
Comparative RNA sequencing reveals substantial genetic variation in endangered primates
- George H. Perry1,7,9,
- Páll Melsted1,7,8,
- John C. Marioni1,7,
- Ying Wang1,7,
- Russell Bainer1,7,
- Joseph K. Pickrell1,
- Katelyn Michelini2,
- Sarah Zehr3,
- Anne D. Yoder3,4,5,
- Matthew Stephens1,6,
- Jonathan K. Pritchard1,2,9 and
- Yoav Gilad1,9
- 1Department of Human Genetics, University of Chicago, Chicago, Illinois 60637, USA;
- 2Howard Hughes Medical Institute, University of Chicago, Chicago, Illinois 60637, USA;
- 3Duke Lemur Center, Duke University, Durham, North Carolina 27705, USA;
- 4Department of Biology, Duke University, Durham, North Carolina 27708, USA;
- 5Department of Evolutionary Anthropology, Duke University, Durham, North Carolina 27708, USA;
- 6Department of Statistics, University of Chicago, Chicago, Illinois 60637, USA
Abstract
Comparative genomic studies in primates have yielded important insights into the evolutionary forces that shape genetic diversity and revealed the likely genetic basis for certain species-specific adaptations. To date, however, these studies have focused on only a small number of species. For the majority of nonhuman primates, including some of the most critically endangered, genome-level data are not yet available. In this study, we have taken the first steps toward addressing this gap by sequencing RNA from the livers of multiple individuals from each of 16 mammalian species, including humans and 11 nonhuman primates. Of the nonhuman primate species, five are lemurs and two are lorisoids, for which little or no genomic data were previously available. To analyze these data, we developed a method for de novo assembly and alignment of orthologous gene sequences across species. We assembled an average of 5721 gene sequences per species and characterized diversity and divergence of both gene sequences and gene expression levels. We identified patterns of variation that are consistent with the action of positive or directional selection, including an 18-fold enrichment of peroxisomal genes among genes whose regulation likely evolved under directional selection in the ancestral primate lineage. Importantly, we found no relationship between genetic diversity and endangered status, with the two most endangered species in our study, the black and white ruffed lemur and the Coquerel's sifaka, having the highest genetic diversity among all primates. Our observations imply that many endangered lemur populations still harbor considerable genetic variation. Timely efforts to conserve these species alongside their habitats have, therefore, strong potential to achieve long-term success.
Comparative genomics is a powerful approach to study evolutionary processes, often used to identify functionally constrained genomic regions (Bejerano et al. 2004; Alexander et al. 2010) or to infer species-specific adaptations and the associated biological mechanisms (Oleksiak et al. 2002; Abzhanov et al. 2006; Gilad et al. 2006; Blekhman et al. 2008). The power of the comparative genomic approach increases with the number of species studied (Drosophila 12 Genomes Consortium 2007). Comparative genomic studies of primates, however, have so far focused mostly on the few species for which complete reference genome sequences are available, namely humans, chimpanzees, orangutans, and rhesus macaques (e.g., Caceres et al. 2003; Khaitovich et al. 2005; The Chimpanzee Sequencing and Analysis Consortium 2005; Gilad et al. 2006; Jiang et al. 2007; Rhesus Macaque Genome Sequencing and Analysis Consortium 2007; Locke et al. 2011).
Genomic data are particularly limited for lemurs (Horvath and Willard 2007), which represent a major primate radiation exclusive to the biodiversity and conservation hotspot of Madagascar (Brooks et al. 2002) and whose habitats have been shrinking rapidly over the past century due to deforestation (Green and Sussman 1990; Harper et al. 2007). Many of the 97 currently recognized lemur species are considered endangered or critically endangered (Mittermeier et al. 2008; International Union for Conservation of Nature 2010). We have very little knowledge of nuclear genetic diversity for any of these endangered species, yet such data are critical for planning conservation efforts because genetic diversity is associated with the risk of extinction (Frankham 2005; Palstra and Ruzzante 2008).
We sought to establish a more comprehensive primate comparative genomic database while simultaneously generating genetic diversity data that would benefit the conservation of endangered species. Since sequencing complete mammalian genomes from a large number of individuals remains prohibitively expensive and because effective DNA capture strategies (e.g., Gnirke et al. 2009)—especially for comparative genomic analysis—require a priori reference genome sequences, we chose an alternative approach for our study. Specifically, we used RNA-sequencing (RNA-seq) combined with a de novo gene assembly strategy to characterize liver transcriptomes from multiple individuals from each of 16 mammalian species, including 12 primates (Fig. 1A). The primates include five lemur species (aye-aye, Coquerel's sifaka, black and white ruffed lemur, crowned lemur, and mongoose lemur) and two other strepsirrhine primates (slow loris and Moholi bushbaby). Since little or no genomic information was previously available for most of the species in our study, we developed a new de novo assembly algorithm, which facilitated comparisons of gene sequences and expression levels within and between individuals and species.

Transcript assembly and phylogenetic reconstruction from RNA-seq data. (A) Typical example of an assembled gene, SNF8, with complete cross-species exon conservation. (Red bars) Identified homologies to the human SNF8 RefSeq coding sequence that were used to isolate the appropriate region of the de Bruijn graph during the assembly process. Divergence times are approximate and based on consensus estimates from previous studies. Photos of strepsirrhine primates were kindly provided by David Haring, Duke Lemur Center. (B) Neighbor-joining trees estimated from nucleotide sequence and gene expression data. Nucleotide sequence distance matrix was computed from concatenated multispecies alignments of coding sequences of 515 genes that were assembled for all 16 species. Gene expression pairwise correlation distance matrix was computed for species mean expression estimates using all genes assembled in at least six species (6494 genes). As expected, the known primate phylogeny was recapitulated perfectly from the nucleotide sequence data (see Supplemental Fig. S7 for the tree, also including bushbaby), with the only discrepancy among nonprimate mammals being the juxtaposition of the mouse and armadillo branches, likely explained by long branch attraction that is a common issue in phylogenetic analyses that include rodents (Cannarozzi et al. 2007). Variation in the expression data also follows a phylogenetic pattern but with slow loris erroneously placed outside all other primates and the misplacement of armadillo.
Our approach, therefore, allowed us to analyze nucleotide sequence, expression level, exon structure, and genetic diversity data from thousands of genes per species, using a cost-effective strategy. Using these data, we were able to identify signatures of positive and directional selection in extant and ancestral primate lineages and to examine the relationship between endangered status and genetic diversity across an extensive primate phylogeny.
Results
To collect comparative genomic diversity data on a large panel of species, we used RNA-seq combined with a de novo gene assembly strategy. We prepared RNA-seq libraries from liver samples from four unrelated individuals for each of 15 species and from two armadillos (Supplemental Methods). Each library was sequenced using one lane of the Illumina Genome Analyzer IIx with paired-end, 76-bp reads (2 × 76 bp). We obtained, on average, 16.4 million (±4.8M) 76-bp paired-end RNA sequencing reads per individual (2.5 Gb of nucleotide sequence per individual). For transcript assembly, we combined the sequence reads from all individuals for each species to generate consensus gene nucleotide sequences. For gene expression and genetic diversity analyses, we considered the sequence data from each individual separately.
De novo transcript assembly
Since sequenced genomes were not available for most of the species in our study, we developed a de Bruijn graph-based approach (Pevzner et al. 2001) for de novo assembly of the transcriptome of each species and simultaneous matching of gene orthologs. Our assembly process is described in detail in the Supplemental Methods (the transcript assembly code is available at http://pritch.bsd.uchicago.edu/software.html). Briefly, for each species we searched the de Bruijn graph for small-scale similarity (in 39-bp windows) to human RefSeq gene sequences (Fig. 1A). These homologous regions were used to set general expectations for transcript coverage levels and to isolate the portion of the graph likely to contain each gene sequence. While this step of our approach relies on the maintenance of sequence similarity between species, simulations demonstrate that our approach is robust to internal exon gains and losses in nonhuman species (Supplemental Methods). Exploring the subgraphs for each gene, we then filtered contigs with lower-than-expected coverage to remove intronic sequences and sequencing errors (Supplemental Fig. S1). We next aligned each remaining path through the graph to the corresponding human RefSeq gene and selected the sequence with the most aligned nucleotides, effectively removing erroneous paths through repetitive elements. We required the presence of at least 50% of the coding region (compared to the corresponding human RefSeq gene) to classify a gene as assembled and to include it in subsequent analyses. Finally, potentially paralogous gene sequences were identified and removed, and the expression levels of the remaining genes were estimated, independently for each sample, based on the number of sequence reads mapped to them. Using this approach, we assembled between 4789 and 5924 gene sequences for 15 species (but only 2680 genes from the bushbaby, probably due to RNA degradation in the bushbaby liver samples [Supplemental Fig. S2]; bushbaby samples were thus excluded from subsequent analyses of the gene expression data).
The availability of high-quality sequenced genomes for six of the species (human, chimpanzee, rhesus macaque, marmoset, mouse, and gray short-tailed opossum) allowed us to test the accuracy of our assembly approach. To do so, we compared gene sequences and estimates of gene expression levels based on the de novo assembly to estimates based on a more conventional genome alignment approach (Supplemental Methods). On average, 98.1% (±1.9%) of the corresponding pairs of de novo assembly and reference genome transcripts had identical or near-identical sequences (≥97%, allowing for polymorphisms) (Supplemental Fig. S3). Estimates of expression levels from the assembled genes and the genome alignment approach were also highly correlated (mean Spearman rank correlation coefficient r > 0.90 for all comparisons) (Supplemental Fig. S4). These observations indicate that the quality of the assembled data is high. This conclusion is further supported by our ability to nearly perfectly recapitulate the known primate phylogeny (Perelman et al. 2011) based on either the sequence data or the estimates of gene expression levels (Fig. 1B; Supplemental Figs. S5–S7).
Genetic diversity and endangered status
We used the RNA-seq data to identify single nucleotide polymorphisms (SNPs) in genes with sequence coverage levels that were sufficient for the accurate identification of heterozygous sites (minimum 15× per strand, 30× total, per individual coverage, for each individual in a species) (Supplemental Methods). On average, we obtained genotypes for 787,744 bp (±341,326 bp) per species from the coding regions of an average of 1170 genes. We used nucleotide diversity at synonymous sites to estimate putatively neutral levels of genetic diversity for each of the 16 species (Supplemental Table S1). To our knowledge, these are the first published estimates of nuclear genome genetic diversity for all but five of the 16 species in our study.
We used several quality control analyses to test the quality of our SNP genotype calls. For human, chimpanzee, rhesus macaque, aye-aye, and mouse, our genetic diversity estimates are generally comparable to those that have been published previously (Yu et al. 2003; Fischer et al. 2004; Voight et al. 2005; Baines and Harr 2007; Hernandez et al. 2007; Perry et al. 2007; Wall et al. 2008; Perry et al. 2010) (Supplemental Methods). We confirmed that our SNP calling strategy is highly accurate (99.4%) by comparing the human genotypes inferred using our approach to genotypes collected using the Illumina 1M-Duo SNP array platform, with the same human samples (Supplemental Methods; Supplemental Fig. S8). Our human sample includes two European Americans and one individual each of East Asian and African ancestry (Supplemental Fig. S9), and as expected, heterozygosity (based on our RNA-seq SNP calls) was highest in the individual of African descent (synonymous sites: 0.126% vs. 0.086%, 0.087%, and 0.091%) (Supplemental Table S2). We also used traditional Sanger sequencing to validate small subsets of SNPs in four species (21/23 human SNPs, 15/16 rhesus macaque SNPs, 21/23 Coquerel's sifaka SNPs, and 15/19 black and white ruffed lemur SNPs were successfully validated [see Supplemental Methods; Supplemental Table S3]), and we evaluated genotype accuracy more generally by assessing consistency among SNPs identified from subsampled sets of reads for each individual of each species (Supplemental Table S4). Finally, we observed an inverse relationship between synonymous site diversity and the ratio of nonsynonymous to synonymous site diversity within each species, as predicted by Nearly Neutral theory (Kimura et al. 1963) (Supplemental Methods; Supplemental Fig. S10). Put together, these analyses suggest that our SNP calling approach performs well.
We then focused on the relationship between nucleotide diversity and conservation status. The conservation status of the species in our study ranges from Least Concern to Critically Endangered, according to the International Union for Conservation of Nature (IUCN) Red List of Threatened Species (International Union for Conservation of Nature 2010). In general, we found no obvious relationship between genetic diversity and conservation status (Fig. 2). The two most endangered primates in our study, the black and white ruffed lemur and the Coquerel's sifaka, have the highest levels of genetic diversity, 3.1 and 5.7 times that of human (synonymous site π = 0.375% from 137,141 synonymous sites, and π = 0.681% from 156,121 sites), respectively. Genetic diversity in humans (π = 0.119% from 167,756 sites) is relatively low compared to other primates. However, the genetic diversity estimate for aye-ayes is substantially lower than that of humans (π = 0.073% from 197,784 sites). Intra-individual estimates of heterozygosity (Supplemental Table S2) for wild-caught animals among each of our Coquerel's sifaka, black and white ruffed lemur, and aye-aye samples suggest that our observations for these species cannot be explained by population structure or by captive population outbreeding strategies (Supplemental Methods).
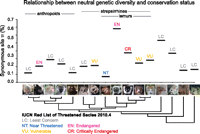
Relationship between genetic diversity and IUCN Red List endangered status. We show average pairwise nucleotide diversity, π, for synonymous sites, as an estimate of neutral levels of genetic diversity for each species. With the exception of the aye-aye, the lemurs in our study tend to have high levels of genetic diversity relative to other primates. The two species in our study considered most endangered by the IUCN, the black and white ruffed lemur and Coquerel's sifaka, have the highest levels of estimated genetic diversity among primates. The relatively low observed genetic diversity estimates for marmoset, armadillo, and opossum may not reflect those that might otherwise be obtained from natural populations, because the individuals from these species in our study are from managed laboratory research colonies.
Gene structure evolution
We proceeded to study patterns of inter-species divergence in exon usage by searching multiple alignments of all available gene sequences across the 16 species for gaps ≥ 50 bp. Since the gene sequences were assembled from RNA sequencing reads, such gaps may indicate fixed inter-species differences in gene/exon structure. Considering the large total divergence time among the species in our study, we were surprised to observe near complete conservation of exon structure among the assembled genes. Specifically, we found only 308 potential exon structure changes across the entire phylogeny. Further analysis of the de Bruijn graph data and multispecies alignments for these genes (Supplemental Methods) suggested that 304 of these gaps were either associated with evidence for alternative splicing or could be explained as alignment artifacts. For example, exon 8 of the KIAA0494 gene was missing from the assembly of all five lemur species in the study, but our analysis of the de Bruijn graph suggested that this result was due to alternative splicing rather than a fixed difference in gene structure between lemurs and other primates. For validation, we sequenced KIAA0494 exon 8 from genomic DNA of lemurs. Alignments of the RNA-seq reads from each species to the predicted exon junctions (Fig. 3A), supported by quantitative PCR experiments (Supplemental Fig. S11), show that exon 8 is usually, but not always, skipped in lemurs, in contrast to the splicing pattern observed in other species.
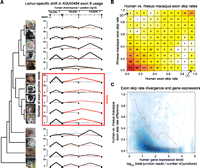
Exon structure divergence and evolution. (A) Phylogenetic shift in splicing and exon usage in the KIAA0494 gene. For each species, the y-axis depicts the number of RNA-seq reads spanning junctions of exons 6–9 (x-axis) based on human reference genome exon positions. Lines representing the number of reads spanning the exon 7 to 9 junction, observed in the overwhelming majority of inferred transcripts in lemurs but only rarely in other species, are highlighted in red. Junctions representing the most common transcript in each species are bolded. (B) Extreme divergence in exon skipping is rare. We mapped our RNA-seq read data against the human and rhesus macaque reference genome sequences to assess patterns of exon usage divergence independently of our assembled gene database (see Supplemental Methods). Shown is a heatmap depicting human vs. rhesus macaque exon skip rates. Included in this plot are all exons with at least 10 reads covering junctions, summed across all individuals of both species, and at least eight reads entering, exiting, or skipping the exon in each species. The number of exons with significant, complete divergence skip rates (i.e., exons always skipped in one species and never skipped in the other; three total), are shown by arrows in the upper left and lower right boxes of the heatmap. (C) Density plot comparing the absolute difference in human versus rhesus macaque exon skip rates to estimated expression levels (human) for the gene containing that exon, for all identified exons with evidence of alternative splicing or differential exon usage, regardless of expression level. Mean and 95th/fifth percentiles are depicted as solid and dashed red lines, respectively. Lower-expressed genes are more likely to harbor exons with larger between-species exon usage differences, reflecting either statistical artifacts or relatively lower constraint on exon structure and splicing on lower-expressed genes, or both.
Thus, using these approaches, we could find only four examples of actual fixed inter-species changes in exon structure in liver-expressed genes, in which certain exons are always skipped in at least one species but never in others. An independent analysis, restricted to species for which sequenced genomes were available, yielded similar results of strong exon structure conservation (Fig. 3B,C; Supplemental Methods). Our results suggest that the absolute gain or loss of individual, nonrepetitive exons has occurred only rarely among single-copy, intermediately and highly expressed genes in primate evolution.
Natural selection at the gene regulatory and sequence levels
Finally, we identified patterns of within- and between-species variation in the sequence and gene expression data that were consistent with the action of positive or directional selection. These analyses were based on lineage-specific ratios of the rates of nonsynonymous to synonymous substitution (dN/dS) estimated by maximum-likelihood (Yang 2007) and by testing for relatively large lineage-specific changes in gene expression levels using a Brownian motion model of gene expression evolution (e.g., Bedford and Hartl 2009), respectively (Supplemental Methods). Importantly, our sampling scheme allowed us to infer the action of natural selection on both external and ancestral branches of the phylogeny (for examples, see Supplemental Fig. S12). Overall, we identified 499 candidate genes whose rapid sequence or regulatory evolution may have played important roles in the adaptations of individual species or the ancestors of subsets of those species (see Supplemental Tables S5, S6 for a complete gene list). While it is unlikely that all 499 candidate genes were subjected to positive or directional selection at the amino acid sequence or regulatory levels, this set of candidates is likely enriched for such genes. Given the important metabolism and detoxification functions of the liver, some of these changes could reflect adaptations related to the extensive dietary diversity among the species in our study.
The relevant fossil record is particularly limited for ancestral primates (Tavare et al. 2002). Therefore, identifying conspicuous signatures of natural selection on this branch was of particular interest. For example, we found a strong signal of positive selection in the ancestral primate lineage in the gamma-glutamyl hydrolase (GGH) gene (Fig. 4A). The GGH enzyme is critical for folate metabolism and homeostasis and was previously shown to have exopeptidase activity in humans but endopeptidase activity in rodents, along with other enzymatic activity differences (Yao et al. 1996). Thus, the human-rodent functional differences in this protein might be explained by adaptive nucleotide substitutions that occurred in ancestral primate lineages. At the gene regulatory level, of the 33 top-ranked genes with relatively large ancestral primate lineage shifts in expression levels, nine are involved in peroxisome functioning, corresponding to an 18-fold enrichment over that expected by chance alone (based on Gene Ontology functional annotations; FDR = 7 × 10−9; genes PEX7, HACL1, IDE, SCP2, PEX13, LONP2, ACOX3, MGST1, and PHYH) (Fig. 4B; Supplemental Fig. S12). Peroxisomes are organelles that function in the breakdown of long-chain fatty acids by β-oxidation, the detoxification of hydrogen peroxide by catalase, the synthesis of bile acids, and cholesterol homeostasis in general (Islinger et al. 2010). We note that we were unable to identify any experimental-based evidence in the literature of peroxisomal functioning for the product of MGST; the GO functional annotation in this case might be erroneous.

Positive and directional selection in the ancestral primate branch. (A) Ratios of the maximum likelihood-estimated (Yang 2007) rates of nonsynonymous (amino acid changing) to synonymous substitution (dN/dS) for the GGH gene shown directly above each branch. Values of dN/dS > 1, highlighted in red and with the number of estimated nonsynonymous (N) and synonymous (S) substitutions shown, are consistent with the past action of positive selection on several ancestral branches of the tree. (B) Relative gene expression branch lengths estimated from 4562 genes without peroxisomal functions and from 60 peroxisomal genes, considering genes with sufficient species representation for analysis of the ancestral primate branch (see Supplemental Methods). The ancestral primate branch, highlighted in red, is relatively 4.4 times longer among the peroxisomal gene set. Nine of the 33 top-ranked genes for patterns of expression consistent with directional selection on the ancestral primate lineage function play roles in the functioning of the peroxisome, significantly more than expected by chance (FDR = 7 × 10−9). The two phylogenies are plotted such that the sums of all branch lengths, excepting the ancestral primate lineage, are equal. The relative lengths of the ancestral primate branches of each phylogeny are shown (the value for the nonperoxisomal genes phylogeny was set to 1.0).
Discussion
We collected RNA-seq data from the liver transcriptomes of multiple individuals from each of 16 mammalian species, including 12 primates, and performed de novo assembly of an average of 5721 genes per species. For many of the primate species in our study, our effort represents the first opportunity to examine nucleotide sequence, gene expression, exon structure, and genetic diversity data on a genomic scale.
We developed a new transcriptome assembly algorithm, primarily because none were available when we initiated our study. Several alternative algorithms that can be used for transcriptome assembly have been released recently, including Trans-ABySS (Robertson et al. 2010) and Oases (http://www.ebi.ac.uk/∼zerbino/oases/), which function by interpreting output from the whole genome assemblers, ABySS (Simpson et al. 2009) and Velvet (Zerbino and Birney 2008), respectively, and Trinity (Grabherr et al. 2011), which directly performs de novo transcriptome assembly. All of these algorithms, including ours, use the de Bruijn graph framework (Pevzner et al. 2001).
We have not evaluated and compared the performance of the different algorithms, as this is beyond the scope of our study. Our assembly method differs from other existing tools in several respects, as described in the Supplemental Methods. In particular, our algorithm was specifically developed to facilitate subsequent comparative genomic analyses; it is unique in its use of a sequence similarity-based comparative assembly approach, thereby establishing multispecies gene orthology as a property of the initial assembly. This aspect of our approach facilitates direct inter-species comparison of gene sequences and expression levels in an evolutionary framework.
Comparative primate genomics
Whereas previous primate comparative genomic studies have focused mainly on humans, apes, and Old World monkeys, we were able to examine the evolutionary histories of gene sequences and expression levels in the context of a relatively comprehensive primate phylogeny. Our sample of species included representatives from both primate suborders: haplorhines (humans, chimpanzees, Old and New World monkeys) and strepsirrhines (lemurs and lorises). Thus, an important property of our study design is that it provided one of the first opportunities to identify evolutionary patterns both among lemurs and in ancestral primate lineages, without the need for full genome sequences from these species.
To limit errors in the de novo assembly and orthologous gene identification process, it was necessary to discard data from duplicated genes. Additionally, we assembled genes from nonhuman species on the basis of small-scale sequence similarity to human RefSeq genes. Our analyses, therefore, were focused on single-copy genes expressed in the liver and present in the human genome. Of such genes, our set of 499 candidate genes provides an important starting point for developing hypotheses concerning the adaptive evolutionary histories of previously unstudied extant species and ancestral primate lineages. For example, our observation of an 18-fold enrichment of peroxisomal genes among those whose regulation possibly evolved under directional selection in the ancestral primate lineage may be of particular interest. While there are known functional differences between macaque and rodent peroxisomes (Hoivik et al. 2004), comparative data from dogs suggested that those differences are likely explained by derived changes in rodents, not primates (Foxworthy et al. 1990). Differences have also been observed in peroxisomal gene functioning and peroxisomal lipid metabolism between apes or humans and other primates (Somel et al. 2008; Keebaugh and Thomas 2010; Watkins et al. 2010). In contrast, our results suggest a different, major biological distinction in the regulation of peroxisome-related genes between all primates and other mammals, possibly driven by adaptive events that occurred in the ancestral primate lineage. Therefore, characterization of the functional consequences of this regulatory difference may ultimately lead to new insights concerning a little understood, but critical, time period in primate evolution.
Lemur genetic diversity
The recent history of rapid deforestation, habitat loss, and political instability in Madagascar has placed many lemurs at particular risk. Prior to this study, nuclear genetic diversity data based on nucleotide sequence data were not available for any lemur besides the aye-aye (Perry et al. 2007), although genetic diversity estimates based on microsatellite data are available for several other species (e.g., Fredsted et al. 2005; Louis et al. 2005; Lawler 2008; Pastorini et al. 2009; Quemere et al. 2010; Razakamaharavo et al. 2010). Genetic diversity data can have high importance in developing informed and effective conservation strategies, due to the association between genetic diversity and the risk of extinction (Frankham 2005; Palstra and Ruzzante 2008). For example, conservation biologists are faced with particular challenges when working with species with low genetic diversity (e.g., the cheetah) (O'Brien et al. 1983, 1985; O'Brien and Johnson 2005).
When we compared levels of neutral genetic diversity estimated from synonymous sites to the conservation status established by the IUCN for each species (International Union for Conservation of Nature 2010), we did not observe a clear pattern of association (Fig. 2). This result is not necessarily a surprise for the lemur species in this study, considering that the most extreme deforestation and habitat loss in Madagascar occurred only in the last 50 yr, likely too recent to alone induce dramatic effects on lemur genetic diversity. Yet, observations of unusually low genetic diversity for lemur species currently considered less endangered or of high genetic diversity for more endangered species may impact conservation priorities and practicalities.
Aye-ayes, considered only Near Threatened by the IUCN, have the lowest estimated genetic diversity of any species in our study. Recently, lemur conservation scientists have recommended that the status of aye-ayes be elevated to Endangered (Mittermeier et al. 2010). We would support this notion based on the combination of the genetic diversity results reported here and our still-limited knowledge of aye-aye behavior. Specifically, while aye-ayes have a broad species distribution across Madagascar, they are largely solitary, with huge individual ranges and low population densities (Ancrenaz et al. 1994)—a potentially ominous demographic profile in the face of continued forest fragmentation and already low genetic diversity.
In contrast, two of the most endangered species, the black and white ruffed lemur and Coquerel's sifaka, have the highest genetic diversity estimates of any primate—3.1 and 5.7 times that of humans, respectively. The Critically Endangered black and white ruffed lemur has experienced rapid population declines in the last quarter century due to habitat disturbance, their ecological reliance on primary forest, and extensive human hunting pressure (International Union for Conservation of Nature 2010). They have a predominantly frugivorous diet and, as major seed dispersers, could be considered critical to the long-term viability of some of Madagascar's forests. Relatively high genetic diversity should benefit black and white ruffed lemur conservation and reintroduction efforts.
Conclusion
With the advent and continued development of new sequencing technologies and assembly methods, we are able to easily characterize natural genetic and regulatory variation in a wide range of species. We are no longer limited to working on species with publicly available, sequenced genomes, which are mostly model organisms relevant to human disease studies. We, therefore, expect large and broad comparative genomic studies to become common. Such studies will increase our understanding of adaptation by allowing us to reconstruct events that occurred on ancestral lineages at unprecedented resolution. This framework also provides an opportunity to truly harness genomic studies in the service of conservation efforts (Allendorf et al. 2010; Frankham 2010).
Methods
Overview
We isolated total RNA from liver tissues harvested within 4 h of death and then stored at −80°C to preserve RNA quality. Following mRNA isolation with oligo-dT magnetic beads (Invitrogen), RNA libraries were prepared and sequenced on an Illumina Genome Analyzer IIx for 76 bp from both ends of each sequence fragment (paired-end; 2 × 76 bp), using one flowcell lane per sample. Since no sequenced genome was available for most of the species in our study, we developed a de Bruijn graph-based approach (Pevzner et al. 2001) for de novo assembly of the transcriptome of each species and simultaneous matching of gene orthologs (described in detail in Supplemental Methods). We generated multispecies alignments of the assembled gene sequences to study the evolution of gene coding sequences (Yang 2007). We also aligned the RNA-seq reads from each individual to the assembled gene sequences of each respective species for SNP analysis and estimation and evolutionary analysis of gene expression levels.
Estimating gene expression levels
To estimate the expression level of each gene, for each sample we first aligned the sequenced reads against a reference containing the sequences of the set of assembled genes for the appropriate species using BWA (Li and Durbin 2009) with default parameters, considering only uniquely mapped reads. For this analysis, we analyzed separately the two reads of each pair. To account for alternative splicing, individual reads not aligned in the first step were evaluated and scored using a gapped alignment approach (Pickrell et al. 2010), described in detail in Supplemental Methods.
For our evolutionary analysis of gene expression levels, we chose to consider orthologous gene regions across species rather than the fully assembled gene sequence from each species. That is, if the full gene sequence was not assembled for every species, then we restricted our analysis to the specific region of the gene that was commonly assembled across species. This approach makes it less likely that our inter-species comparison of gene expression levels would be affected by sequencing biases or the inclusion of alternatively spliced exons in some species only. To do so, we performed a multispecies alignment (Bradley et al. 2009) and identified the maximum orthologous region that was fully aligned across all species. Reads contributing to a gene's expression level were restricted to those falling in the maximum orthologous region, which was itself constrained to exclude noncoding regions (i.e., UTRs were not included in the gene expression analysis). We used the total number of reads mapping to the identified orthologous region of a transcript as a measure of its expression level. The data were then normalized and adjusted for GC content using procedures described in full in Supplemental Methods.
SNP identification
We aligned all reads from each individual to the database of consensus sequence transcripts that was assembled for the relevant species, using the default parameters of BWA (Li and Durbin 2009). In the final preparation step of the RNA-seq libraries, there is a PCR amplification step that uses the ligated adapter sequences as primer sites for consistent amplification. To help limit any bias from PCR amplification in the SNP identification process, we performed a filtering step to consider only one read pair from each uniquely aligned starting position and strand. Specifically, if two paired reads each had the same start position for read 1 but different start positions from read 2, then these reads were considered to have originated independently and were both kept in the analysis. When more than one paired read had identical aligned start positions (at both ends), we kept one read at random and excluded the remaining reads from further analysis. For this filtering decision, we ignored the alignment quality score, as single nucleotide differences from the consensus sequence due to true SNPs could have subtle effects on that score. That said, we did not consider any base call with a phred-scaled quality score lower than 30.
To establish SNP identification criteria, we systematically assessed genotyping accuracy as a function of multiple different per-strand coverage requirements and “SNP call definitions” based on the proportion of the most common nucleotide at each site. By “SNP call definition,” we mean the threshold at which a heterozygous site would be called, when the proportion of reads with the most common nucleotide at a given position was at or below that threshold (for reads aligning to both strands). By requiring the SNP definition to be met by reads mapped to each strand, we limited the effects of potential strand-specific sequencing biases (Nakamura et al. 2011). Examples of SNP call definitions that we considered were ≤0.6, ≤0.65, ≤0.7, ≤0.75, etc.
To determine the coverage requirement and SNP call definition thresholds, we compared SNP genotypes from the 1M-Duo Illumina SNP array platform data collected for each of the four human samples in the study to the variants inferred from the RNA-seq data using our method (Supplemental Fig. S8). Based on this analysis, we chose to assess all sites covered by a minimum of 15 sequence reads per strand (minimum of 30 total reads), and, of such sites, we classified as heterozygous those for which the proportion of the most common nucleotide was ≤0.7 on each strand. This approach for SNP calling is generally similar to that which we previously used with genomic DNA sequencing data and found to result in highly accurate SNP identification (Perry et al. 2010).
Finally, we performed a subsampling analysis with the reads from each individual. For this analysis, reads were randomly distributed into two subsets. SNPs were identified from each subset of the data using the coverage and SNP call definition threshold criteria described above. We then determined the consistency of SNP inferences in the subsampled data within each individual. We removed three samples—one chimpanzee and two aye-ayes—from further SNP analysis due to relatively low concordance in heterozygous site identification in the subsample analysis (Supplemental Table S4).
For each species, we estimated genotypes for all sites with sufficient coverage for SNP identification in all individuals (n = 2 for armadillo and aye-aye, n = 3 for chimpanzee, n = 4 for all other species). We classified all heterozygous positions as well as any sites with homozygous differences between individuals as SNPs. Species-level estimates of genetic diversity π (average pairwise genetic distance) and θ (sample-size corrected proportion of segregating sites) were computed for all genes with at least 100 sites with sufficient coverage for SNP identification in each individual of that species and are provided in Supplemental Table S1.
Data access
Paired end 76 × 76-bp sequencing data obtained in this study have been submitted to the NCBI Sequence Read Archive (SRA) (http://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi) under accession number SRA046085. The transcript assembly code used in this study is available at http://pritch.bsd.uchicago.edu/software.html. The full database of assembled gene sequences, full gene multispecies alignments, orthologous coding region multispecies alignments, lineage-specific dN/dS results, normalized gene expression estimates, log likelihood ratios for lineage-specific expression level changes, and the identified SNPs and genotype data for each species are available as a Supplemental Database file on the Genome Research website and at http://giladlab.uchicago.edu/data.html.
Acknowledgments
We thank the Duke Lemur Center, National Disease Research Interchange, Yerkes National Primate Research Center, Southwest Foundation for Biomedical Research, Alpha Genesis, David Fitzpatrick, Julie Heiner, Matt Dean, Michael Nachman, and Richard Truman for providing the samples used in this study. The lemur, loris, and bushbaby photographs in the phylogeny figures were provided by David Haring, Duke Lemur Center. Marmoset, treeshrew, mouse, armadillo, and opossum photographs are from Wikimedia Commons. We thank Z. Gauhar, P. Gagneux, E. Louis, and O. Ryder for useful discussions and/or comments on the manuscript. This work was funded by the Howard Hughes Medical Institute to J.K.P., and by NIH grant GM077959 to Y.G. G.H.P. was supported by N.I.H. fellowship F32GM085998.
Footnotes
-
↵9 Corresponding authors.
E-mail ghp3{at}psu.edu.
E-mail pritch{at}uchicago.edu.
E-mail gilad{at}uchicago.edu.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.130468.111.
- Received August 10, 2011.
- Accepted December 2, 2011.
- Copyright © 2012 by Cold Spring Harbor Laboratory Press
Freely available online through the Genome Research Open Access option.








