome sequencing demonstrated effective structural variant discovery with our approach, which involved (1) generation of high-complexity, long-insert mate-pair libraries by a novel combination of methods; and (2) algorithm development and implementation of a new structural variant caller specifically for this type of data. Although previous studies have used large-insert libraries for low-level physical coverage (below 230×) of normal (Williams et al. 2012) and tumor genomes (Hillmer et al. 2011), our study achieves the deepest—to date—physical coverage (750×) as well as high sequence coverage (58×) with a single mate-pair library. The great degree of physical coverage afforded by long-insert mate-pair libraries facilitated our proof-of-concept demonstration that structural variants can be discovered even in very low-tumor content samples: In the case of NPC-5421, two of three gene fusion variants were inferred from the mate pair data to be present at only 6% allele frequency; both were validated by Sanger sequencing. Thus, although our study, like most other studies of genetic variation in unique samples, cannot estimate sensitivity of detection, we conclude that even very-low tumor content samples are amenable to genomic analysis. Library complexity could have supported even deeper sequencing than what we performed, so we speculate that more structural variants could have been discovered in NPC-5421, had the need existed, for example, in a clinical application of our approach.
The higher-tumor content case, NPC-5989, harbored two instances of a novel type of structural variant, which we term “coupled inversion.” It is possible that this particular tumor type is prone to such events, or that previous approaches did not discover them in other tumors because the methodology utilized did not support their discovery. Either way, the molecular mechanism leading to coupled inversions is puzzling, given that the three causative double-stranded breaks are repaired in a coordinated fashion, with neither of the eventually inverted two fragments getting lost in the process. It is conceivable that (1) the two outer breaks occur first, producing a fragment encompassing the entire eventually rearranged region; (2) this fragment is repaired into a circle; (3) the circle is then broken by and fused to one of the loose chromosome ends; and (4) the two loose chromosome ends, one of which contains the rearranged fragment, are joined to reconstruct a full chromosome. At least one coupled inversion on Chr11 is consistent with this model, because of the sequence homology between the outer breakpoint regions (Supplemental Material), but other scenarios are also possible.
Another intriguing aspect of these NPC cases was the gene fusions we discovered. Three of the four gene fusions were in frame, and all, even the out-of-frame fusion, were expressed in the tumor cells. We were able to confirm by tissue microarray analysis that three of the involved genes (MAML2, PTK2, and SP3), were recurrently rearranged in a small number of other NPC cases.
MAML2 is a member of the Mastermind gene family, whose members were shown to be involved in Notch signaling (Wu et al. 2000). MAML2 was previously found to participate in a fusion with CRTC1 in salivary mucoepidermoid carcinoma (Tonon et al. 2003) and Warthin's tumor of salivary glands (Martins et al. 2004), but its involvement in NPC has not been previously reported. MAML2 contains a C-terminal transactivation domain and an N-terminal Notch interaction domain that is lost in the YAP1-MAML2 fusion (Fig. 4A,B). Similarly to the CRTC1-MAML2 fusion (Tonon et al. 2003), YAP1-MAML2 may therefore be able to activate target genes in a Notch-independent manner (Wu et al. 2000). The target specificity may be provided by the YAP1 portion of the fusion (Fig. 4A), which contains a domain that interacts with the pluripotency transcription factor TEAD1 (Li et al. 2010). In particular, the YAP1-TEAD1 complex was demonstrated to bind promoters of many genes important for ES cells, which are also targets of polycomb group proteins, NANOG, POU5F1, and SOX2 (Lian et al. 2010). YAP1 is also critical to cellular reprogramming and promotes cellular de-differentiation. Therefore, one possible model for oncogenic function of YAP1-MAML2 fusion is that the fusion protein is recruited to TEAD1 target pluripotency genes and is able to activate gene expression in a Notch-independent manner resulting in cellular dedifferentiation and increased proliferation (Fig. 4C).
PTK2, a partner in the PTK2-SP3 fusion reported here, is also known as FAK. This gene has been extensively implicated in cancer-cell migration, survival, and metastasis (Frame et al. 2010). In NPC, the fibronectin extra domain (EDA) has been shown to correlate with tumor aggressiveness and radiation resistance via engagement of the FAK pathway (Ou et al. 2012). In EBV-mediated gastric carcinoma, EBV infection resulted in enhanced cell migration and invasion via increased FAK phosphorylation (Kassis et al. 2002). Lastly, although the transcription factor SP3 itself has not been implicated in NPC, one of its target genes, RASSF1, is a tumor suppressor and plays an important role in NPC development (Lee et al. 2009). Therefore MAML2, PTK2, and SP3 are candidate oncogenes that may act in a dominant fashion to help drive NPC tumor initiation or progression.
Methods
FISH analysis
PTK2, SP3, and MAML2 were tested on NPC tissue arrays with NPC tissue cores from 196 patients. Locus specific FISH analysis was performed using the following bacterial artificial chromosomes (BACs) from the Human BAC Library RPCI-11 (BACPAC Resources Center, Children's Hospital Oakland Research Institute) unless otherwise noted. YAP1: RP11-90M3, RP11-11N20; MAML2: RP11-77A22, RP11-12E16, RP11-13L14; PTPLB: RP11-816C20, RP11-295I13; RSRC1: RP11-1120M18, RP11-1012D12; SP3: RP11-262E6, RP11-627M13; and PTK2: RP11-1123G22, RP11-195E4. BACs were directly labeled with either Spectrum Green or Spectrum Orange (Vysis). The chromosomal locations of all BACs were validated using normal metaphases. Probe labeling and FISH was performed using Vysis reagents according to manufacturer's protocols (Vysis). Slides were counterstained with 4,6-diamidino 2-phenylindole (DAPI) for microscopy. For all slides, FISH signals and patterns were identified on a Zeiss Axioplan epifluorescent microscope. Signals were interpreted manually, and images were captured using Metasystems Isis FISH imaging software (MetaSystems Group, Inc.). A cutoff of equal to or greater than 20 breaks per 100 nuclei was selected for a positive score.
SMASH breakpoint analysis
All reads were aligned using Novocraft Novoalign and NovoalignCS mapping software on an IBM Smartcloud cluster (details in Supplemental Material). The following parameters were used with Novoalign: (-S 4000 -s 10 -p 7,10 0.4,2 -t 120). Reads with MAPQ scores with 90 or above were used to retain tumor sample reads, and reads with MAPQ scores with 30 or above were used to retain matched normal sample reads. Empirical insert distribution of mate pairs was used to filter out concordant tumor mate pairs that were within three standard deviations from the empirical mean and whose ends were on the same chromosome in consistent orientation. The resulting candidate discordant mate pairs were used to detect tumor breakpoints as described in the text. Tumor breakpoints were scanned against all normal mate pairs to eliminate breakpoints supported by at least one normal mate pair. The regions immediately adjacent to the breakpoint were analyzed for sequence similarity using the Smith-Waterman algorithm in order to identify potential false positive breakpoints. Specifically, we compared 3-kb regions immediately adjacent to the breakpoint and eliminated breakpoints with 70% identity over at least 700 bps. For the split-read analysis, reads were split into two equal-sized portions and aligned independently using Novoalign allowing for soft-clipping. The resulting pairs were filtered to identify discordant halves, which were compared against somatic breakpoints, and full read sequences were used for breakpoint refinement based on the soft-clipping boundaries.
Library construction and DNA sequencing
All samples were obtained under a Stanford IRB-approved protocol. Genomic DNA from tumor and normal samples was extracted using standard techniques (Qiagen kits 10223, 80224, 13362, 80204). Library construction was performed according to the SOLiD mate-pair library protocol with the following modifications: (1) The low efficient on-beads reaction was used for adapter ligation only; (2) the ligation efficiency was increased by using overhang T-A ligation; (3) Illumina PE adapters were used so that sequencing could be performed on the Illumina HiSeq platform to generate longer (100 bp) and more accurate sequencing reads, facilitating better read mapping; and (4) no gel purification was performed after PCR amplification of the library. Twenty micrograms of genomic DNA was sheared with HydroShear (Standard Shearing Assembly) to 3–4 kb size following the manufacturer's instructions and then purified with the LifeTech PureLink column. The sheared DNA was blunted using T4 DNA Pol and dNTPs followed by phosphorylation using T4 PNK and ATP. CAP adapters were formed by annealing short oligos of 9 nucleotides and 7 nucleotides. The short 7-nucleotide oligo lacks the 5′ phosphate. CAP adapters were ligated at 100× molar ratio to the genomic DNA using T4 DNA Ligase (Life Technologies 15224-017) in a 200-µL reaction volume using 50 units of enzyme (incubated for 30 min at room temperature), followed by size selection using an agarose gel to a range of 3–4 kb. Constructs were then circularized (at 3× molar ratio to the sheared DNA) using biotin-labeled internal adapter with matching overhangs (560-µL reaction with 70 units of T4 DNA ligase for each 1 µg of sheared DNA and incubated at room temperature for 30 min). The resulting circular DNA contained nicks on both strands at the sites of adaptor ligation due to the missing phosphate group on the CAP adaptor. Linear DNA was then removed by digesting with Plasmid-Safe DNA nuclease. The nicks on both strands were translated into the genomic DNA region using E. coli DNA polymerase I. The sizes of sequencing tags are controlled by digestion time to allow nicks to penetrate by ∼150 bp into the insert DNA. T7 exonuclease was used to digest nicked dsDNA. The exposed single strand was then digested by S1 nuclease. The resulting construct was end-repaired by T4 DNA Pol and T4 PNK, and A-tailed by Klenow exo-Polymerase. Constructs containing internal adapters were isolated by binding to magnetic streptavidin-coated beads (Dynabeads M-280) using a magnetic rack (DynaMag2). Illumina sequencing primers were ligated to the purified library on-beads and PCR amplified to produce a final library. Paired-end sequencing was performed using the HiSeq 2 × 101-bp protocol.
PCR validation
PCR validation of somatic breakpoints was performed by designing PCR primer pairs within 100–500 bp of the predicted breakpoint boundaries. PCR was performed using tumor and matched normal DNA. PCR products were analyzed by agarose gel elecrophoresis to determine specific PCR products stemming from breakpoint amplicons. Breakpoint amplicons were subjected to Sanger sequencing to determine the exact breakpoint sequences and identify precise breakpoint boundaries. Fusion transcripts in the samples were detected using standard RT-PCR by designing RT primer and amplification primers that were placed into the exons of SMASH-predicted fusion partners next to the breakpoint positions. The resulting RT-PCR products were analyzed by agarose gel electrophoresis to investigate expression of the fusion product in the tumor and control samples. Specific bands were Sanger-sequenced to infer the exact sequence of the fusion transcripts bordering the fusion junction.
Data access
Raw sequencing data have been submitted to the NCBI BioProject (http://www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA207396. SMASH source code is available for download from http://www-hsc.usc.edu/∼valouev/SMASH/SMASH.html.
Competing interest statement
Anton Valouev is an author of a provisional patent application that includes the SMASH SV detection algorithm.
Acknowledgments
The authors would like to thank Paul Thomas for feedback and critical reading of the manuscript, Zayed Albertyn and Colin Hercus for their help with Novoalign, Darin Briskman for his help with IBM SmartCloud, Jason Merker for evaluation of SMASH results, LifeTech for collaborative use of a SOLiD 4 sequencer, and members of the Sidow and West laboratories for valuable discussions and comments.
Footnotes
-
↵6 Corresponding authors
E-mail valouev{at}usc.edu
E-mail arend{at}stanford.edu
E-mail rbwest{at}stanford.edu
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.156224.113.
- Received February 14, 2013.
- Accepted October 7, 2013.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported), as described at http://creativecommons.org/licenses/by-nc/3.0/.








