
Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer
- Han Liang1,9,11,
- Lydia W.T. Cheung2,9,
- Jie Li2,
- Zhenlin Ju1,
- Shuangxing Yu2,
- Katherine Stemke-Hale2,
- Turgut Dogruluk3,
- Yiling Lu2,
- Xiuping Liu4,
- Chao Gu2,
- Wei Guo2,
- Steven E. Scherer5,
- Hannah Carter6,
- Shannon N. Westin7,
- Mary D. Dyer2,
- Roeland G.W. Verhaak1,
- Fan Zhang2,
- Rachel Karchin6,
- Chang-Gong Liu4,
- Karen H. Lu7,
- Russell R. Broaddus8,
- Kenneth L. Scott3,
- Bryan T. Hennessy2,10 and
- Gordon B. Mills2
- 1Department of Bioinformatics and Computational Biology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA;
- 2Department of Systems Biology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA;
- 3Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, Texas 77030, USA;
- 4Department of Experimental Therapeutics, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA;
- 5Human Genome Sequencing Center, Baylor College of Medicine, Houston, Texas 77030, USA;
- 6Department of Biomedical Engineering and Institute for Computational Medicine, Johns Hopkins University, Baltimore, Maryland 21205, USA;
- 7Department of Gynecologic Oncology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA;
- 8Department of Pathology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA
-
↵9 These authors contributed equally to this work.
Abstract
Endometrial cancer is the most common gynecological malignancy, with more than 280,000 cases occurring annually worldwide. Although previous studies have identified important common somatic mutations in endometrial cancer, they have primarily focused on a small set of known cancer genes and have thus provided a limited view of the molecular basis underlying this disease. Here we have developed an integrated systems-biology approach to identifying novel cancer genes contributing to endometrial tumorigenesis. We first performed whole-exome sequencing on 13 endometrial cancers and matched normal samples, systematically identifying somatic alterations with high precision and sensitivity. We then combined bioinformatics prioritization with high-throughput screening (including both shRNA-mediated knockdown and expression of wild-type and mutant constructs) in a highly sensitive cell viability assay. Our results revealed 12 potential driver cancer genes including 10 tumor-suppressor candidates (ARID1A, INHBA, KMO, TTLL5, GRM8, IGFBP3, AKTIP, PHKA2, TRPS1, and WNT11) and two oncogene candidates (ERBB3 and RPS6KC1). The results in the “sensor” cell line were recapitulated by siRNA-mediated knockdown in endometrial cancer cell lines. Focusing on ARID1A, we integrated mutation profiles with functional proteomics in 222 endometrial cancer samples, demonstrating that ARID1A mutations frequently co-occur with mutations in the phosphatidylinositol 3-kinase (PI3K) pathway and are associated with PI3K pathway activation. siRNA knockdown in endometrial cancer cell lines increased AKT phosphorylation supporting ARID1A as a novel regulator of PI3K pathway activity. Our study presents the first unbiased view of somatic coding mutations in endometrial cancer and provides functional evidence for diverse driver genes and mutations in this disease.
Endometrial cancer is the most common gynecological malignancy and the fourth most common cancer among women in Western countries, with more than 280,000 cases occurring annually worldwide (Ferlay et al. 2010). Although the prognosis is favorable for patients identified with early-stage disease, the outcomes for patients with metastatic or recurrent endometrial cancer remain abysmal, with median survival of <1 yr. For effective cancer prevention and treatment, it is necessary to identify critical genetic changes that initiate endometrial cancer and contribute to its progression. Over the last decade, multiple groups have identified common somatic mutations of several important genes in endometrial cancer (Dedes et al. 2011). For example, PIK3CA mutations occur in ∼30% of endometrial cancers, with the mutation frequency varying according to histological grade (Oda et al. 2005; Velasco et al. 2006; Catasus et al. 2008; Miyake et al. 2008; Hayes et al. 2009; Cheung et al. 2011). PTEN, a major negative regulator of the phosphatidylinositol 3-kinase (PI3K) pathway, is frequently mutated in endometrial tumors (Risinger et al. 1998; Mutter et al. 2000; Sun et al. 2001; Salvesen et al. 2004). Other cancer genes such as TP53, KRAS, AKT, and CTNNB1 are also frequently mutated in this disease (Lax et al. 2000; Schlosshauer et al. 2000; Moreno-Bueno et al. 2002; Stefansson et al. 2004; Byron et al. 2008; Jia et al. 2008; Catasus et al. 2009; Shoji et al. 2009). More recently, we have demonstrated that PI3K pathway aberrations occur in >80% of endometrial cancers, often with co-mutations of multiple pathway members (Cheung et al. 2011). Although these studies provide important insights into the molecular basis of endometrial cancer, they have primarily focused on a limited set of well-known cancer genes. To date, a comprehensive view of the mutation spectrum in this disease has not been achieved.
Recent advances in next-generation sequencing technology have enabled the unprecedented characterization of a full spectrum of somatic alterations in cancer genomes (Mardis 2011). In particular, through target enrichment, whole-exome sequencing represents a cost-effective strategy to identify mutations in protein-coding exons in the human genome. In the past several years, this approach has been successfully applied to several human cancers (Cancer Genome Atlas Research Network 2008, 2011; Jones et al. 2010; Gui et al. 2011; Wang et al. 2011). Given the large numbers of somatic mutations typically detected by this approach, a key challenge in the downstream analysis is to distinguish “drivers” that functionally contribute to tumorigenesis from “passengers” that occur as the consequence of genomic instability. So far, most of the published studies have characterized at most one or two genes of interest, leaving the vast majority of mutated genes unexplored.
Here we implemented a systems-biology approach to identify driver cancer genes in endometrial cancers. We first performed whole-exome sequencing on 13 endometrial cancers along with matched normal samples to detect somatic alterations with high precision and sensitivity. For validated mutated genes, we combined bioinformatics prioritization with high-throughput screening of short-hairpin RNA (shRNA) and overexpression constructs in a highly sensitive cell viability assay to identify candidate driver cancer genes. The impact of candidate driver genes from the initial screen was then assessed in endometrial cancer cell lines by small interfering RNA (siRNA) knockdown. Finally, we focused on one candidate driver gene, ARID1A, for mechanistic investigation. Figure 1 shows an overview of our approach. Our study thus provides an integrated approach to identifying and characterizing novel cancer genes that have not been implicated in endometrial tumorigenesis.
Overview of our systems-biology approach to identifying cancer driver genes in endometrial cancer.
Results
Landscape of somatic mutations in the exomes of endometrial cancer
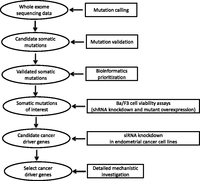
To gain an unbiased view of somatic alterations that contribute to the pathogenesis of endometrial cancer, we performed whole-exome sequencing of 14 endometrial tumor samples (one tumor with an unusually high mutation frequency and low estimated tumor content was excluded from further analysis) and matched normal DNA samples using the SOLiD platform. Supplemental Table 1 provides the histologic information and other clinical characteristics of these samples. On average, we obtained 185 million 50-nt single reads for tumor samples and 62 million reads for normal samples (Table 1). The Agilent SureSelect capture reagent used for enrichment targets 170,843 exons (∼1.2% of the human genome); for the targeted regions, the average coverage was 94× and 42× for tumor and normal samples, respectively. We developed a computational pipeline to detect somatic mutations by comparing single-nucleotide variations in tumor and normal samples (Supplemental Figs. 1, 2; Supplemental Methods). At the depth of sequencing used and with the majority of tumors being low grade (grade 2), we identified a mean of 89.5 somatic point mutations (range 34–263) per tumor corresponding to an average of 3.7 mutations per megabase (range 0.98–12.0) (Table 1). As shown in Figure 2, the most common mutations were transitions in the CpG context (that is, G>A/C>T), which parallels several other cancer lineages (Ding et al. 2010; Chapman et al. 2011; Puente et al. 2011). In addition, the tumors contained a mean of 14.2 somatic coding small insertion/deletions (indels) (Table 1; Supplemental Table 2; Supplemental Methods).
Landscape of somatic mutations in the exomes of 13 endometrial tumors
Mutation profile of somatic mutations in the exomes of endometrial cancer. The frequency of six classes of mutations is shown for all mutations in the exomes, nonsilent coding mutations, and silent mutations, respectively.
Focusing on somatic mutations that potentially affect protein function, across the 13 tumor samples, we identified 576 unique nonsynonymous mutations and 24 nonsense mutations in 566 genes (Supplemental Table 2). The most perturbed biological pathways were integrin, angiopoietin, complement system, and PTEN signaling (Fisher exact test, P < 2.8 × 10−3, false discovery rate [FDR] < 0.05, Supplemental Table 3). Based on potential biological interest, we selected 97 mutation sites for Sequenom MassARRAY validation and obtained a true-positive rate of 81%, leading to 69 genes with validated mutations and a prediction of 487 nonsynonymous mutations and 18 nonsense mutations in the 13 tumors (Supplemental Table 2; Supplemental Methods). To estimate the sensitivity of our mutation calling algorithm, we examined full-length sequences of nine genes with concurrent Sanger sequencing and Sequenom MassARRAY detection. The estimated sensitivity was 80%, with false negatives occurring primarily due to low coverage (Supplemental Methods). These results indicate that the identified mutated gene set largely characterizes the landscape of somatic coding mutations in the 13 endometrial cancers assessed.
Novel candidate driver cancer genes revealed by Ba/F3 functional assays
Several of the validated mutated genes have previously been shown to be targeted in endometrial cancers, including TP53, PTEN, CTNNB1, PIK3CA, PIK3R1, and PIK3R2 (Lax et al. 2000; Sun et al. 2001; Moreno-Bueno et al. 2002; Oda et al. 2005; Cheung et al. 2011). We recently reported their mutation profiles in a large collection of well-characterized endometrial tumors, demonstrating that aberrations, including frequent co-mutations, of the PI3K pathway occur in ∼80% of endometrial cancers (Cheung et al. 2011). However, the functional roles of the remaining mutated genes in endometrial cancer are largely unknown. To identify potential driver cancer genes for further characterization, we performed bioinformatics analyses and selected 30 genes using the following criteria: (1) if the mutation in a gene is predicted to have a high impact on the protein function by CHASM (FDR of impact score <30%) (Carter et al. 2010) or MutationAssessor (high impact) (Reva et al. 2011); or (2) a gene contains multiple or recurrent mutations (Methods). Supplemental Table 4 provides details on selection of candidates for further analysis. We then evaluated the functional impact of these selected genes in murine Ba/F3 cells (Warmuth et al. 2007). This cell line is a murine bone marrow-derived pro-B-cell line that depends on interleukin-3 (IL-3) for proliferation, but readily becomes IL3-independent in the presence of an oncogene or oncogenic event. Thus, the Ba/F3 cells represent a sensitive tool for measuring the effect of an introduced perturbation on cell proliferation and survival, not only for kinase genes but also non-kinase genes (Lutz et al. 2000; Warmuth et al. 2007; Yoda et al. 2010; Cheung et al. 2011).
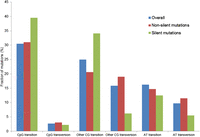
We first performed a “loss-of-function” analysis on the 30 candidate genes using shRNA gene silencing. As shown in Figure 3A, compared with parental cells and cells transfected with empty vector or nonspecific shRNAs, inhibiting the expression of eight genes (INHBA, KMO, PHKA2, TTLL5, AKTIP, IGFBP3, GRM8, and ARID1A) with two independent shRNAs was sufficient to promote the IL3-independent survival of Ba/F3, compatible with these genes having tumor-suppressor-like activity. In contrast, the introduction of ERBB3 and RPS6KC1 shRNAs remarkably inhibited IL3-independent cell survival, compatible with oncogene-like activity. These observations were confirmed by at least one of two additional independent shRNAs in a secondary screen (data not shown). We confirmed shRNA knockdown efficacy by Western blotting for eight proteins where appropriate antibodies were available (Fig. 3C). Nine genes with efficient shRNA knockdown had no effect in the Ba/F3 cells with four (NOS1AP, CDK17, C1orf198, and PHKG1) shown in Figure 3A and Supplemental Figure 3a. One of two shRNAs in the initial screen for two genes (TRPS1 and WNT11) promoted IL3-independent Ba/F3 survival. The activity was confirmed by two additional independent shRNAs (Fig. 3B; Supplemental Fig. 3b). Significant knockdown was not observed or antibodies were unavailable for the remaining nine genes. Taken together, the shRNA-mediated knockdown screen revealed 12 potential driver cancer genes including 10 tumor suppressors and two oncogene candidates from the 30 genes assessed in the Ba/F3 survival assay.
Novel candidate driver cancer genes identified by shRNA screening in Ba/F3 viability assay. (A) Ba/F3 cells were transfected with short-hairpin RNAs (shRNA) targeting indicated genes. Empty vector (pGIPZ) and nonspecific shRNA served as the control. Cells were cultured without IL-3 for 4 wk and harvested for viability assay. Cell viability relative to Ba/F3 parental cells was shown. (*) P < 0.05, compared with Ba/F3 parental control. (B) Whole cell lysates were also collected for Western blotting with indicated antibodies, and ERK2 was used as the loading control.
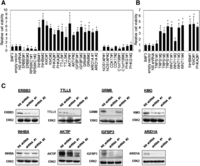
To complement the shRNA screen, we performed a “gain-of-function” analysis on 17 out of the 30 genes by overexpressing the wild-type or mutated gene in the Ba/F3 cells. Among the 17 genes examined, six genes were scored positive in the Ba/F3 shRNA screen. As shown in Figure 4A, overexpression of wild-type ERBB3 significantly increased cell survival, consistent with its proposed role as an oncogene, but the P590L mutation showed no difference relative to the wild-type, suggesting that this mutation is not functional, albeit in terms of survival in the Ba/F3 assay. Compared with controls, overexpression of wild-type AKTIP and INHBA significantly reduced cell survival, consistent with their potential roles as tumor suppressors. Interestingly, their corresponding mutations (Q281K in AKITP and R310Q in INHBA) significantly increased the Ba/F3 survival compared to the wild-type constructs (P < 0.05), suggesting a critical inactivating effect of these mutations and strongly supporting AKTIP and INHBA as bona fide tumor suppressors. In contrast, upon overexpression, wild-type or mutants of the GRM8, PHKA2, and WNT11 genes which scored positive in the shRNA screen showed no significant effect on survival of Ba/F3, suggesting that the effects of these genes may be dosage-independent and are not evident in the presence of the wild-type gene in Ba/F3 cells. Concordant with the lack of effect in the Ba/F3 shRNA screen, there was no significant change in Ba/F3 survival with overexpression of the remaining 11 genes or their mutants (Fig. 4B).
Candidate driver cancer genes confirmed by overexpression of wild-type genes or mutants in Ba/F3 viability assay. Ba/F3 cells were transfected with wild-type (WT) or corresponding mutant(s) (mutation sites indicated) of (A) six positive genes in the shRNA screen and (B) 11 genes inactive in the screen. Cells transfected with shRNA were included in the assay as reference. pGIPZ vector is the empty vector carrying shRNA while LacZ corresponds to β-galactosidase in the pLenti6.3 vector. Cells were cultured without IL-3 for 4 wk and harvested for viability assay. Cell viability relative to Ba/F3 parental cells was shown. (*) P < 0.05, compared with Ba/F3 parental control. (#) A significant difference in cell viability between WT and mutant-transfected cells (P < 0.05).
Effect of candidate driver cancer genes by siRNA knockdown in endometrial cancer cell lines
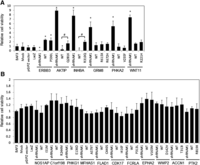
To extend our observations in Ba/F3 cells to human endometrial cancer cells, we introduced siRNAs targeting the 12 candidate driver cancer genes into four human endometrial cancer cell lines and assessed effects on cell viability. We included an siRNA targeting the established PTEN tumor-suppressor gene to validate the approach. As shown in Figure 5 and Supplemental Figure 4, decreased PTEN protein and increased AKT phosphorylation was accompanied by significant increased cell viability in PTEN siRNA-transfected cells carrying a wild-type PTEN gene and expressing PTEN protein, but not in PTEN protein-negative cell lines, supporting the utility of the approach. Importantly, KLE, in which all genes assessed were wild-type, exhibited the same responses to the corresponding shRNA as Ba/F3 except for IGFBP3 siRNA (Fig. 5B). The responses of EFE184 (all genes tested are wild-type except ARID1A with undetectable protein expression), SK-UT-2 (ARID1A is mutated with detectable protein; mutation status of others is unknown), and SNG-II (ARID1A is mutated with undetectable protein; the mutation status of the others is unknown) were more variable (Supplemental Fig. 4). ARID1A siRNA only demonstrated altered viability of KLE and SK-UT-2 where the ARID1A protein is expressed (Fig. 5B; Supplemental Fig. 4). Importantly, AKTIP siRNA significantly increased viability in all cell lines, consistent with its potential tumor-suppressor role suggested in the Ba/F3 system. The siRNAs for WNT11 and INHBA increased viability in three cell lines, whereas IGFBP3 siRNA increased cell viability in two cell lines. In contrast, ERBB3 and RPS6KC1 siRNAs inhibited cell viability in all cell lines at least at one time point. siRNAs targeting NOS1AP, PHKG1, and C1orf198, which had no effect in Ba/F3 survival, did not alter cell viability in all of the endometrial cell lines we examined, providing additional support for the validity of the Ba/F3 model and the pipeline of characterization of mutations. Collectively, the effect of silencing candidate driver genes in endometrial cancer cell lines largely recapitulated the results obtained in Ba/F3 cells.
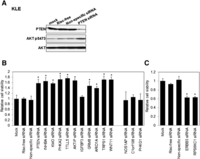
Functional effect of candidate driver cancer genes by siRNA-mediated gene silencing in KLE endometrial cancer cell line. KLE cells were transfected with siRNAs targeting the indicated genes. Mock, risc-free siRNA, and nonspecific siRNA served as controls. (A) Efficacy of PTEN siRNA on AKT phosphorylation was determined by Western blotting. Cells transfected with indicated siRNAs were assayed for cell viability (B) 7 d or (C) 5 d post-transfection. Cell viability relative to mock-transfected cells was shown. (*) P < 0.05, compared with mock control.
Regulation of PI3K pathway activation by ARID1A
ARID1A (AT-rich interactive domain 1A; also known as BAF250) is a key member of the SWI/SNF chromatin-modeling complex, and the gene has recently been reported to be frequently mutated in a wide variety of cancer types (Jones et al. 2010; Wiegand et al. 2010, 2011; Guan et al. 2011b; Gui et al. 2011; Wang et al. 2011; Mamo et al. 2012). While previous studies and our present results suggest its potential role as a tumor suppressor, the molecular mechanism underlying its functional role in cancer is largely unknown. To explore how ARID1A contributes to the pathogenesis of endometrial cancer, we first resequenced this gene in the same cohort of tumor samples (n = 222) as in our previous study (Cheung et al. 2011) and observed a (nonsilent) mutation frequency of 41.6% (Fig. 6A; Supplemental Fig. 5). Strikingly, ARID1A mutations co-occur with mutations in PTEN (Fisher exact test, P < 1.2 × 10−5, Bonferroni corrected P < 1.0 × 10−4) and PIK3CA (P < 2.3 × 10−3, Bonferroni corrected P < 1.8 × 10−2) (Fig. 6A; Supplemental Table 5), as well as with overall PI3K pathway aberration (P < 1.1 × 10−3, Bonferroni corrected P < 8.6 × 10−3). Since coordinate PI3K pathway mutations are common in endometrial cancer relative to other cancer lineages, the concordance of aberrations in the PI3K pathway and ARID1A could represent coordinate targeting of the PI3K pathway or a mutually exclusive function for ARID1A and the PI3K pathway. We thus examined the effect of ARID1A mutations on protein and phosphoprotein levels of core members in the PI3K pathway using reverse-phase protein arrays and found that the phosphorylation of several downstream targets (PDK1, AKT, GSK3, TSC2, p70S6K, and ACC) are significantly up-regulated in tumors with ARID1A mutations (two-sided t-test, P < 0.05, FDR < 0.1; Fig. 6B). Since (1) PTEN loss has a dominant effect on the activation of the PI3K pathway (Hollander et al. 2011) and (2) ARID1A shows co-mutation patterns with PTEN and PIK3CA, we then focused on a subset of tumor samples in which both PTEN and PIK3CA genes were wild-type, and also PTEN expression was retained (n = 47). Strikingly, in these samples, phosphorylation of AKTpS473 and p70S6KpS371, two key PI3K pathway proteins, remained significantly up-regulated in ARID1A-mutated samples (Fig. 6C) as compared with samples where ARID1A, PTEN, and PIK3CA are wild-type, indicating that the activation of PI3K pathway by ARID1A mutations is not due to co-occurrence of ARID1A mutations with aberrations in PTEN or PIK3CA.
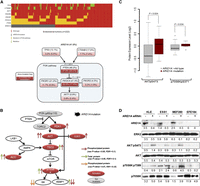
Mutational and functional analysis of ARID1A on the activation of the PI3K pathway. (A) Co-mutation patterns of ARID1A and key genes related to the PI3K pathway. (Upper panel) Mutation diagram in the full set of endometrial tumor samples (n = 222). Each column represents a tumor, and each row corresponds to a single gene. (Lower panel) Mutation or co-mutation frequencies are expressed as a percentage of all the samples, and the co-mutation frequencies from random expectation are shown in parentheses for comparison. (Dark red) Genes with statistically significant co-mutations, accompanied with Bonferroni-corrected P-values. (B) The functional effect of ARID1A mutation on protein expression of the PI3K pathway. Each arrow represents a protein marker with significant differential expression between ARID1A wild-type and mutated samples: (red arrows) phosphorylated proteins and (green arrows) total proteins with P < 0.05 (two-sided t-test, FDR < 0.1); (orange arrows) phosphorylated proteins with marginal significance P < 0.07 (FDR < 0.13). (Solid red) Activated genes; (solid gray) genes without available protein expression data. (C) The functional effect of ARID1A mutation on the phosphorylation of AKT and p70S6K in tumor samples in which both PTEN and PIK3CA genes are wild-type, and also PTEN expression is retained (n = 47). P-values were calculated based on a two-sided t-test. (Boxes) The distribution of individual values from the lower 25th percentile to the upper 75th percentile; (solid line in the middle) median values; (lower and upper whisker) fifth and 95th percentiles; (small circles) outlier data points. (D) Four endometrial cancer cell lines were transfected with 20 nM ARID1A siRNA or nonspecific siRNA and harvested after 72 h for Western blotting with the indicated antibodies. Numerical values below each lane of the immunoblots represent the quantification of the relative protein level by densitometry.
We determined whether ARID1A regulated PI3K pathway activity in endometrial cancer cell lines. Consistent with RPPA analysis from the large endometrial cancer sample cohort, knockdown of ARID1A significantly elevated AKT phosphorylation levels in three cell lines (KLE, ESS1, and MFE280) expressing wild-type ARID1A (Fig. 6D). In contrast, up-regulation of AKT phosphorylation was not observed in the EFE184 cell line in which ARID1A protein is not present (Fig. 6D). These results are consistent with inhibition of the PI3K pathway contributing to the tumor-suppressor activity of ARID1A.
Discussion
Our study represents the first unbiased view of somatic mutations in endometrial cancer, which strongly complements previous gene- and pathway-focused studies. More importantly, our results provide substantial functional evidence for a diversity of novel candidate drivers, suggesting key insights into the pathogenesis of endometrial cancer with implications for the development of targeted therapy.
Next-generation sequencing technology has facilitated characterization of the full spectrum of aberrations in cancer genomes in a cost-effective and timely manner (Mardis 2011). However, there is still a great gap between creating a catalog of mutations and alterations and identifying a short list of “actionable” elements (Chin et al. 2011). Here we developed a systems-biology approach to filling this gap: We combined computation-prediction-based prioritization with functional screening in a highly sensitive cell viability assay. This approach allows the identification of a large number of candidate driver cancer genes in an efficient way. Focusing on a candidate driver gene of high interest, we further examined the underlying molecular mechanisms through an integrative analysis of mutation profile and protein expression on a large, well-characterized sample set and “hypothesis-driven” functional studies in endometrial cancer cell lines. These methods and the related experimental systems can be readily applied to other cancer types.
Through the combination of gene silencing and overexpression strategies in Ba/F3 and endometrial cancer cell lines, we revealed 12 potential driver cancer genes in endometrial cancer. Among these genes, ARID1A has attracted wide interest recently. Frequent mutations throughout the gene sequence including multiple truncation mutations have been reported in ovarian (Jones et al. 2010; Wiegand et al. 2010), gynecological (Guan et al. 2011b), bladder (Gui et al. 2011), gastric (Wang et al. 2011), breast (Mamo et al. 2012), and endometrial cancers (Guan et al. 2011a; Wiegand et al. 2011), suggesting a role as a tumor suppressor. ARID1A encodes BAF250a, a nuclear protein and a key component of the SWI/SNF chromatin-remodeling complex that functions as a regulator of gene expression and chromatin dynamics (Wu et al. 2009). However, to date, the mechanisms by which loss of ARID1A function contributes to cancer pathophysiology remains poorly understood. ARID1A has been suggested to suppress cell proliferation of ovarian and endometrial cancer cell lines through physically interacting with p53 to coordinately regulate the transcription of cell cycle–related genes (Guan et al. 2011b), linking its function to nuclear localization. Here we not only confirmed a high mutation frequency in a large cohort of endometrial tumors, but also for the first time, demonstrated that ARID1A can regulate PI3K pathway activity, consistent with inhibition of the PI3K pathway contributing to ARID1A tumor-suppressor activity. Since the PI3K pathway represents a promising target for therapy (Hennessy et al. 2005), these results have direct implications for clinical translation. At present, it remains unclear which molecules in the pathway are targeted by ARID1A, and further studies are required to elucidate mechanistic details.
Among the other candidate tumor suppressors identified, AKTIP, INHBA, and WNT11 appear to be the most promising ones. AKTIP and INHBA are particularly likely to represent tumor suppressors because knockdown and overexpression demonstrated opposite effects in Ba/F3, and, critically, patient-derived mutations abrogated the effects of the wild-type expression construct (Figs. 3, 4). AKTIP was first identified as an AKT binding partner (Remy and Michnick 2004). Exogenous overexpression of AKTIP enhanced AKT phosphorylation, but this activation induced apoptosis for unknown reasons (Remy and Michnick 2004). Based on data from the Cancer Genome Atlas, homologous deletion of AKTIP occurs in multiple cancer lineages including breast (2%), ovary (0.9%), and prostate (1%), consistent with a tumor-suppressor role. However, the functional role of AKTIP could be tumor type-specific, since it has been proposed as a putative oncoprotein in cervical cancer but a tumor suppressor in ovarian cancer (Cinghu et al. 2011; Notaridou et al. 2011). INHBA encodes inhibin beta A, a subunit of both the activin and inhibin receptors of the transforming growth factor (TGF-beta) superfamily (Risbridger et al. 2001). Inhibin beta A, like TGF-beta, can inhibit or stimulate cell growth dependent on the cellular context. For example, INHBA substantially inhibited tumor growth and angiogenesis in in vivo gastric cancer and neuroblastoma models (Schramm et al. 2005; Kaneda et al. 2011). Meanwhile, INHBA mRNA was up-regulated in lung cancer and may result in the promotion of cell proliferation (Seder et al. 2009). In endometrial cancer cell lines, the role of INHBA is controversial (Di Simone et al. 2002; Tanaka et al. 2003). It is possible that the contribution of INHBA to the tumorigenesis is determined by the relative expression levels of other receptor subunits and their interacting partners. WNT11 is a key member of the Wingless-type (Wnt) signaling pathway whose deregulation has been implicated in endometrioid endometrial tumors as evidence by beta-catenin mutations and aberrant nuclear accumulation (Ikeda et al. 2000; Moreno-Bueno et al. 2002). The canonical Wnt cascade is mediated by nuclear beta-catenin binding to T-cell factor transcription factors to activate genes relevant to tumorigenesis, while noncanonical Wnt signaling is beta-catenin independent (Bejsovec 2005). Interestingly, WNT11 is down-regulated in hepatocellular carcinoma, and it can modulate both canonical and noncanonical Wnt pathways to execute its tumor-suppressor actions (Toyama et al. 2010). Thus, the role of WNT11 in Wnt signaling in endometrial cancer warrants further investigation.
ERBB3 and RPS6KC1 are potential oncogenes identified in our study. ERBB3 (HER3) belongs to the epidermal growth factor receptor family and has been implicated in cancer, previously. ERBB2, a partner of ERBB3, as well as ERBB3 itself is often amplified and overexpressed in breast, ovarian, prostate, and lung cancers (for review, see Sithanandam and Anderson 2008). Overexpression of ERBB3 promotes tumorigenesis through multiple mechanisms including cell cycle progression, stimulation of cell migration, and invasion primarily via activation of the PI3K pathway (Sithanandam and Anderson 2008). ERBB3 is highly expressed in endometrial cancer, but the functional role remains unclear (Srinivasan et al. 1999; Ejskjaer et al. 2007). RPS6KC1 (encoding ribosomal protein S6 kinase polypeptide 1) is another potential oncogene. RPS6KC1 mutations have been previously found in breast, ovary, and lung cancers (Davies et al. 2005; Stephens et al. 2005; Cancer Genome Atlas Research Network 2011). Two independent studies show that RPS6KC1 does not have kinase activity (Hayashi et al. 2002; Liu et al. 2005), but, instead, likely functions as an adaptor molecule to recruit binding partners (sphingosine kinase-1 and peroxiredoxin-3) to early endosomes (Hayashi et al. 2002; Liu et al. 2005). These trafficking pathways within the endosomal system play a fundamental role in regulating protein degradation, recycling, secretion, and compartmentalization; and, indeed, defective vesicular trafficking is a hallmark of malignant transformation (Mosesson et al. 2008).
Our study has several limitations. First, somatic alterations in noncoding regions, genome rearrangements, and copy number variations would have been missed. Second, the number of samples sequenced is relatively small, thus sequencing of additional samples will be necessary to establish a comprehensive genomic landscape for endometrial cancer. Finally, additional functional and clinical studies will be required before the insights from the genomic analysis can be translated into effective personalized therapy for endometrial cancer.
Methods
Sample collection
All studies of 222 patients diagnosed at the University of Texas MD Anderson Cancer Center (Houston, TX, USA) from 1998 to 2009 were approved by the Institutional Review Board. Tumor content (≥80%), histological classification, grade, and stage were reviewed by two independent pathologists. Genomic DNA from frozen tumor resections was extracted by the MD Anderson Bioextraction core using the QIAamp DNA Mini Kit (QIAGEN); normal DNA was extracted from peripheral blood leukocytes using the QIAamp Blood kit (QIAGEN).
Exome sequencing and detection of somatic alterations
Whole-exome libraries (captured by Agilent SureSelect All Exon Kit) were constructed and sequenced on a SOLiD V3.0 Genome Analyzer using 50-nt single reads. Sequencing reads were processed, mapped to the human genome (hg19), and analyzed using Bioscope (version 1.21) combined with in-house scripts. Supplemental Figure 1 shows the overall computational pipeline for detecting somatic mutations. Mutation validation was performed by Sequenom MassARRAY genotyping, and whole-gene resequencing on the large sample cohort was performed by Sanger sequencing. A complete description is provided in the Supplemental Methods.
Mutational analysis
Mutation data of ARID1A in 222 endometrial tumors were obtained as previously described (Cheung et al. 2011). A two-sided Fisher exact test was used to assess whether (nonsilent) mutations in ARID1A tend to co-occur with mutations in other genes (or PI3K pathway aberrations) in the tumor samples. PI3K pathway aberrations were defined as PTEN loss or mutations in any pathway genes (including PTEN, PIK3CA, PIK3R1, PIK3R2, and AKT1). A Bonferroni correction was used to control multiple testing.
RPPA data analysis
To examine the effect of ARID1A mutations on the PI3K pathway, high-throughput RPPA data for 24 PI3K-pathway-related proteins in the 222 samples were obtained from our previous study (Cheung et al. 2011). RPPA data were processed and normalized as previously described (Park et al. 2010). A two-sided t-test was used to test whether the expression of a given protein in ARID1A wild-type samples was significantly different from that in ARID1A mutated samples, and a false discovery rate (Benjamini and Hochberg 1995) was used to control multiple testing.
Bioinformatics analysis
Pathway enrichment analysis was performed with Ingenuity Pathways Analysis (IPA) (version 8.0). The status of PTEN was determined as in our previous study (Cheung et al. 2011). A two-sided Fisher exact test was used to assess whether ARID1A mutations were correlated with the status of PTEN (loss or retained). To select mutated genes for shRNA studies, functional effects of somatic mutations were predicted with two programs: (1) CHASM (Carter et al. 2010), mutations with FDR < 30% were selected; (2) MutationAssessor (Reva et al. 2011), mutations with high functional effect were selected.
Ba/F3 viability assays
The interleukin-3 (IL-3)–dependent prolymphoid cell line Ba/F3 was maintained in RPMI1640 medium containing 10% fetal bovine serum supplemented with 5 ng/mL IL-3 at 37°C in a 5% CO2 atmosphere. Constructs containing wild-type genes or their corresponding mutants cloned into pLenti6.3 (Invitrogen) and shRNAs in pGIPZ vector (Open Biosystems) were transfected into Ba/F3 cells using the Neon electroporation system according to the manufacturer's instructions (Invitrogen). At 96 h post-transfection, the cells were resuspended in medium without IL-3. Cells (5 × 103) were plated in 96-well plates and cultured for 4 wk. Cell viability was evaluated using Cell Titer Blue (Promega) for mitochondrial dehydrogenase activity. Statistical analysis was performed using ANOVA followed by Tukey's post hoc test (GraphPad Software). P < 0.05 was considered statistically significant.
siRNA transfection in endometrial cancer cell lines
Endometrial cell lines EFE-184, ESS-1, and MFE280 were purchased from DSMZ-German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). KLE was provided by Dr. Russell Broaddus (MD Anderson Cancer Center, Houston, TX), and SK-UT-2 was kindly provided by Dr. Bo R. Rueda (Massachusetts General Hospital, Boston). All cell lines were cultured in media according to the suppliers' instructions at 37°C in a 5% CO2 atmosphere. Cells were transfected with 20 nM siRNAs (Dharmacon) using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturers' instructions. The cells were harvested at time points as indicated in the figure legends and processed for either cell viability assay using Cell Titer Blue (Promega) or Western blot analysis.
Western blotting
Whole-cell lysates were extracted with RIPA lysis buffer (1% NP-40, 5 mM EDTA, 1 mM sodium orthovanadate, 1% phenylmethylsulphonyl fluoride, and complete protease inhibitor cocktail). The protein concentrations were determined with DC Protein Assay Reagent (Bio-Rad Laboratories). Cell lysates (25 μg) were loaded onto SDS/PAGE and transferred to a Hybond-ECL nitrocellulose membrane (Amersham Biosciences). The membrane was blocked with 5% nonfat milk and incubated with primary antibody overnight at 4°C. Protein expression was visualized with an ECL plus kit (Amersham Biosciences).
Data access
The sequencing data from this study can be accessed at the European Genome-phenome Archive (EGA) (http://www.ebi.ac.uk/ega/) under accession number EGAS00001000318.
Acknowledgments
This work was supported by a Stand Up to Cancer Dream Team Translational Research Grant, a Program of the Entertainment Industry Foundation (SU2C-AACR-DT0209) to G.B.M.; a Uterine Cancer SPORE grant (NIH/NCI P50 CA098258) to R.R.B, K.H.L., and G.B.M.; U01 CA168394 to G.B.M.; a Career Development Award from the Conquer Cancer Foundation of the American Society of Clinical Oncology to B.T.H.; NIH/NCI R21CA152432 to R.K.; a training fellowship from the Keck Center Computational Cancer Biology Training Program of the Gulf Coast Consortia (CPRIT Grant No. RP101489) to L.W.T.C.; grants from the G.S. Hogan Gastrointestinal Research Fund and the Lorraine Dell Program in Bioinformatics for Personalization of Cancer Medicine to H.L.; TCGA GDAC Grant (NIH/NCI U24 CA143883) to G.B.M. and H.L.; and an Institutional Core grant (NIH/NCI P30 CA016672).
Author contributions: H.L., G.B.M., and B.T.H. designed the research; L.W.T.C., J.L., S.Y., K.S.H., T.D., Y.L., C.G., W.G., S.E.S., and K.L.S. performed the experimental work, including Sequenom, Sanger sequencing, Ba/F3 viability assays, and Western blots; X.L. and C.G.L. performed library preparation and SOLiD sequencing; H.L., L.W.T.C., J.L., Z.J., H.C., S.N.W., M.D., R.G.W.V., F.Z., R.K., and G.B.M. performed data analyses; K.H.L., R.R.B., and B.T.H. provided patient samples; H.L., L.W.T.C., J.L., and G.B.M. produced the text and figures; H.L. and G.B.M. provided leadership for the project. All authors contributed to the final manuscript.
Footnotes
-
↵11 Corresponding author
E-mail hliang1{at}mdanderson.org
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.137596.112.
- Received January 11, 2012.
- Accepted July 2, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








