
Genetic variation in radiation-induced cell death
- Denis A. Smirnov1,
- Lauren Brady2,
- Krzysztof Halasa3,
- Michael Morley3,
- Sonia Solomon3 and
- Vivian G. Cheung1,3,4,5,6
- 1Howard Hughes Medical Institute, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA;
- 2Cell & Molecular Biology Graduate Program, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA;
- 3The Children's Hospital of Philadelphia, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA;
- 4Department of Genetics, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA;
- 5Department of Pediatrics, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA
Abstract
Radiation exposure through environmental, medical, and occupational settings is increasingly common. While radiation has harmful effects, it has utility in many applications such as radiotherapy for cancer. To increase the efficacy of radiation treatment and minimize its risks, a better understanding of the individual differences in radiosensitivity and the molecular basis of radiation response is needed. Here, we integrated human genetic and functional genomic approaches to study the response of human cells to radiation. We measured radiation-induced changes in gene expression and cell death in B cells from normal individuals. We found extensive individual variation in gene expression and cellular responses. To understand the genetic basis of this variation, we mapped the DNA sequence variants that influence expression response to radiation. We also identified radiation-responsive genes that regulate cell death; silencing of these genes by small interfering RNA led to an increase in radiation-induced cell death in human B cells, colorectal and prostate cancer cells. Together these results uncovered DNA variants that contribute to radiosensitivity and identified genes that can be targeted to increase the sensitivity of tumors to radiation.
Radiation exposure is increasingly common. Medical diagnostic tools such as the X-ray and computed tomography imaging expose patients to ionizing radiation (IR), which can cause DNA damage and increase one's risk of malignancies. However, these radiation-based devices have greatly improved the diagnosis and treatment of many diseases. Thus, the solution is not to eliminate radiation exposure but to protect individuals who are the most sensitive to radiation and to minimize dose and exposure to all individuals (Barnett et al. 2009).
Pharmacogenetics has made significant contributions in maximizing therapeutic gains while minimizing side effects; however, those studies have focused mainly on chemicals as therapeutics and have not included radiation. The exclusion of radiation in pharmacogenetics is not surprising since radiation presents a unique set of challenges. Most people are exposed to radiation in nonmedical settings in addition to medical exposures, thus complicating the monitoring of exposure. Safety trials of radiation are impossible given its known toxic effects. Third, most drugs are developed for one or a few diseases. In contrast, radiation is used in a wide range of treatment; over 50% of all cancer treatment protocols include the use of radiation. Target tissues range from skin to skeletal muscles and bone marrow; each tissue type has special cellular components that influence the absorbed radiation dose, and manifests side effects differently.
In recent years, cell-based and genetic studies have improved our understanding of the molecular and genetic basis of radiosensitivity by identifying the genes and pathways that are involved in radiation response (Amundson et al. 2001, 2008; Smirnov et al. 2009; Efimova et al. 2010; Niu et al. 2010; Noon et al. 2010). In this study, we focused on radiation-induced cell death. We measured changes in gene expression and quantified cell death in irradiated B cells from 99 normal individuals and found extensive individual variation in these measurements. We took advantage of this variation and mapped the sequence variants that influence responses to radiation. We also identified radio-responsive genes that regulate cell death. By using RNA interference in human B cells and cancer cells, we showed that the knockdown of genes such as INSIG1 and FAM65B increased radiation-induced cell death. These results promote the development of individualized radiotherapy that accounts for patient sensitivity to radiation and lowers necessary treatment dose by sensitizing the tumors to radiation.
Results
Individual variability in radiation-induced apoptosis
To assess cellular response to radiation, we exposed B cells from 99 individuals to 10 Gy of IR and measured cell death by two assays: caspase 3/7 levels and cytotoxicity. These measurements were made before radiation and 24 h after exposure. Radiation induced cell death in all individuals; however, the level of cell death showed extensive individual variability. Some individuals showed only minimal increases in caspase activation, while others showed as much as a 700% increase in caspase activity (average, 294%; range, 121%–722%) (Fig. 1A). Similar results were obtained from the cytotoxicity assays; the correlation between the two measurements was high (r = 0.62, P < 10−7).
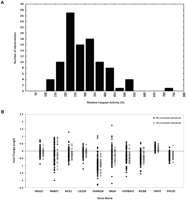
Variability in cellular responses to radiation exposure. (A) Relative caspase 3/7 activity varies in cultured B cells from 99 unrelated individuals following exposure to IR. For each individual, relative caspase activity is a mean of two independent measurements. (B) Fold changes in 10 radiation-responsive genes at 2 h following radiation exposure; data for the 99 unrelated individuals in this study are shown as black diamonds, data for the 30 unrelated individuals from our previous study (Smirnov et al. 2009) as gray diamonds. TNIP2 and PHF20 are examples of radiation-responsive genes that show less individual variability.
Individual variation in gene expression response to radiation
Gene expression underlies phenotypic manifestations like cell death. To gain insights into radiation response, we used microarrays to measure gene expression levels in B cells from the same 99 individuals before and at 2 and 6 h after exposure to IR (10 Gy). We defined genes as IR-responsive if they showed a 1.5-fold or more increase or decrease in gene expression at 2 h and/or 6 h following radiation exposure in at least 20% of the individuals. Following these criteria, 3117 genes (represented by 3839 probes on the microarray) were found to be IR responsive. Of these genes, 41% were induced and 59% were repressed at 2 h following radiation, and similarly, 51% were induced and 49% were repressed at 6 h following radiation. Among the IR responsive genes are ATF3, GDF15, CDKN1A, GADD45A, CDC25A, and JUN. Some of these genes were previously identified by us and others to be involved in the radiation response (Amundson et al. 2001; Jen and Cheung 2003; Smirnov et al. 2009; Turtoi et al. 2010). More than 2000 genes (2464 genes) (see Supplemental Table S1; Supplemental Fig. S1) were identified as radiation responsive in this and our previous studies (Smirnov et al. 2009). These results provide strong evidence that these few thousand genes are important in the response of human cells to radiation. Table 1 gives examples of genes that showed the greatest fold change in expression at 2 and 6 h following radiation exposure.
Examples of genes whose expression levels were induced or repressed in cultured B cells of 99 individuals at 2 h (A) and 6 h (B) after IR exposure
While there are a large number of genes that are activated or repressed following radiation exposure, individuals differ in their expression responses to radiation. The extent of the changes in gene expression varies extensively among individuals. Among these variable genes are well-characterized transcriptional regulators, including ID2, the inhibitor of DNA binding (average fold change, 2.1; range, −2.6 to 12), an oncogene MDM2 (average fold change, 3.1; range, −1.8 to 11), and a gene involved in cell cycle regulation that encodes a serine/threonine kinase, PLK1 (average fold change, −2.9; range, −8.8 to 1.4). Previously, we reported individual variability in the gene expression response to radiation when we had studied cells from 30 individuals (Smirnov et al. 2009). Here, we replicated those findings with 99 individuals. Figure 1B shows examples of genes with similar patterns of variability in the two studies. These genes were chosen as examples because they are the focus of this study, not just for their patterns of expression. The cells from these two studies were processed and analyzed independently; the highly similar patterns increase our confidence in the observations.
Radiation-responsive genes and correlation with cell death
Since individuals differ in both radiation-induced gene expression and cell death, we reasoned that some of the variability in expression response may contribute to individual differences in cell death. To identify genes that play a role in radiation-induced cell death, we looked for genes whose changes in expression levels correlated with changes in caspase 3/7 activity across the 99 individuals. Our sample size enabled us to obtain a good estimate of the correlations between gene expression and cell death. As an initial step in our analysis, we set the correlation coefficient threshold at 0.2, in order to include many genes for the subsequent follow-up. At this threshold, changes in the expression levels of 335 of the IR-responsive genes at 2 and/or 6 h following radiation were found to correlate with relative caspase activity (Supplemental Table S2). There are 26 genes that showed significant correlation with relative caspase activities at both time-points. We do not expect much overlap between these two time-points since radiation response is highly dynamic; by 6 h, many of the early response genes have already returned to their baseline levels; for example, RIC8B, JUN, and NFKBIA were activated soon after radiation, but their expression levels had returned to baseline by the 6-h time-point. In contrast, genes such as DUSP4, INHBE, and CEBPB were late responders. Gene ontology analysis (Ashburner et al. 2000) shows that the genes correlated with caspase activity are significantly enriched (Pc < 0.001) for functional roles in transcription and cellular response to stress. Although genes grouped as “cell death” were not significantly enriched after correction for multiple testing in the ontology analysis, many of the genes such as BBC3, BCL2, and JUN in the transcription and response to stress categories play critical roles in the cell death pathways.
DNA variants that affect radiation-induced gene expression and cell death
Next, to study individual variability in expression and cellular responses to radiation, we carried out genetic mapping to identify the sequence variants that contribute to the variation. We focused on the radiation response of the 335 genes that are correlated with relative caspase activity following IR exposure. We began with genome-wide linkage analyses in 30 extended pedigrees. By using microarrays and B cells from members of 30 CEPH families, we obtained expression levels of these genes at baseline and 2 and 6 h after radiation exposure. With these expression measurements, we calculated the fold change of genes at the two time-points. Then, we treated the 2-h and 6-h expression changes as quantitative phenotypes and combined them with SNP genotypes of the same individuals in genome-wide linkage scans (Haseman and Elston 1972; Shete et al. 2003). At a threshold of LOD > 3.4 (genome-wide corrected significance level of ∼0.05), the expression response of 94 phenotypes (53 two-hour, 41 six-hour phenotypes) showed significant evidence of linkage (Supplemental Table S3). Some of the phenotypes segregated with two or more regions in the genome; in total, there are 108 candidate regions that reached our threshold of (corrected P) Pc < 0.05. Many of these results greatly exceed the significance threshold. Figure 2 shows examples of the linkage plots. We then examined the linkage results to identify the candidate locations of the polymorphic regulators for these radiation-induced expression phenotypes. Only one of the linkage peaks, that for KLF12, was close (within 5 Mb) to the target genes, suggesting a cis-acting regulation; the remaining linkage peaks were far away from the genomic locations of the target genes, suggesting that they harbor regulators that act in trans to influence the expression of the target genes (Rockman and Kruglyak 2006). For these distal linkages, 105 out of 107 peaks were found on different chromosomes as the target genes. Results from our previous study (Smirnov et al. 2009) and those from model organisms (Li et al. 2006; Smith and Kruglyak 2008) have shown that the individual variation in the responses to cellular perturbations is mostly due to polymorphic trans-acting factors.

Linkage results of four IR-responsive genes whose expression levels correlate with cell death. (A–D) Chromosomal location is plotted on the horizontal axis with evidence of linkage represented by P-value (−log10) on the vertical axis.
Family-based and population association analyses
To confirm the linkage results and fine map the candidate regulatory regions, we carried out family-based and population-based association analyses.
Proximal (likely cis) linkage
We confirmed the proximal linkage of KLF12 expression response by association analysis. Quantitative transmission disequilibrium test (QTDT) (Abecasis et al. 2000a,b) showed significant evidence of linkage and association of SNPs near KLF12 (rs9318214, P = 0.01) with its radiation-induced expression response. By using data from 57 unrelated individuals, we tested SNPs in KLF12 for allelic association with its expression response after radiation. Despite the small sample size, a significant association (P = 0.01, rs9573321) with the expression of KLF12 was detected, thus further supporting the linkage finding that the response of KLF12 to radiation is cis-regulated. The result suggests that the effect of the cis-acting polymorphism on the expression response of KLF12 is relatively large; otherwise, we would not be able to detect it with the small sample size.
Distal (trans) linkages
We followed up results of the 94 phenotypes with significant distal linkage findings. Thirteen of the phenotypes had two or more significant linkage regions. We carried out QTDT with SNPs in these 107 distal linkage peaks (t > 4). The candidate regions usually include several genes; we examined only the candidates that are expressed in B cells. We tested sequence variants in and 10 kb upstream of and downstream from these expressed genes for evidence of linkage and association with their candidate target genes in members of the 30 CEPH families. From the 107 linkage peaks, we found significant linkage and association (P < 0.01, FDR < 0.5%) between sequence variants in 48 genes with 46 radiation-induced expression phenotypes. At a less stringent threshold of P < 0.05 (FDR < 1%), sequence variants in 83 regulators are linked and associated with the expression response of 73 genes. The transcription regulator BZW2 influences the radiation response of two different target genes: MFHAS1, which encodes a leucine-zipper protein; and BRIP1, a RNA helicase that interacts with BRCA1. Overall, we uncovered 84 polymorphic regulator–target gene pairs. The 20 most significant regulator–target gene pairs are shown in Table 2 (for a complete list, see Supplemental Table S2).
Examples of polymorphic regulator–target gene pairs identified in linkage and association analysis in members of 30 CEPH families
Population association of target gene expression
To further confirm our mapping results, we tested DNA variants in the 83 regulators (P < 0.05, QTDT) for allelic association with expression response of their target genes following radiation using data from 57 unrelated individuals. By use of regression analysis, we determined the association of the radiation-induced expression response with the genotypes of the regulators. Despite a small sample size, SNPs in 20 of 83 regulators showed a significant association with expression response of their target genes (nominal P < 0.05) (Supplemental Table S3). We then estimated the phenotypic variation explained by these variants by calculating the R2 of the linear regression analysis. The results showed that the polymorphisms in these regulators have a relatively large influence on individual differences in expression response to radiation. Polymorphisms in seven of the 20 regulators explain 10% or more of the variability (mean, 10%, range, 7%–16%).
Gene knockdown leads to increased radiation-induced apoptosis
Genetic analyses allowed us to identify the sequence variants that contribute to individual variation in response to radiation. Next we carried out gene knockdown to study the genes whose radiation-induced expression levels correlate with cell death. Our goal is to determine if these genes not only correlate with but also regulate cell death.
For these studies, we focused on the 205 genes whose changes in expression levels following radiation were negatively correlated with caspase activity. We chose these genes because the negative correlations suggest that if we decrease their expression, we could increase radiation-induced cell death. To test this, we knocked down the expression of 12 of these genes in B cells from three individuals using small interfering RNAs (siRNAs) and then measured radiation-induced cell death. These genes were picked because they span a range of correlation thresholds and because of practical reasons such as availability of reagents (siRNAs). Following siRNA knockdown of five genes (RIC8B, HSPBAP1, FAM65B, INSIG1, and MGA), we observed a significant increase (P < 0.05) in caspase 3/7 levels (Fig. 3A; Supplemental Table S4). For the remaining genes, the knockdown of NCK1, TSPAN12, and KDM4B in human B cells also led to an increase in radiation-induced cell death; however, the extent of increase did not reach statistical significance.

Increased radiation-induced cell death following gene knockdown. After siRNA transfection and exposure to radiation, various measurements were made in different cell types: (A) relative changes in caspase 3/7 activity in cultured B cells; (B) relative changes in cellular viability by MTS assay in colorectal cancer cells, HT29 (black bars), and in prostate cancer cells, PC3 (white bars); and (C) relative changes in cellular viability by colony-formation assay in colorectal cancer cells, HT29. Data are shown as mean ± SEM of two or more independent transfections (for raw data, see Supplemental Tables S4, S5).
To test whether the increase in radiosensitivity following siRNA knockdown in B cells is also observed in cancer cells, we knocked down expression of the five genes in colorectal cancer (HT29) and prostate cancer (PC3) cell lines. These cancer cells are more radio-resistant than B cells so instead of using caspase assays to measure cell death, we used the MTS assay, an enzyme-based test of viability. In the colorectal cancer cells (HT29), knockdown of all five genes resulted in a decrease in viability or an increase in cell death (Fig. 3B; Supplemental Table S4). In the prostate cancer cell line PC3, siRNA against three (RIC8B, HSPBAP1, and INSIG1) of the five genes also led to a decrease in viability (Fig. 3B; Supplemental Table S4).
In addition to using the MTS assay to assess cell death, we tested the effect of gene knockdown using a colony formation assay. In colorectal cancer cells (HT29), siRNA silencing of four genes (HSPBAP1, FAM65B, INSIG1, and MGA) led to a decrease in ability of the cells to form colonies (Fig. 3C; Supplemental Table S5). These results show that we can increase the sensitivity of normal and cancer cells to radiation by silencing these radio-responsive genes.
DNA variants in genes that regulate cell death
To further examine the five genes whose expression responses correlate with cell death in irradiated cells and, when knocked down, increase cellular radiosensitivity, we turned to the genetic mapping data. Those results identified the polymorphic regulators of radiation response for four of the five genes (Table 3). Sequence variants in NTHL1, CD226, C17orf85, and GALNT7 influence the radiation-induced expression of RIC8B, FAM65B, INSIG1, and MGA, respectively. Isoforms of these regulators have differential effects on their target genes and subsequently on radiation-induced cell death.
Polymorphic regulators of genes that affect radiation-induced cell death
Discussion
Radiation exposure is common and has significant health impact. Since the 1940s, by use of population studies, Stern (Spencer and Stern 1948), Neel (Neel 1958), and others have shown the effects of radiation on human health. More recently, others have documented the consequences of radiation exposure in medical settings (Berrington de Gonzalez and Darby 2004; Brenner and Hall 2007). Molecular studies have provided mechanistic details on the cellular effects of radiation by identifying pathways, such as TP53, that play a role in radiation-induced cell death (Kuerbitz et al. 1992; Clarke et al. 1993). Despite the potential harm, radiation has contributed significantly to improving the diagnosis and treatment of diseases through technologies such as computer tomography and radiotherapy for cancer. Faced with this dichotomy, the field will continue to use radiation, but it needs to identify ways to minimize harm.
To begin to develop approaches that will maximize therapeutic gains and minimize side effects, we focused on identifying the genetic contributors to radiosensitivity and ways to alter the sensitivity of cells to radiation. Our ultimate goal is to determine a person's sensitivity to radiation in order to assess exposure risks, and to develop ways to minimize the dose requirement by sensitizing tumor cells to radiation. Traditionally, genetic studies of radiosensitivity have been difficult because of the heterogeneous manifestations from rash to vasculitis and tumorigenesis. The genetic etiologies are also very complex. To study such a complex system, one can either simplify the genetics by looking at model organisms with smaller and simpler genomes or decrease the complexity of the phenotypes. Our approach is to maintain the genetic complexity but simplify the phenotypes by looking at the intermediate steps of response, in particular gene and cellular changes. We posit that since we are interested in the genetics, we should not simplify it; rather we can examine less complex phenotypes that can be measured reproducibly in a large number of individuals. Gene expression and various cell death measurements such as caspase activities are attractive because we and others have already shown that they are genetically regulated (Cheung and Spielman 2002; Schadt et al. 2003; Smirnov et al. 2009) and because advances in genome technology have allowed high-throughput analyses.
Previously, we studied the genetic regulation of radiation-responsive genes without focusing on genes with specific functions (Smirnov et al. 2009). Here, since our interest is the genetics of radiation-induced cell death, we focused on those genes that correlate with cell death. Although in the previous study, we identified sequence variants that influence genes in cell death pathways, in this study, using a more targeted approach, we uncovered over 80 polymorphic regulators for genes involved in cell death.
Examining these regulators have improved our understanding of the pathways involved in radiation-induced cell death. First, we looked for literature evidence for roles in cell death pathways. Among the 83 regulators, 52 have literature support for involvement with cell death. Often the functions of one or both genes in the regulatory pairs that we identified are unknown or their roles in radiation response are poorly characterized. For each regulatory pair, if we know the function of one gene in radiation response, we can use that information to infer the function of its partner. For example, we identified XAF1 as a regulator of CLDN10 and identified WWOX as the regulator of ZC3H12A. XAF1 mediates cell death by inhibiting the caspase-activity of XIAP (Liston et al. 2001). The role of XAF1's target gene, CLDN10, in radiation response and the basis of the correlation between CLDN10 expression and cell death are unknown. Our data allowed us to infer that CLDN10 plays a role in radiation-induced cell death through the caspase-mediated pathway. Similarly, WWOX and its target gene ZC3H12A have been implicated in cell death, but their roles in radiation response are poorly known. WWOX is involved in TNF-mediated apoptosis (Chang et al. 2003, 2005); we infer that the correlation of ZC3H12A with cell death may also be driven by tumor necrosis factors.
In most of the analyses, we studied one gene at a time; to get a more integrated view of how regulators interact with each other to mount response to radiation, we carried out a network analysis. With the Ingenuity system, we built a network that included 30 of the regulators (Supplemental Fig. S2). The analysis allows us to connect these regulators with genes such as TP53 and VEGF that are well known to play a role in radiation response. Identifying the regulatory relationships and investigating the interactions among the regulators are initial steps toward understanding how human cells coordinate genes and pathways to respond to radiation exposure.
In addition to our genetic mapping analyses, we also examined the data for genes whose expression levels correlated with cell death. We found a few hundred such genes and focused on the genes whose expression negatively correlated with cell death. We reasoned that decreasing the expression levels of these genes may increase the radiosensitivity of cells. By knocking down expression in some of these genes, first in B cells and then in colorectal and prostate cancer cells, we increased radiation-induced cell death as expected. These genes have not been previously identified as radiation-responsive; in this study, the large sample size allowed us to identify them. They function in diverse ways, from RIC8B in G-protein signaling (Nagai et al. 2010) and INSIG1 in SREBP-mediated signaling (Yang et al. 2002) to HSPBAP1, which plays a role in stress response by binding to heat shock proteins (Jiang et al. 2001). Our finding reveals the diverse pathways used by cells to induce cell death following radiation exposure.
In conclusion, by focusing on individual variation and by using genetic and molecular methods, we uncovered genes that are involved in radiation-induced cell death and sequence variants that influence these pathways. Extension of such systems approaches will improve our basic understanding of radiosensitivity, which can then be translated into clinical practice.
Methods
Samples
For cell death and gene expression analyses, we used immortalized/cultured B cells from 99 unrelated individuals in the CEPH collection (Dausset et al. 1990). The immortalized B cells were seeded at a density of 5 × 105 cells/mL in RPMI 1640 with 15% fetal bovine serum, 2 mM L-glutamine, and 100 U/mL penicillin-streptomycin. The colorectal cancer cell line HT29 and prostate cancer cell line PC3 were cultured in RPMI 1640 with 10% fetal bovine serum, 2 mM L-glutamine, and 100 U/mL penicillin-streptomycin. The cancer cell lines were passaged every 3–4 d, with subcultivation ratios ranging from 1:4–1:6.
For genetic analyses, we studied individuals from 30 CEPH families (CEPH 1333, 1341, 1346, 1362, 1408, 1416, 1420, 1421, 1423, 1424, 1444, 1447, 1451, 1454, 1582, 1331, 1332, 1344, 1347, 1349, 1354, 1356, 1357, 1358, 1413, 1418, 1456, 1458, 1463, 1477). On average, each family has eight offspring per sibship (range, 7–9).
Gene expression experiments
For gene expression studies, immortalized B cells were seeded at 5 × 105 cells/mL 18 h before exposure to IR (10 Gy in a 137Cs irradiator). Many genes show dose-dependent changes in gene expression (see Supplemental Fig. S5). At 10 Gy, the changes in gene expression are often the largest; thus this is an optimal dose for studying individual differences in gene expression.
Cells were harvested prior to radiation and at 2 and 6 h after IR. RNA was extracted from the cells, labeled, and hybridized onto Affymetrix Human U133A 2.0 Arrays. For our genetic analysis, we used a random number generator to determine the order in which the cells were grown and array hybridizations were carried out; cells from family members were not processed together except by chance. For each sample, the baseline, 2-h IR, and 6-h IR were processed together. The cRNA samples were prepared in a total of eight batches (about 96 samples per batch). Hybridizations were performed in batches of 48 samples. The samples from unrelated individuals were processed in three batches of about 90 samples per batch. Expression intensity was scaled to 500 using the global scaling method implemented in the Expression Console software from Affymetrix and log transformed. We defined IR-responsive genes as those that showed at least a 1.5-fold change in gene expression levels in 20% or more of unrelated individuals at any time-point. Expression data from the 15 CEPH families (CEPH 1333, 1341, 1346, 1362, 1408, 1416, 1420, 1421, 1423, 1424, 1444, 1447, 1451, 1454, 1582) were those from our previous study (Smirnov et al. 2009). Gene ontology analysis was conducted using DAVID Bioinformatics Resources 6.7, with functional annotation clustering.
Analysis of linkage and association
Genetic analysis was performed as in our previous studies (Smirnov et al. 2009; Cheung et al. 2010). Briefly, multipoint genome-wide linkage analysis was done by SIBPAL in S.A.G.E (Haseman and Elston 1972) using the W4 option (Shete et al. 2003). SIBPAL determines evidence for the linkage at each SNP from regression of the phenotype difference between siblings on the estimated proportion of marker alleles shared identical-by-descent between siblings; the result is reported as a t-value with corresponding significance. Family-based association analysis with SNPs near and within the target genes or candidate regulators was carried out using QTDT (Abecasis et al. 2000a,b). We used the orthogonal (ao) model and variance component options (wega). Only candidate regulators with an 80% present call from the Affymetrix microarrays were defined as expressed and used in association analyses. For population association analysis, expression phenotypes or relative caspase activities were used as dependent variables and regressed on SNP genotypes (coded 0, 1, or 2). R2 was estimated for each phenotype–SNP combination as the ratio of the regression sum of squares to the total sum of squares. Gene network analysis was conducted using Ingenuity Systems software, IPA version 9.0 (www.ingenuity.com).
Caspase and cytotoxicity assays
Cultured B cells from 99 unrelated individuals were seeded at a density of 5 × 105 cells/mL and irradiated at 10 Gy in a 137Cs irradiator. The cellular response to radiation exposure was measured 24 h after irradiation using the Caspase-Glo 3/7 assay (Promega) or the MultiTox-Fluor Multiplex Cytotoxicity assay (Promega). These measurements were compared to those in cells that received mock radiation treatment. At 24 h, we observed increased cell death after irradiation, but at later time-points, we begin to see similar Caspase 3/7 activity in nonirradiated control cells, as the cells are likely becoming too confluent (Supplemental Fig. S3).
HT29 and PC3 cells were analyzed using CellTiter 96 Aqueous One Solution Cell Proliferation Assay (MTS) (Promega) 1 wk following irradiation at 5 Gy. To perform colony formation assay, HT29 cells at 500 cells/well were seeded into six-well plates and irradiated with 5 Gy. Colonies were counted 2 wk after irradiation using the Molecular Imager ChemiDoc XRS+ System (Bio-Rad). To measure the effect of siRNA knockdown on radiosensitivity, cells were transfected using methods described below and incubated for 72 h prior to irradiation.
Knockdown of candidate regulators
Two or more cultured B-cell lines were transfected with Accell siRNAs (Dharmacon) directed against candidate genes or a nontarget control according to the manufacturer's instructions. For each regulator, we used a pool of siRNAs to target the regulators in order to minimize off-target effects (Myers et al. 2006). Cancer cell lines were transfected with Silencer Selected siRNAs (Applied Biosystems) against candidate genes or a nontarget control using the RNAiMAX reagent according to the manufacturer's instructions (Invitrogen). RNA was harvested 72 h after transfection to assess knockdown efficiency (Supplemental Fig. S4). Effect of siRNA on gene expression was analyzed by quantitative PCR (7900HT Analyzer, Applied Biosystems). Expression of beta-actin was used as a control for normalization, and changes in expression were calculated relative to cells transfected with nontarget control siRNA. Sequences of PCR primers and siRNAs are presented in Supplemental Table S6.
Data access
The microarray data from this study have been deposited in the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE26835.
Acknowledgments
We thank Dr. Alan Bruzel for suggestions and comments and Derek Oldridge for discussion and help with data analysis. This work is supported by grants from the National Institutes of Health (to V.G.C.) and funds from the Howard Hughes Medical Institute.
Footnotes
-
↵6 Corresponding author.
E-mail vcheung{at}mail.med.upenn.edu.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.122044.111.
- Received February 9, 2011.
- Accepted June 28, 2011.
- Copyright © 2012 by Cold Spring Harbor Laboratory Press








