
Bisulfite sequencing of chromatin immunoprecipitated DNA (BisChIP-seq) directly informs methylation status of histone-modified DNA
- Aaron L. Statham1,6,
- Mark D. Robinson1,2,3,6,
- Jenny Z. Song1,
- Marcel W. Coolen1,4,
- Clare Stirzaker1,5,7 and
- Susan J. Clark1,5,7,8
- 1Cancer Program, Garvan Institute of Medical Research, Sydney 2010, New South Wales, Australia;
- 2Bioinformatics Division, Walter and Eliza Hall Institute, Parkville 3052, Victoria, Australia;
- 3Department of Medical Biology, University of Melbourne, Parkville 3050, Victoria, Australia;
- 4Department of Human Genetics, Nijmegen Centre for Molecular Life Sciences (NCMLS), Radboud University Nijmegen Medical Centre, 6500 HB, Nijmegen, The Netherlands;
- 5St. Vincent's Clinical School, University of NSW, Sydney 2010, New South Wales, Australia
Abstract
The complex relationship between DNA methylation, chromatin modification, and underlying DNA sequence is often difficult to unravel with existing technologies. Here, we describe a novel technique based on high-throughput sequencing of bisulfite-treated chromatin immunoprecipitated DNA (BisChIP-seq), which can directly interrogate genetic and epigenetic processes that occur in normal and diseased cells. Unlike most previous reports based on correlative techniques, we found using direct bisulfite sequencing of Polycomb H3K27me3-enriched DNA from normal and prostate cancer cells that DNA methylation and H3K27me3-marked histones are not always mutually exclusive, but can co-occur in a genomic region-dependent manner. Notably, in cancer, the co-dependency of marks is largely redistributed with an increase of the dual repressive marks at CpG islands and transcription start sites of silent genes. In contrast, there is a loss of DNA methylation in intergenic H3K27me3-marked regions. Allele-specific methylation status derived from the BisChIP-seq data clearly showed that both methylated and unmethylated alleles can simultaneously be associated with H3K27me3 histones, highlighting that DNA methylation status in these regions is not dependent on Polycomb chromatin status. BisChIP-seq is a novel approach that can be widely applied to directly interrogate the genomic relationship between allele-specific DNA methylation, histone modification, or other important epigenetic regulators.
Epigenetic-based mechanisms play a critical role in gene expression and cellular differentiation, in both development and disease, including cancer. The genome-wide distribution of DNA methylation and chromatin modifications is now being revealed by large-scale sequencing studies; however, these techniques only permit correlative studies between chromatin marks and the underlying DNA methylation status. To provide further insights into the complex interactions between different epigenomic states, we developed a direct genome-wide sequencing approach, to interrogate at base-resolution allele-specific DNA methylation of all regions marked with a specific histone modification.
Understanding the direct interplay of DNA methylation and chromatin modification and how these epigenetic marks change during cellular differentiation and disease is a still a major challenge in cancer biology. In particular, a key question is what triggers DNA methylation and how the epigenome is remodeled in cancer cells. CpG island-promoter genes, associated with pluripotency of embryonic stem (hES) and progenitor cells, are often marked with active H3K4 trimethylation (H3K4me3) and repressive H3K27 trimethylation (H3K27me3) histones to form a bivalent state. Although this pattern was initially reported to be embryonic stem (ES) cell specific, bivalent domains have also been found in differentiated somatic cells (Mikkelsen et al. 2007; Mohn et al. 2008). The CpG-island promoters of bivalent genes in hES cells constitute a significant fraction of hypermethylated DNA in cancer cells, leading to the hypothesis that a stem cell signature and loss of H3K27me3 may trigger aberrant DNA methylation in malignancy (Ohm et al. 2007; Schlesinger et al. 2007; Widschwendter et al. 2007). Indeed, DNA methylation and H3K27me3 occupancy have been reported to be mutually exclusive in hES cells and cancer cells, using genome-wide approaches (Gal-Yam et al. 2008; Hahn et al. 2008; Takeshima et al. 2009). However, we (Coolen et al. 2010) and others (Gal-Yam et al. 2008; Meissner et al. 2008; Hawkins et al. 2010) have also identified a subset of genes in cancer that appear to harbor both repressive epigenetic marks.
Genome-wide chromatin modification studies are commonly performed using chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) (Pellegrini and Ferrari 2012). Several methods, however, have been developed to map global DNA methylation status; most of these are based on one of three techniques: digestion with methylation-sensitive restriction enzymes, affinity enrichment of methylated DNA, or chemical conversion with sodium bisulfite (for review, see Widschwendter et al. 2007; Laird 2010). The “gold-standard” bisulfite conversion protocol is the only technique that allows the methylation state of each cytosine residue in the target sequence to be defined. Whole-genome bisulfite sequencing is being applied to organisms with larger genomes, including mammals (Lister et al. 2009; Laurent et al. 2010), but the prohibitive cost makes DNA methylation-based affinity enrichment and reduced representation protocols followed by sequencing a favorable alternative (Meissner et al. 2008; Gu et al. 2010). The direct relationship between chromatin modification and DNA methylation at single genes has been studied by combining ChIP and bisulfite PCR genomic sequencing analysis (ChIP-BA) (Matarazzo et al. 2004; Collas 2010; Angrisano et al. 2011; Li and Tollefsbol 2011). However, due to the technical challenges of limited DNA generated after ChIP, epigenome-wide integration studies are still based on “overlaying” independent chromatin modification and DNA methylation maps (Gal-Yam et al. 2008; Kondo et al. 2008; Hawkins et al. 2010).
Here, we undertook a novel approach to directly address the relationship between Polycomb-bound chromatin and DNA methylation by performing genome-wide bisulfite sequencing on H3K27me3-ChIP DNA (Fig. 1A). We resolve the challenges of performing the bisulfite reaction on small amounts of sonicated formaldehyde-fixed ChIP DNA, in order to minimize degradation and maximize recovery of enough DNA to enable successful library generation and sequencing. A custom data analysis pipeline was also developed that identifies marked genomic regions and calculates their methylation status. Using BisChIP-seq, we directly show for the first time that H3K27me3-marked histones can bind to both methylated and unmethylated DNA and that this association is dependent on genomic location, and in cancer the codependency of marks is largely redistributed. BisChIP-seq is therefore a novel approach that can be widely applied to study the direct relationship between DNA methylation and other important epigenetic regulators.
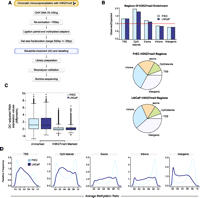
BisChIP-seq DNA methylation profiles of H3K27me3-enriched DNA from normal PrEC and cancer LNCaP cells. (A) Flowchart of BisChIP-seq protocol to perform bisulfite treatment and library preparation on H3K27me3-ChIP DNA. (B) Distribution of H3K27me3-enrichment genome-wide relative to observed over expected and pie charts showing relative distributions across the genome. (C) Affymetrix Gene 1.0 ST expression values for H3K27me3-marked and -unmarked genes in PrEC and LNCaP cells. (D) Distribution frequency of CpG methylation levels at H3K27me3-marked regions that fall into each regional annotation category from low (0%) to high (100%) methylation (0.0–1.0).
Results
Optimization of BisChIP-seq protocol
We developed the BisChIP-seq protocol to directly address the relationship between Polycomb-bound chromatin and DNA methylation. One of the challenges of bisulfite sequencing is the amount and quality of DNA required for optimal conversion (Clark et al. 1994; Clark et al. 2006). Therefore, before proceeding with bisulfite treatment of H3K27me3-ChIP DNA, we performed optimizations using 100 ng of sonicated ChIP input DNA isolated from formaldehyde-fixed cells. First, methylated adaptors (Illumina) were ligated to the DNA, followed by gel elution size selection (200 ± 20 bp) (as required by Illumina sequencing), prior to bisulfite treatment. Two bisulfite methods were compared: the QIAGEN EpiTect Bisulfite Kit with a 5-h bisulfite treatment and the modified method of Clark et al. (2006) with a 4-h bisulfite conversion on very small amounts of DNA (as expected after gel elution size fractionation). A methylation-specific headloop suppression PCR (MSH-PCR) (Rand et al. 2005) was used to assay the yield of positive controls, GSTP1 and EN1. We found that the Clark et al. method with Microcon YM50 desalting procedure achieved the greatest yield of bisulfite-converted DNA (Supplemental Fig. 1a,b). Next, we optimized the steps necessary to enable successful library generation for Illumina sequencing. Following bisulfite treatment, the efficiency of the library prep PCR was tested using 5 μL, 10 μL, and 22.5 μL of adaptor-modified bisulfite-treated DNA (60 μL) in a 50-μL library PCR reaction with paired-end primers PE 1.0 and PE 2.0 and PfuTurbo Cx hotstart DNA polymerase, as per the Illumina protocol and 18 PCR cycles. We showed that too much bisulfite-treated input DNA (22.5 μL) had an inhibitory effect on the library preparation yield (Supplemental Fig. 1c). The number of PCR cycles was also optimized to give sufficient yield in the library preparation without compromising the complexity of the library by overamplification. We tested 10, 12, 14, and 18 PCR cycles, and 14 cycles gives adequate yield for high-throughput sequencing (Supplemental Fig. 1d).
Chromatin immunoprecipitation with H3K27me3 antibody was performed on a prostate cancer cell line (LNCaP) and normal prostate epithelial cells (PrEC) and validation of H3K27me3 enrichment was confirmed by qPCR of known candidate genes (Supplemental Fig. 2; Coolen et al. 2007). For the BisChIP-seq protocol (Fig. 1A), the H3K27me3 ChIP DNA (75–100 ng, respectively) was resonicated and checked on an Agilent Technologies 2100 Bioanalyser to ensure the maximum yield of DNA in the 150-bp size range, followed by ligation of methylated paired-end adaptors (Illumina) and size selection by gel elution near 200 bp. Bisulfite treatment was performed for 4 h on the gel-eluted H3K27me3 ChIP DNA as per the Clark et al. (2006) protocol with optimizations (described in Methods), and sequencing libraries were generated (using optimized conditions, described above). A sample (1 μL) from the library preparation was checked on an Agilent Technologies 2100 Bioanalyser (Supplemental Fig. 1e) to confirm successful library generation after bisulfite treatment and with sufficient yield for Illumina sequencing, before cluster generation and Illumina GAIIx sequencing (75-bp paired-end, three lanes of PrEC and four lanes of LNCaP) (Supplemental Table 1). Using the methodology described above, we show that we can perform bisulfite treatment on ChIP DNA and successfully generate a library for sequencing. The amount of DNA obtained from any ChIP will clearly depend on the type of antibody used and the abundance of the mark in each cell type, and here we show that <100 ng of DNA is sufficient for Illumina bisulfite sequencing.
Analysis of BisChIP-seq data
Sequences from the bisulfite-treated H3K27me3-ChIP DNA were mapped using a custom pipeline for the alignment of paired-end bisulfite ChIP reads, adapted from the procedure described in Lister et al. (2009) (see Methods). A total of 38,403,614 and 70,682,755 reads were obtained for PrEC and LNCaP, respectively, with a bisulfite conversion rate of 99.7% for PrEC and 99.8% for LNCaP (Supplemental Table 1). We used ChromaBlocks, a procedure for detecting large regions of low enrichment, as expected for H3K27me3 (Hawkins et al. 2010). In total, 53,749 and 52,677 regions were enriched (FDR < 0.001), covering 148.6 Mb and 139.9 Mb in PrEC and LNCaP (Table 1), respectively, comparable to the results of Hawkins et al. (2010). H3K27me3-marked DNA was enriched preferentially at transcription start sites (TSSs) and CpG islands in both cell types, while exons, introns, and intergenic regions were not significantly enriched in these regions (Fig. 1B; Table 1). In total, 5029 and 4639 TSSs were marked by H3K27me3 in PrEC and LNCaP, respectively, and expression of these genes was at basal levels, correlating with the role of H3K27me3 in gene repression (Fig. 1C; Table 1).
ChromaBlocks analysis of BisChIP-Seq data
The proportion of cytosine base calls that are not bisulfite-converted at CpG dinucleotides is used to determine DNA methylation levels. Using a minimum of 10 reads, the methylation status on the plus and minus strand of individual CpG sites was shown to be highly concordant (Supplemental Fig. 3) (r = 0.94 and 0.91 in PrEC and LNCaP, respectively). Therefore, the two strands were pooled, allowing us to interrogate 2,482,996 and 2,552,762 CpG sites in PrEC and LNCaP, respectively (Table 2); <1% of H3K27me3-enriched regions had insufficient coverage to determine methylation levels. Methylation levels assessed from the H3K27me3-enriched regions are also highly concordant (r = 0.932, PrEC; r = 0.925, LNCaP) with lower-resolution Infinium 450K array methylation data obtained from native LNCaP and PrEC DNA (Supplemental Fig. 4a) (see Methods). Example comparisons of BisChIP-seq methylation and clonal bisulfite sequencing from native DNA show that bisulfite-based DNA methylation results are not affected by prior cross-linking (Supplemental Fig. 4b).
DNA methylation levels of H3K27me3-enriched regions
Bimodal DNA methylation profiles of H3K27me3-enriched regions are redistributed in cancer
In normal prostate PrEC cells, the H3K27me3-enriched regions, which overlap with the TSSs or CpG islands, show bimodal DNA methylation profiles; that is, the regions are predominantly associated with either unmethylated or methylated DNA (Fig. 1D). In contrast, exons, introns, and intergenic H3K27me3-enriched regions are primarily associated with methylated DNA (Fig. 1D). Surprisingly in the prostate cancer cells, H3K27me3-enriched regions are associated with different DNA methylation profiles to normal prostate cells, and this redistribution of dual repressive marks in cancer is dependent on genomic location and density of methylation. Exons, introns, and intergenic H3K27me3-enriched regions are comparatively depleted in highly methylated DNA. In contrast, H3K27me3-enriched TSSs and CpG islands show an increased association with medium to highly methylated DNA and a reduced binding to lowly methylated DNA (Fig. 1D; Table 2). For example, 3330 (∼71.7%) of the LNCaP TSS regions show medium or high levels of DNA methylation, whereas only 2866 TSS regions (57.0%) in normal cells were marked by both H3K27me3 and medium or high levels of DNA methylation (χ2 test, p < 1 × 10−15). One example region that shows a cancer-associated redistribution of marks is the RCSD1 CpG island promoter, where similar levels of H3K27me3 enrichment are observed in normal and cancer cells, and yet the same RCSD1 CpG island is unmethylated in the normal cell and hypermethylated in the cancer cell (Fig. 2A). This pattern of dual repressive marks is observed in a subset of normally silent CpG island promoters, where the gain of extensive DNA methylation in cancer is not associated with a loss of H3K27me3 (Supplemental Fig. 5a–d). These data directly show that DNA methylation and H3K27me3 are not mutually exclusive, but can co-occur at TSSs. Interestingly, all of the H3K27me3-associated genes were repressed regardless of the level of DNA methylation (Supplemental Fig. 6).
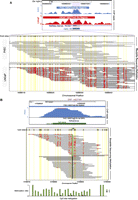
BisChIP-seq examples showing differential methylation and allele-specific methylation in H3K27me3-enriched ChIP DNA. (A) UCSC Genome Browser screen shot of BisChIP-seq data showing the RCSD1 TSS and CpG island, where H3K27me3-modified histones are enriched in both PrEC and LNCaP. (Purple shading) In PrEC cells the CpG island is unmethylated, whereas in LNCaP cells the island becomes extensively DNA methylated without losing the H3K27me3 mark. Individual bisulfite methylation sequencing reads are shown with CpG sites (black circles) in yellow shading for each molecule. (Red circles) CpG DNA methylation. (B) Example of allele-specific methylation in PrEC cells at rs637481 on chromosome 1. UCSC Genome Browser screen shot of BisChIP-seq data indicates regions of significant H3K27me3-enrichment called by ChromaBlocks. (Purple line) Position of the A/G SNP at rs637481. Individual bisulfite molecule sequencing reads are shown with all CpG sites in the sequence (black circles) in yellow shading for each molecule. (Red circles) BisChIP-seq readout of CpG DNA methylation. The allele-specific methylation ratio is indicated by bar graphs.
Allele-specific DNA methylation at H3K27me3-marked regions
Using BisChIP-seq, we could directly interrogate allele-specific DNA methylation at H3K27me3-marked regions that contain heterozygous single nucleotide polymorphisms (SNPs). Approximately 106,887 SNPs and 215,628 SNPS had sufficient coverage in the H3K27-ChIP bisulfite sequencing data (at least 20 reads) from PrEC and LNCaP, respectively, to make genotype calls, resulting in 6472 and 13,034 clear heterozygous SNPs (see Methods). Of these, 762 and 1195 exhibited strong evidence of allele-specific differential methylation (difference-in-proportions test, FDR < 0.05) in H3K27me3-enriched regions; that is, both methylated and unmethylated alleles were equally bound by the Polycomb mark. For example, SNP rs637481 shows either high or low levels of methylation on the A and G allele, respectively, in PrEC (Fig. 2B); more allele-specific differential methylation examples of H3K27me3-enriched regions in both PrEC and LNCaP are shown in Supplemental Figure 7a and b. These data directly show that in normal cells, H3K27me3 marks many regions independent of DNA methylation and the underlying sequence.
Discussion
Here, we report a novel method, which combines bisulfite-conversion methodology with chromatin immunoprecipitation and deep sequencing, that allows a direct genome-wide interrogation of two epigenetic marks for the first time on the same DNA molecule. Previous methods by themselves have only allowed correlative studies of the epigenome to be assessed that do not take into account potential allele-specific differences. We show that H3K27me3 polycomb-bound histones are not always mutually exclusive with DNA methylation but can co-occur in a genomic region-dependent manner and this co-occurrence is remodeled in cancer.
It is intriguing that in the normal somatic epithelial cell, we find a discrete bimodal distribution of H3K27me3, with either fully methylated or unmethylated DNA at TSS and CpG islands, whereas in exonic, intronic, or intergenic regions, H3K27me3 primarily associates only with methylated DNA. By overlaying chromatin-modification profiles and DNA methylomes in hESCs and primary fibroblasts, Hawkins et al. (2010) also found that nearly one-third of the genome differs in chromatin structure on differentiation, with dramatic redistributions of repressive H3K27me3 marks. However, in contrast to our findings, they found that H3K27me3 is primarily associated with unmethylated DNA at promoters in IMR90 cells, but outside of promoters, H3K27me3 was associated with DNA methylation. These results may reflect different cell type–specific relationships between DNA methylation and histone modifications, or may highlight the fact that direct bisulfite methylation sequencing of ChIP DNA allows a more sensitive analysis of a subset of these two epigenetic marks.
In cancer, we found an extensive alteration of the distribution H3K27me3-modified histones and the relationship with DNA methylation status. Notably, there was an increase in regions with the dual repressive marks at TSSs and CpG islands and a loss or decrease in the extent of DNA methylation in H3K27me3-enriched intronic and exonic sequences. Interestingly, there was also a pronounced shift in preference for H3K27me3 to bind unmethylated DNA in intergenic regions. It is possible that the gross alteration of patterns that are observed here may simply reflect that changes in DNA hypomethylation and hypermethylation are independent of H3K27me3 occupancy in cancer; that is, H3K27me3-mediated silencing is mechanistically distinct from DNA methylation-associated silencing (Kondo et al. 2008). Indeed, we found that all H3K27me3-marked regions were repressed independent of the level of DNA methylation, and therefore the relationship between H3K27me3 and DNA methylation levels may be more complex than previously proposed (Suzuki and Bird 2008; Cedar and Bergman 2009), in both normal differentiation and malignancy.
One of the other primary advantages of BisChIP-seq is the use of single-molecule bisulfite sequencing to directly interrogate allele-specific methylation of ChIP DNA. Interestingly, using this technique, we demonstrate that H3K27me3-modified histones can bind directly to both methylated and unmethylated alleles in the same cell, further supporting the concept that H3K27me3 and DNA methylation are not mutually exclusive and can either work independently and/or cooperatively to enforce gene silencing. The concept of what establishes allele-specific methylation in a normal cell, however, is still an open question, but clearly in some genomic contexts appears to be independent of the presence of H3K27me3-modified chromatin.
In summary, we demonstrate that BisChIP-seq is a cost-effective novel approach that may be applied to directly interrogate the interactions between DNA methylation and other histone modifications, as well as other important epigenetic regulators, such as transcription factors that can be enriched by genome-wide immunoprecipitation. Specifically, using BisChip-seq, we showed that both methylated and unmethylated alleles can be associated with H3K27me3-enriched DNA and that in cancer the relationship of the bimodal repressive marks is altered in a regional-dependent manner. Our results highlight the importance of studying allele-specific DNA methylation and chromatin marks directly, because these may have different patterns in different sequence and cellular contexts.
Methods
Cell culture
LNCaP prostate cancer cells were cultured as described previously (Song et al. 2002). Normal prostate epithelial cells (PrECs) (Cambrex Bio Science cat. no. CC-2555) were cultured according to the manufacturer's instructions in Prostate Epithelial Growth Media (PrEGM) (Cambrex Bio Science cat. no. CC-3166).
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation (ChIP) assays were performed according to the manufacturer's instructions (Millipore). Briefly, ∼1 × 106 cells, in a 10-cm dish, were fixed by adding formaldehyde to a final concentration of 1% and incubating for 10 min at 37°C. The cells were washed twice with ice-cold PBS containing protease inhibitors (1 mM phenylmethylsulfonyl fluoride [PMSF], 1 μg/mL aprotinin, and 1 μg/mL pepstatin A), harvested, and treated with SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris at pH 8.1) for 10 min on ice. The resulting lysates were sonicated to shear the DNA to fragment lengths of 200–500 bp. The complexes were immunoprecipitated with antibodies specific for tri-methyl-histone H3(lys27) (Millipore #07-449/lot number #DAM 1514011; this antibody lot was previously [Egelhofer et al. 2011] shown to be 100% specific with no cross-reactivity using a panel of modified peptides on a dot blot assay [http://compbio.med.harvard.edu/antibodies/antibodies/56]). Ten microliters of antibody was used for each immunoprecipitation according to the manufacturer's instructions. A no-antibody control was included for each ChIP assay, and this showed a lack of non-specific precipitation by quantitative Real-Time PCR analysis. Input samples were processed in parallel. The antibody/protein complexes were collected by Protein A/G PLUS agarose beads (Santa Cruz sc-2003) and washed several times following the manufacturer's instructions. The immune complexes were eluted with 1% SDS and 0.1 M NaHCO3; samples were treated with proteinase K for 1 h; and DNA was purified by phenol:chloroform extraction and ethanol precipitation and resuspended in 30 μL of H2O. qPCR validation of H3K27me3 enrichment using known candidate genes was performed (ChIP primers in Supplemental Table 2).
Preparation of ChIP DNA for bisulfite treatment and Illumina Genome Analyzer
H3K27me3-ChIP DNA was pooled from three to five ChIP assays to obtain 100 ng of DNA for adaptor ligation and gel-size elution, followed by bisulfite treatment. The ChIP DNA was concentrated by ethanol precipitation to give a final yield of 100 ng of ChIP DNA in 40 μL of water. The ChIP DNA was further sonicated using a Bioruptor (High, 30 sec on and 30 sec off for 25 min) to ensure the maximum yield of DNA in the size range of 150–200 bp and checked on a bioanalyzer. End repair and addition of A bases to the 3′ end of the DNA fragments were performed according to the “Preparing Samples for ChIP Sequencing of DNA” (Illumina Part #11257047 Rev.A). Methylated paired-end adaptors undiluted (Illumina Part #1005560) were then ligated onto the ChIP DNA using 1 μL of undiluted methylated adaptor oligo mix, 4 μL of DNA ligase (Illumina Part #1000522), in a total reaction volume of 10 μL for 15 min at room temperature. The reactions were cleaned up using the MinElute PCR Purification Kit (QIAGEN Part #28004) and eluted in 10 μL of EB buffer following the manufacturer's instructions. Size selection of the library was performed according to the Illumina protocol for “Preparing Samples for ChIP Sequencing of DNA” (Illumina Part #11257047 Rev.A). Two microliters of loading buffer was added to 10 μL of DNA, and the entire sample was loaded onto a single lane of the gel. A gel slice in the range of 200 ± 20 bp was excised and eluted using the QIAGEN Gel Extraction Kit (QIAGEN Part #28704) and eluted in 40 μL of EB buffer.
Bisulfite treatment of ChIP DNA
Bisulfite conversion was performed with minor modifications of the Clark et al. (2006) protocol. The bisulfite reactions were done in duplicate. One microliter of tRNA (10 μg/μL) was added to the 20 μL of DNA in a final volume of 21 μL. Then 2.33 μL of 3 M NaOH was added to each 21-μL DNA sample and incubated for 15 min at 37°C and 2 min at 90°C, placed on ice, and then centrifuged briefly. Next, 208 μL of saturated sodium metabisulfite (pH 5.0) (7.6 g of Na2S2O5 with 464 μL of 10 M NaOH made up to 15 mL with water) and 12 μL of 10 mM quinol were added, vortexed, briefly centrifuged, and incubated for 4 h at 55°C in a PCR machine. The bisulfite reaction was cleaned up using the Microcon YM50 device (Millipore). The duplicate bisulfite reactions were combined in a new tube, and 1 μL of tRNA (10 μg/μL) was added before transferring to a Microcon YM50 column. The column was spun at 14,000g for 25 min at room temperature. The filtrate was discarded, and 350 μL of H2O was added to the column and spun for a further 12 min at room temperature. This wash step was repeated. After discarding the filtrate, 350 μL of 0.1 M NaOH (freshly prepared) was added to the column for the desulfonation step and again spun at 14,000g for 10 min at room temperature; a final wash of 350 μL of H2O was followed by a 12-min spin at room temperature at 14,000g. The column was placed in a fresh tube, and 60 μL of H2O was added and gently pipetted up and down and allowed to stand for 5 min before being inverted and spun at 1000g for 3 min to transfer the bisulfite-treated DNA into the microfuge tube. The bisulfite-treated DNA was stored at −20°C. The QIAGEN EpiTect Bisulfite Kit (cat. no. 59104) was performed according to the manufacturer's instructions for bisulfite treatment of “low concentrations of DNA.” Quantitation of bisulfite conversion and DNA yield was compared using Methylation Specific Headloop PCR (MSH-PCR). MSH-PCR primers, probes, and conditions for EN1 and GSTP1 are as described previously (Rand et al. 2005).
Library preparation of bisulfite-treated ChIP DNA
Five microliters of bisulfite-treated DNA (from above) was used in the PCR amplification step for library preparation. Triplicate 50-μL PCR reactions were set up as follows: 5 μL of DNA, 1.25 μL of dNTPs (10 mM) (Illumina #1000564), 1 μL of PCR primer PE 1.0 (Illumina Part #1001783), and 1 μL of PCR primer PE 2.0 (Illumina Part #1001784), in 1× PfuTurbo Cx reaction buffer (Stratagene #6000410) and 1 μL of PfuTurbo Cx Hotstart DNA polymerase (2.5 U/μL; Stratagene #600410). The PCR reaction was performed for 5 min at 95°C, 30 sec at 98°C, followed by 14 cycles of 10 sec at 98°C, 30 sec at 65°C, 30 sec at 72°C, for 14 cycles, then 5 min at 72°C; and hold at 4°C. The reactions were cleaned up following the instructions of the MinElute PCR Purification Kit, eluting each reaction in 15 μL of EB buffer, and pooling the triplicate reactions to give a final volume of 45 μL. One microliter was checked on an Agilent Technologies 2100 Bioanalyzer. Twenty microliters was sent to Illumina for cluster generation and GAIIx sequencing.
Alignment of bisulfite-treated sequencing reads
A custom pipeline was written for the alignment of paired-end bisulfite ChIP reads, adapted from the procedure described in Lister et al. (2009) (available from http://github.com/astatham/Bisulfite-seq-pipeline). Briefly, methylation information was removed from reads (C's in read 1 replaced with T's; G's in read 2 replaced with A's), and reads were then mapped separately to both strands of the bisulfite-converted hg18 genome using Bowtie with the following parameters (-solexa1.3-quals -nomaqround -n 3 -l 24 -e 300 -y -k 10). Reads from the two strands were collated and ranked by the number of mismatches against the reference; only reads with fewer than six mismatches that were three mismatches closer to the reference than the next best hit were retained as unique (for mapping statistics, see Supplemental Table 1). Since the median fragment size for our paired-end libraries (119-bp PrEC and 114-bp LNCaP) was less than the read length (150 bp), the majority of the fragment sequences overlapped in the middle; to avoid doubling up on base calls, these fragments were merged into single contig reads, with disagreements between base calls decided by higher-quality scores; ties were decided randomly. The number of methylated (C) and unmethylated (T) base calls at each CpG site within the genome was then extracted and imported into R for downstream analysis.
Detection of H3K27me3-enriched regions
Regions of enrichment were determined using an adaptation of the ChromaBlocks algorithm (Hawkins et al. 2010) for identifying both strong peaks and regions of broad enrichment, as implemented in the Repitools package (Statham et al. 2010).
Gene expression array data
Affymetrix Gene 1.0 ST array data for LNCaP and PrEC cells is available at NCBI's Gene Expression Omnibus (GEO) under accession number GSE19726. Robust multi-chip analysis (RMA) was used to summarize probe-level data, and probe GC content effects were removed by subtracting the mode of RMA expression values in bins of average probe GC content.
Infinium HumanMethylation450 BeadChips
Genomic DNA was isolated from PrEC and LNCaP cells using the QIAamp Mini kit (QIAGEN) following the manufacturer's instructions. Hybridizations to Infinium HumanMethylation450 BeadChips (Illumina) were performed in triplicate as a service by the Australian Genome Research Facility. The “minfi” Bioconductor package was used to process raw data into methylation “beta” values using the “preprocessIllumina” function with default options for background correction and normalization.
Detection of allele-specific methylation (ASM)
To detect allele-specific methylation (ASM), all known single-nucleotide variant positions in hg18 were extracted from dbSNP 130 using the UCSC Table Browser. The number of A, C, G, and T base calls at each SNP position was extracted from aligned bisulfite reads using samtools (Li et al. 2009); SNPs with <20× coverage were discarded. The remaining SNP positions were deemed potentially heterozygous when the proportion of base calls containing the reference was not significantly different from 0.5 (p > 0.05) (prop.test in R). Full-length bisulfite reads overlapping heterozygous SNPs were extracted, and the change in proportion of (surrounding) methylated CpGs was tested using the difference-in-proportions test (FDR < 0.05). SNPs that overlapped a CpG site were excluded from this analysis.
Data access
The 450k array and BisK27ChIP-seq data from this study have been submitted to NCBI's Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) under accession nos. GSE34340 and GSE30558, respectively. The SuperSeries accession that connects data is GSE34403.
Acknowledgments
We thank Dr. Kate Patterson for reviewing the manuscript and for help with the figures. We thank Illumina for the Fasttrack Genetic Analysis Services collaboration for BisChIP-seq experiments. This work is supported by a CINSW Fellowship (M.W.C.), a CINSW Student Scholarship (A.L.S.), and National Health and Medical Research Council (NH&MRC; APP1011447) (S.J.C.).
Author contributions: S.J.C., C.S., and M.W.C. conceived and designed the experiments; J.Z.S. performed the experiments; A.L.S. and M.D.R. analyzed the data; and C.S., M.D.R., and S.J.C. wrote the paper.
Footnotes
-
↵8 Corresponding author.
E-mail s.clark{at}garvan.org.au.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.132076.111.
- Received September 15, 2011.
- Accepted January 30, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








