
Polygenic cis-regulatory adaptation in the evolution of yeast pathogenicity
- Hunter B. Fraser1,7,
- Sasha Levy2,6,
- Arun Chavan3,6,
- Hiral B. Shah3,
- J. Christian Perez4,
- Yiqi Zhou1,
- Mark L. Siegal5 and
- Himanshu Sinha3
- 1Department of Biology, Stanford University, Stanford, California 94305, USA;
- 2Department of Genetics, Stanford University, Stanford, California 94305, USA;
- 3Department of Biological Sciences, Tata Institute of Fundamental Research, Mumbai 400005, India;
- 4Department of Microbiology and Immunology, University of California, San Francisco, California 94103, USA;
- 5Center for Genomics and Systems Biology, Department of Biology, New York University, New York, New York 10003, USA
-
↵6 These authors contributed equally to this work.
Abstract
The acquisition of new genes, via horizontal transfer or gene duplication/diversification, has been the dominant mechanism thus far implicated in the evolution of microbial pathogenicity. In contrast, the role of many other modes of evolution—such as changes in gene expression regulation—remains unknown. A transition to a pathogenic lifestyle has recently taken place in some lineages of the budding yeast Saccharomyces cerevisiae. Here we identify a module of physically interacting proteins involved in endocytosis that has experienced selective sweeps for multiple cis-regulatory mutations that down-regulate gene expression levels in a pathogenic yeast. To test if these adaptations affect virulence, we created a panel of single-allele knockout strains whose hemizygous state mimics the genes' adaptive down-regulations, and measured their virulence in a mammalian host. Despite having no growth advantage in standard laboratory conditions, nearly all of the strains were more virulent than their wild-type progenitor, suggesting that these adaptations likely played a role in the evolution of pathogenicity. Furthermore, genetic variants at these loci were associated with clinical origin across 88 diverse yeast strains, suggesting the adaptations may have contributed to the virulence of a wide range of clinical isolates. We also detected pleiotropic effects of these adaptations on a wide range of morphological traits, which appear to have been mitigated by compensatory mutations at other loci. These results suggest that cis-regulatory adaptation can occur at the level of physically interacting modules and that one such polygenic adaptation led to increased virulence during the evolution of a pathogenic yeast.
A great deal of research has focused on identifying genes contributing to pathogenicity. In most cases, these genes have been implicated as a result of their absence (or lower copy number) in the pathogens' free-living relatives (Baquero et al. 2008; Moran et al. 2011). While this comparative gene-content approach has been quite informative, it cannot be applied to changes in gene expression. As a result, gene expression adaptations have been far more difficult to link to pathogenicity than gene gain/loss, and no gene expression adaptations have been linked to the evolution of pathogenicity in any species.
Although it is now well established that evolutionary changes in gene expression are widespread—both within and between species—much less is known about the role of natural selection in shaping this variation (Gilad et al. 2006; Fay and Wittkopp 2008; Fraser 2011). Negative (or purifying) selection has been found to be the dominant selective force acting on new mutations affecting gene expression (Denver et al. 2005; Rifkin et al. 2005; Gilad et al. 2006; Fay and Wittkopp 2008; Fraser 2011), but among those mutations not purged from the population, identifying which have been subject to positive selection (as opposed to the random genetic drift of neutral mutations) has traditionally been quite challenging.
Two classes of methods have successfully identified gene expression adaptations (Fraser 2011). The first identifies single genes whose expression affects an adaptive trait, often via QTL mapping of the trait followed by detailed investigation of candidate genes. This approach has led to several fascinating examples of gene expression adaptations (Shapiro et al. 2004; Colosimo et al. 2005; Gompel et al. 2005; Miller et al. 2007; Prud'homme et al. 2007; Wray 2007; Rebeiz et al. 2009; Manceau et al. 2011). However, the amount of effort required—as well as the need for previously characterized candidate genes within each QTL region—has limited its general applicability. The second method is genome-wide scans for entire sets of genes (e.g., pathways or protein complexes) undergoing coordinate adaptation (Fraser 2011). This approach has several attractive features: Unlike QTL mapping, it is not limited to previously selected and measured traits, does not require genotyping and phenotyping of hundreds of individuals, and it directly identifies the specific genes under selection; unlike most other methods for detecting natural selection, it makes no assumptions about population demography or a subset of neutral sites; it can provide genome-wide (lower-bound) estimates of the extent of gene expression adaptation; and it can be applied in minutes to an appropriate data set, including many published data sets. Its main limitations are that it can only be applied to known gene sets and that further work is required to determine the phenotypes affected by selection. Previous applications of this approach have identified hundreds of gene expression adaptations, including the first example of gene expression adaptation occurring at the pathway level (Fraser et al. 2010), as well as the first examples of gene expression adaptations affecting behavioral traits (Fraser et al. 2011). Many other studies have identified suggestive evidence of gene expression adaptations (Bullard et al. 2010; Fraser 2011) or evidence of gene expression changes affecting traits whose adaptive significance is unknown (Wray 2007).
Here we apply a systematic scan for gene expression adaptations to Saccharomyces cerevisiae, an ideal species in which to study the evolution of pathogenicity. Although it has historically been generally recognized as safe (GRAS), an increasing number of life-threatening infections have been reported, primarily in immunocompromised patients (Enache-Angoulvant and Hennequin 2005). While specific virulence factors are yet to be determined, both high-temperature growth (Htg) and pseudohyphal morphology are associated with pathogenicity (Clemons et al. 1994; McCusker et al. 1994a,b). Determining the genetic changes underlying the transition to a pathogenic lifestyle is greatly facilitated by the fact that such transitions have occurred recently and in multiple diverse lineages (Muller and McCusker 2009), providing natural replication. Furthermore, the unrivaled genetic and genomic resources associated with budding yeast enables more detailed characterization of cis-regulatory adaptations than has been possible in any other species.
Results
Scanning for selection on yeast cis-regulation
We and others recently developed a test for lineage-specific natural selection acting on the cis-regulation of entire sets of genes (Fig. 1; Bullard et al. 2010; Fraser et al. 2010, 2011; Fraser 2011). Input for the test is genome-wide allele-specific expression (ASE) data from a hybrid between two diverged lineages (denoted A and B) and a catalog of gene sets to be tested (sets can be based on any type of relationship, e.g., shared functional annotation, physical interaction, etc.). The genes in each set are then tested for biased ASE directionality—i.e., the number of genes with increased expression from A-alleles, as opposed to B-alleles—compared to the genome as a whole. Since each gene's ASE typically represents an independent cis-regulatory difference between the two parental lineages (Bullard et al. 2010; Fraser et al. 2011), the expectation under neutrality is that in any gene set, the fraction of genes with ASE favoring expression from allele A is approximately equal to the overall fraction of all ASE genes that favor allele A (Fig. 1). Departure from this neutral expectation indicates lineage-specific selection (Fig. 1): either positive selection for multiple cis-regulatory changes in one lineage, or a relaxation of negative selection against cis-regulatory changes in one lineage.
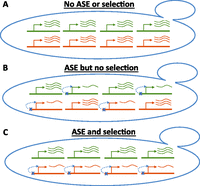
Illustration of approach for finding cis-regulatory selection. Each yeast cell represents a hybrid between two diverged strains. Four genes from a single gene set are shown, with two alleles per gene (red and green). The number of curved lines next to each allele represents that allele's expression level. (A) All four genes are expressed equally from each allele, so there is no allele-specific expression (ASE) or evidence for selection. (B) Each gene has a down-regulating cis-regulatory mutation (blue x), but these act on alleles from each parent with equal frequency, so there is still no evidence for selection. (C) All four genes have ASE in the same direction (down-regulating, red), a pattern indicating lineage-specific selection on cis-regulation (though more than four genes are required to achieve statistical significance).
We applied this test to microarray-based ASE data (Gagneur et al. 2009) from a hybrid between two diverged strains of S. cerevisiae: a common laboratory strain (S288c, or “S”), and a pathogenic strain derived from an isolate from the lungs of an AIDS patient (YJM789, or “Y”) (McCusker et al. 1994a). Their genomes differ by ∼60,000 SNPs and ∼324 kb of insertions/deletions, together accounting for ∼3.3% of the genome (Wei et al. 2007). To measure ASE, transcribed polymorphisms are required to differentiate the two alleles; 2765 genes had at least eight ASE-informative microarray probes overlapping such polymorphisms. Of these, 1425 (51.5%) genes had detectable ASE (Gagneur et al. 2009).
With these genes, we tested two categories of gene sets for selection on cis-regulation: functional annotations and protein complexes. Testing 215 Gene Ontology categories, we did not observe any with significant bias in ASE directionality. However among 99 protein complexes, we observed one complex with a highly significant bias (see Methods). The complex consisted of 17 genes, of which 10 exhibited S+ ASE (higher expression from S allele; top 10 rows of Table 1), while none had Y+ ASE. The probability of observing at least 10/17 genes with ASE all in the same direction is 4.3 × 10−5 (P = 0.013 after Bonferroni correction). ASE for eight of these 10 genes (Table 1, bold) is further supported by pyrosequencing of cDNA from a Y/S hybrid strain. This ASE directionality bias indicates that these genes likely experienced lineage-specific selection on cis-regulation (Bullard et al. 2010; Fraser 2011; Fraser et al. 2011), though it does not establish in which lineage the selection occurred.
Summary of ASE and phenotyping data
This complex is highly enriched for proteins involved in the early stages of endocytosis (13/17 proteins) (Table 1). It was first detected in a high-throughput proteomics study (Gavin et al. 2002); subsequent studies have supported most of the interactions, although they have split them into several different modules that cooperate to form endocytic vesicles (Kaksonen et al. 2005). Thus the complex is likely composed of distinct modules that transiently associate with one another during vesicle formation.
Population-genetic tests of positive selection
To determine whether these genes' ASE was in fact due to positive selection, as opposed to relaxed negative selection, we employed a population-genetic test for selective sweeps (Fraser et al. 2010). This test relies on the fact that the region surrounding a recent selective sweep will be devoid of genetic variation among the descendants of the swept population. We compared variation at our candidate selected genes to a large set of control genes to account for demographic history (Fraser et al. 2010). Although a deficit of variation can have many causes—e.g. low mutation rate, background selection, or purifying selection—these other potential causes are expected to persist throughout the phylogeny, since they are not due to stochastic/rare events like the occurrence/sweeping of an adaptive mutation. Therefore at a particular locus, a marked deficit of genetic variation specific to a subset of related strains suggests that a selective sweep occurred in an ancestor of those strains (in contrast, a deficit of variation observed at a locus in all strains would be consistent with alternatives such as low mutation rate). Relaxed negative selection, the alternative possibility based on our ASE selection test, would (if anything) increase genetic variation in the previously constrained cis-regulatory regions (Fraser et al. 2010)—the opposite effect of a selective sweep.
Our test genes were the eight with confirmed ASE (Table 1), and our controls were all 971 genes with S+ ASE. To quantify genetic variation, we utilized genome-wide genotypes from 63 diverse strains of S. cerevisiae (Schacherer et al. 2009). For each gene, we split the 63 strains into three sets of 21 strains each: those most closely related to the focal strain (S or Y) at that locus, those of intermediate relation, and those most distantly related. We quantified the difference between the test and control genes by aligning their 5′ ends and comparing their genetic variation (within each set of 21 strains) in a 1-kb moving window. Since the majority of yeast cis-regulatory information is present within several hundred bases upstream of each gene's 5′ end, sweeps on cis-regulatory variants should be specific to just one or two windows close to the 5′ end, as we observed previously (Fraser et al. 2010).
Performing this test using polymorphism data from the strains closest to Y, we observed a significant deficit of variation specifically at the 5′ ends of the eight confirmed ASE genes (P = 0.0051) (Fig. 2A), as expected from selective sweeps on cis-regulatory variants in their promoters. We did not observe such a deficit, either in the central window or any other, in three controls utilizing polymorphism data from other sets of 21 strains: those most distant from Y, those of intermediate distance from Y, and those closest to S (Fig. 2B). This suggests that most or all of the eight confirmed ASE genes underwent sweeps for expression down-regulation in the pathogenic Y lineage.
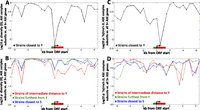
Population-genetic analysis. In all panels, patterns of genetic variation at the eight confirmed ASE genes are compared with the variation at all 971 genes showing ASE in the same direction. Variants are compared in a 1-kb moving window (500-bp step size), with all genes aligned by their 5′ ends and oriented in the same direction; the average ORF length is shown in red, and approximate promoter length is shown in green. Comparisons of either genetic diversity θ (A,B) or Tajima's D (C,D) are shown, with P-values computed using the nonparametric Wilcoxon test. The test is one-sided so that low P-values specifically indicate lower θ or D values among the eight ASE genes—as expected from a selective sweep—compared with the 971 control genes. Comparisons use either the 21 strains most closely related to Y (A,C), or one of three sets of 21 control strains (B,D): those most closely related to S (blue), those of intermediate distance to Y (red), or those of greatest distance to Y (green).
In addition to reducing the number of genetic variants in a region, selective sweeps can lead to a skew in the allele frequency distribution, since new mutations that arise after the sweep are initially at very low frequency and only slowly become common. To test for such a skew, we employed Tajima's D, which tests for an excess of rare variants compared to the neutral expectation (Tajima 1989)—again comparing candidate genes with control genes to account for demographic history. We observed significantly lower D values in the eight candidate genes than in the 971 controls among the 21 strains closest to Y (P = 0.0049) (Fig. 2C), further supporting the idea that they experienced a selective sweep in this lineage. As above, we did not find a similar pattern either in windows adjacent to the genes or in any of our three controls (Fig. 2D), indicating that the allele frequency skew is specific to Y's closest relatives and is thus unlikely to be due to phenomena such as mutation rate variation or background/purifying selection. Collectively, our population-genetic tests (Fig. 2) strongly support the idea that most or all of the eight ASE genes underwent recent positive selection for cis-acting down-regulation in an ancestor of the pathogenic Y strain.
Fitness effects of the cis-regulatory adaptations
To investigate the phenotypic consequences of divergence in the endocytosis complex, we created reciprocal hemizygous (RH) strain pairs for the eight confirmed ASE genes, as well as four additional complex subunits. RH strains are diploid hybrids (in this case, between S and Y) in which a single allele of a gene has been deleted from the genome. RH strains are typically compared to one another to assess phenotypic effects of each allele separately. They can also be compared to their wild-type S/Y hybrid progenitor to assess the effects of a ∼50% reduction in each gene's transcription—roughly mimicking the adaptive down-regulations these genes experienced in the Y lineage (see Methods).
Our finding that the down-regulation of these genes' expression levels in the Y lineage was likely due to positive selection implies that this down-regulation conferred a fitness advantage. To investigate if dosage of the endocytosis module genes affects fitness in standard laboratory conditions, we measured their growth at 30°C. The strains showed almost no fitness variation on either solid or liquid media (Supplemental Figs. 1, 2).
However the most important environment for pathogen evolution is within the host. To measure fitness in vivo, we pooled all 24 RH strains and the wild-type S/Y hybrid and injected the pool into the tail veins of three immunocompromised mice; 5 d later these mice were killed, and live yeast were isolated from their kidneys. We then quantified the abundance of each strain in the starting pool and kidneys of each mouse by qPCR; their ratio reflects the relative fitness of each strain in vivo. Strikingly, of the 21 RH strains differing in fitness from the wild-type hybrid in vivo, 20 were fitter than the wild-type hybrid (Fig. 3A). Such a consistent advantage of RH strains is unlikely to occur by chance (binomial P = 10−5), suggesting that reduced expression of these genes confers a significant fitness advantage in a mammalian host and that these adaptive down-regulations have increased the virulence (defined here as the capacity of a pathogen to invade its host) of the Y strain.
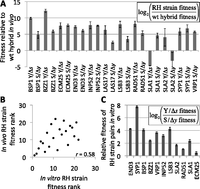
Fitness of reciprocal hemizygous (RH) strains. (A) Fitness in vivo. RH strains and the wild-type Y/S hybrid were competed for 5 d in immunocompromised mice. Twenty out of 21 strains with fitness values significantly different from the wild type were fitter than the wild type (P = 10−5), despite showing no measurable growth differences at 30°C in vitro (Supplemental Figs. 1, 2). All but the two SLA2 RH strains differ significantly from the wild type. Error bars, ±1 SE. (B) High-temperature (37°C) growth in vitro predicts fitness in vivo. Each strain's relative fitness rank is plotted both in vivo and in vitro; a significant (P = 0.002) correlation is observed. (C) Fitness in vitro. Y-alleles yield higher fitness than S-alleles at 37°C in vitro for 11 of 11 RH strain pairs. Genes are ordered by decreasing significance of the Y-allele advantage. Error bars, ±1 SE.
A phenotype known to be associated with virulence is Htg (37°C–41°C) in vitro, likely because a strain must be able to grow at mammalian body temperature to be pathogenic (McCusker et al. 1994a,b). To determine whether the growth advantage of our RH strains in vivo can be explained by variation in Htg, we performed growth competitions between each RH strain and the wild-type S/Y hybrid at 37°C. Similar to our results in vivo, we found that 20/22 RH strains were fitter than the wild-type hybrid (Supplemental Fig. 3). Notably, the only strains not fitter than the hybrid were the two RH strains for SLA2, which lacks ASE and whose RH strains were the only ones whose fitness was indistinguishable from the wild type in vivo (Fig. 3A). Moreover, a strain's in vitro fitness was a good predictor of its in vivo fitness (Spearman ρ = 0.58, P = 0.002) (Fig. 3B), suggesting that the RH strains' increased virulence may be due in part to their improved ability to grow at physiological temperature.
In addition to comparisons to the wild-type S/Y hybrid, RH strains can be compared to one another: Any differences between a pair of RH strains deleted for different alleles of the same gene must be due to divergence between the two alleles (this approach has previously been used to implicate one of our candidate genes, END3, in Htg) (see Supplemental Text). A clear trend emerged after 48 h of Htg competition in vitro: The pathogenic Y-allele yielded higher fitness than the S allele for 11/11 genes (Fig. 3C; Table 1). Because this trend held even for the four genes without detectable ASE, it suggests either that they may have condition-specific ASE not detected by our pyrosequencing, or that divergence in their protein sequences (the four genes each differ by one to nine amino acids between S and Y) or translation may have similar impacts on Htg as the divergence in the eight genes with detectable ASE. This result is notable, considering that <17% of all yeast genes have evolved detectable differences in their effects on Htg between S and Y (see Methods), so among 11 genes only one to two RH pairs would be expected to show divergent Htg by chance (probability of observing 11/11 = 3 × 10−9). In vivo, the same trend was apparent: the Y-allele was fitter for seven genes, whereas the S allele was favored for one gene, SLA1 (Table 1). This greater relative fitness of the Y-alleles at high temperatures suggests that their divergence in Htg is the result of lineage-specific selection, since neutral divergence would not result in such a biased directionality (the same logic that was applied in the S/Y ASE data selection scan above).
Association of genetic variants at the candidate loci with clinical origin
Because our RH strains were all derived from the S/Y hybrid, they cannot establish whether these genes may play a role in the pathogenicity of other clinical isolates (i.e., strains isolated from human patients) of S. cerevisiae. If they did play such a role, then the alleles at these loci may differ between clinical versus nonclinical strains. Because pathogenicity has likely emerged multiple times in diverse strains of S. cerevisiae (Muller and McCusker 2009), genetic differences are unlikely to consistently associate with clinical origin simply by chance.
A recent study reported a genome-wide association study (GWAS) among 88 diverse strains of yeast (including 44 clinical isolates) genotyped at over 130,000 genetic markers (Muller et al. 2011). They identified two loci that associated with clinical origin with genome-wide significance; one of these was a haplotype in VRP1, one of our eight ASE genes. To test if more of our candidate genes were associated with clinical origin as well, we calculated the strength of association at each of the eight loci. At a permissive cutoff (nominal P < 5 × 10−4), we observed six of our eight candidates (75%) (Table 1) compared with 23.3% of our 971 control genes (with S+ ASE). This 3.2-fold enrichment is unlikely to occur by chance (binomial P = 0.0029) and suggests that several of these are likely to be bona fide associations. Interestingly, among the nine endocytosis complex subunits without confirmed ASE, only two (22.2%) (Table 1) were associated with clinical origin, consistent with the random expectation. These genetic associations suggest that the eight complex subunits with ASE harbor genetic variants that contribute to pathogenicity not only in the Y strain but across many diverse clinical isolates.
Pleiotropic effects of the cis-regulatory adaptations
These adaptations could potentially have pleiotropic effects on traits other than virulence and Htg, which could either constrain or promote their fixation. To detect pleiotropic effects, we examined cellular morphology, a complex trait affected by many endocytosis genes (Naqvi et al. 1998; Care et al. 2004). We stained our RH strains and their parents for three subcellular landmarks (cell wall, actin, and nuclear DNA) (Fig. 4A,B) and used these to extract 220 quantitative morphological phenotypes per cell (Ohya et al. 2005). For example, quantifying each strain's average cell size, we observed significant variation between RH strain pairs (Fig. 4C). In fact, despite differing by only five to 35 nucleotides, most RH pairs showed substantially greater difference in size than the Y and S parental strains (whose genomes differ by >350 kb).
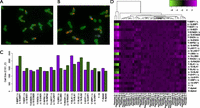
Pleiotropic effects on morphology. (A,B) Representative micrographs from a hemizygous deletion pair, LSB3 Δy/S (A), and LSB3 Y/Δs (B). Cells are stained with FITC- concanavalin A (green), Alexa Fluor 594 phalloidin (red), and DAPI (blue). (C) Barplot of a single morphological trait, cell size of budded yeast with two nuclei (C101_C), for all RH strain pairs. Error bars, ±1 SE. The work by Ohya et al. (2005) contains a full list of phenotypes and their descriptions. (D) Heatmap of hierarchically clustered nonredundant morphological phenotypes for all RH pairs. Phenotypes are the top principal components of 220 quantitative morphological traits of unbudded (A), budded with one nucleus (A1B), and budded with two nuclei (C) cells. The three leftmost phenotypes primarily correspond to cell size of yeast from each of the three cell types (A, A1B, and C).
To systematically assess the pleiotropic effects of these alleles on other morphological traits, we applied principal components analysis to the data, resulting in a set of independent phenotypes (see Methods). We observed that many traits in addition to cell size differed between paired RH strains: Among 43 phenotypes in 12 RH pairs (516 comparisons) (Fig. 4D), 313 (60.7%) differed at Wilcoxon P < 0.05; overall, only 2/12 RH pairs differed less than the Y and S parent strains. These differences did not correlate with ASE, Htg in vitro, or fitness in vivo, implying complex multifactorial effects of these alleles' divergence. Considering that the Y and S strains differ in >350 kb of genomic sequence—over 10,000-fold greater divergence than any of our RH strain pairs—their greater morphological similarity is striking. This lack of correspondence between genetic and morphological divergence suggests that the pleiotropic effects of these adaptations on cell size and other morphological traits were deleterious, but have been mitigated by compensatory mutations.
Discussion
Collectively, our results suggest that this polygenic adaptation played a role in the evolution of yeast pathogenicity. The four main points supporting this interpretation are that (1) the selective sweeps occurred in the pathogenic lineage; (2) a fitness advantage of down-regulation is seen in vivo and at 37°C in vitro, but not at 30°C; (3) the genes are highly enriched for divergence in Htg favoring Y-alleles; and (4) genetic variants at the loci are associated with clinical origin. Although this is the first example of a gene expression adaptation affecting pathogenicity, it may prove to be a common mode of pathogen evolution, considering the prevalence of gene expression adaptation in species where genome-wide scans have been conducted (Fraser 2011).
Even with such detailed genetic and phenotypic characterization, it remains a challenge to determine what was the actual selection pressure that gave rise to these adaptations. For example, it is entirely possible that the selective force was not for virulence but for some related trait—such as Htg in a free-living environment—and that the increased virulence did not come into play until after the adaptations had swept through the population. It is also possible that most of the adaptations were selected in response to an initial subunit's down-regulation to restore proper stoichiometry of physical interactions or to compensate for pleiotropic effects. But regardless of the specific selection pressures involved, our data support the idea that these adaptations facilitated the evolution of pathogenicity. Although the biological mechanism by which reducing expression of these genes increases virulence remains to be elucidated, it appears to be based in part on increased Htg.
This polygenic adaptation stands in contrast to previously reported cis-regulatory adaptations in several respects. For most known cis-regulatory adaptations, pleiotropy is thought to be minimized as a result of their tissue-specificity (Shapiro et al. 2004; Colosimo et al. 2005; Gompel et al. 2005; Miller et al. 2007; Prud'homme et al. 2007; Wray 2007; Rebeiz et al. 2009, Manceau et al. 2011). Since unicellular species such as yeast cannot reduce pleiotropy in this way, they may be dependent on compensatory mutations to mitigate any unavoidable deleterious pleiotropic effects. Indeed, this is what we observed for the endocytosis complex adaptations, whose pleiotropic effects on morphology have apparently been compensated by other loci. Another point of contrast is that nearly all previous studies of cis-regulatory adaptation have focused on single genes (with a few exceptions [Fraser 2011]) (Shapiro et al. 2004; Colosimo et al. 2005; Gompel et al. 2005; Miller et al. 2007; Prud'homme et al. 2007; Wray 2007; Rebeiz et al. 2009, Manceau et al. 2011). Our results highlight a previously unknown mode of evolution: polygenic cis-regulatory adaptation acting at the level of a physically interacting module.
Methods
Scan for selection on gene expression
We performed a selection scan as previously described (Fraser et al. 2011), using the hypergeometric test for enrichment within the ASE genes (false discovery rate [FDR] < 0.3 as previously estimated) (Gagneur et al. 2009) with higher expression from S-alleles and separately for those with higher expression from Y-alleles. We chose gene sets from the Gene Ontology (http://www.geneontology.org) and from the MIPS protein complex catalog (http://mips.helmholtz-muenchen.de/genre/proj/mpact/) with at least 10 members that had ASE at FDR < 0.3, since this is the minimal number needed to remain significant after Bonferroni correction for multiple testing. The endocytosis-related complex was the only gene set to remain significant after this multiple test correction.
Because adjacent genes can share cis-regulatory information and thus may not be independent in terms of our test, we examined the genomic locations of the genes in the endocytosis complex. The closest pair was separated by over 20 genes, indicating that they are unlikely to share any cis-regulatory elements.
We expect that positive selection on the expression levels of a gene set will result in more frequent ASE within the set than expected by chance; a simple binomial comparing the fraction of S+ to Y+ would fail to capture this. In order to capture both this enrichment of ASE, as well as its biased directionality, we calculated the probability of observing at least 10/17 genes in the endocytosis complex with ASE all in the same direction, as follows. Out of 5988 genes on the array, 974 had S+ ASE at FDR < 0.3, and 451 had Y+ ASE at FDR < 0.3. The probability of a random set of 17 genes containing n genes with S+ ASE and 17 – n without ASE is (974/5988)n × ((5988 − 974 − 451)/5988)17-n × 17!/(n! × (17 − n)!). Summing from n = 10 to 17 yields P = 4.3 × 10−5. The equivalent calculation for Y+ ASE yields 1.8 × 10−8, and summing the two P-values (to make the test two-sided) gives the final probability of 4.3 × 10−5. Another way to arrive at precisely the same P-value is to calculate [probability of n out of 17 randomly chosen genes having ASE] × [probability of all n being S+, or all Y+]. This is: ((974 + 451)/5988)n × ((5988 − 974 − 451)/5988)17-n × 17!/(n! × (17 − n)!) × [(974/(974 + 451))n + (451/(974 + 451))n]. Summing from n = 10 to 17 yields P = 4.3 × 10−5.
Our subsequent discovery that two of the 10 genes did not have observable ASE in the pyrosequencing data was not surprising, considering the estimated FDRs of ASE among the 10 (previously estimated) (Gagneur et al. 2009). The sum of FDRs is an estimate of the number of false positives expected; for the 10 genes this sum is 1.2, indicating that one to two false positives are likely. The discovery of two false positives does not require us to recalculate the significance of the original enrichment, because that would be “penalizing” only those genes with more accurate pyrosequencing data (if we had pyrosequencing data for all genes, we could recalculate the enrichments, but with such data for only a subset this is not possible).
Pyrosequencing
cDNA was generated from the RNA of the S/Y hybrid yeast grown overnight at 30°C in YPD media. We performed pyrosequencing on a Pyromark Q24 (Qiagen), following the manufacturer's protocols. In all cases we verified ASE using multiple SNPs in the gene and/or multiple primer sets. All primer sequences are listed in Supplemental Table 2.
Population-genetic test of selective sweeps
Because most strains of S. cerevisiae lack clear population structure (as assessed by methods such as STRUCTURE) (Schacherer et al. 2009), we decided not to assign strains to discrete populations. Instead, we stratified the 63 strains with genotype data (Schacherer et al. 2009) into three groups of 21 strains each, with different levels of relatedness to a given strain, separately at each gene in the genome.
To define the three sets of 21 strains representing three levels of divergence from a given focal strain, we first summed the total number of sequence differences between the focal strain (Y or S) and each of the other 62 strains in a 1-kb window centered on a particular ORF's 5′ end (i.e., start codon). This sum was our metric of genetic distance: The 21 strains (including the focal strain) with the smallest sum were assigned as the most closely related, etc. We repeated the distance calculation separately for each gene in our analysis (971 S+ ASE genes, including our eight test genes; three additional S+ ASE genes lacked polymorphism data), because individual gene trees can be quite different from the “average” gene tree.
After defining each set of 21 strains at each individual gene, the genetic diversity (θ, also referred to as π) was calculated in a moving window by summing the number of polymorphisms differing between all 210 possible strain pairs within each set of 21 strains in each 1-kb window and then dividing by 2.1 × 105 (the number of allele pairs tested multiplied by the window size). Tajima's D was calculated as described (Tajima 1989). The distribution of θ or D values for the eight candidate genes and the 971 controls (raw data shown in Supplemental Fig. 6) were then compared using a one-sided Wilcoxon test, reflecting our expectation that lower θ or D values among the eight candidate genes would result from a selective sweep. Similar results were seen when using all 10 endocytosis complex ASE genes inferred from the microarray (Table 1) or when using all S/Y ASE genes as controls (θ test minimum P = 0.0055, Tajima's D test minimum P = 0.0054) or when using different numbers of strains (the central window was significant in the θ test at P < 0.01 for all Y-centered strain sets from size 17–35).
The correlations between P-values for different controls—particularly apparent in Figure 2B—may result from two factors. First, the strains closest to S (blue lines in Fig. 2) overlap extensively with the strains furthest from Y (green lines) and also partially with those of intermediate distance to Y (red lines), so their P-values are not independent. Second, genomic properties that influence the density of polymorphisms and are conserved across diverse yeast strains—such as regional variation in mutation/recombination rates, background/purifying selection, etc.—may give rise to correlations even between nonoverlapping sets of strains (e.g., the green and red lines in Fig. 2).
Construction of RH strains
The genetic background of our RH strains would ideally approximate the ancestral strain in which the cis-regulatory mutations of interest arose. Since it is impossible to determine the genotype of this ancestral strain, we chose to model it as closely as possible in two important respects. First, it should have a genotype intermediate between S and Y, as it would have been closely related to the last common ancestor of these two strains; and second, it should have a great deal of heterozygosity, similar to most clinical strains characterized to date (Muller and McCusker 2009). The S/Y hybrid therefore was an ideal choice in both respects since it is the closest possible approximation to the S/Y ancestor, and it is highly heterozygous.
Heterozygous deletion strains in S288c-background (S1002) and YJM145/YJM789-background (YJM385), lys2 and lys5 mutants, respectively, were constructed by replacing specific ORFs with one of kanMX4, hphMX4, or natMX4 dominant drug-resistance cassettes. These cassettes were amplified from plasmids by PCR according to the method described by Goldstein and McCusker (1999). The PCR products were purified and introduced into yeast by lithium acetate–based transformation (Geitz et al. 1995). The transformants were selected on YPD medium, containing G418 (200 μg mL−1), hygromycin B (300 μg mL−1), or nourseothricin (100 μg mL−1) for kanMX4, hphMX4, or natMX4, respectively. Homologous integration was verified by colony PCR at both ends of the integrated cassettes. Sporulation was induced in heterozygous deletion and wild-type strains by growing the strains in YPD overnight at 30°C, followed by incubation for 48 h at 30°C in 0.5% (w/v) potassium acetate. For each gene, the spores of the heterozygous strain were crossed with the spores of the complementary lys mutant wild-type strain (e.g., YFG1-288/yfg1Δ x YFG1-145/YFG1-145 and YFG1-288/YFG1-288 x YFG1-145/yfg1Δ) to make a pair of RH strains. The RH hybrids were selected on SDC-Lys plates by Lys+ complementation. All strains are listed in Supplemental Table 1, and all primer sequences are listed in Supplemental Table 2.
In the absence of ASE and transcriptional feedback (Springer et al. 2010), RH strains are expected have 50% of the wild-type expression level of the hemizygous gene. For our genes of interest, ASE ranges from ∼1.0- to 1.7-fold (Table 1), so RH strains are expected have ∼37%–63% of the diploid hybrid's transcription. In most cases, this is greater than the amount of down-regulation we observed between S and Y, so it may overestimate the effects of each individual down-regulation; however, since only one gene is being down-regulated in each RH strain, compared to at least eight of the endocytosis complex genes in the Y strain, we are likely to underestimate the composite effects of this polygenic adaptation (as long as reducing the level of one subunit has a less extreme effect than reducing the level of multiple subunits).
Measuring fitness in vivo
The 24 RH strains and the Y/S diploid hybrid were grown overnight in YPD media, spun down, resuspended in 0.9 M NaCl, and pooled in equal amounts at a final density of 1.7 × 108 cells/mL, as determined by counting in a hemocytometer. Part of this pool was frozen for subsequent genomic DNA isolation and strain quantification.
The tail veins of six 30-d-old female CD-1 nude mice (Charles River Laboratories, Wilmington MA) were each injected with 100 μL of the pooled yeast, according to the method described by Noble et al. (2010). Three mice were killed after 5 d and the other three after 14 d, and yeast were isolated from kidney homogenates according to the method described by Noble et al. (2010). No yeast were detected in the 14-d samples, but thousands of colonies formed from the 5-d samples.
The abundance of each strain before and after growth in the mice was quantified by qPCR. Genomic DNA was isolated from each pool, and unique regions of each strain (spanning the junction between the selectable marker used in strain construction, and the flanking genomic sequence) were amplified by qPCR. Each primer pair was tested for specificity, and 24/25 strains could be accurately quantified based on control experiments (the exception being VRP1 Y/Δs) (for primer sequences, see Supplemental Table 2). The relative fitness of each strain was then estimated as the difference in the number of PCR cycles required to reach a threshold fluorescence value for the pre- and post-growth competition pools. All RH strains were tested at least in triplicate, and the wild-type Y/S hybrid strain was tested 36 times (three biological replicates, each with 12 technical replicates).
Htg assay
Y/Δs, Y/S, and S/Δy strains were mixed in 1:1:1 ratio and grown in YPD broth for 48 h at 37°C. Aliquots were taken from the growing cultures at 0 h, 24 h, and 48 h. Growth kinetics of these aliquots were followed independently in media containing one of three antibiotics (hyg, kan, and nat) at 30°C to estimate the amount of viable cells of Y/Δs, Y/S, and S/Δy, respectively, at the time-points mentioned (all strains grew equally well at 30°C in both liquid and solid media [Supplemental Figs. 1, 2] so measuring their growth kinetics at this temperature quantifies the abundance of each strain). Growth curves were plotted for all the strains (time on the x-axis, OD on the y-axis). For each replicate, the growth curves at 24 h and 48 h were compared with its growth curve at 0 h. Shift of the growth curve along the x-axis is representative of the strains' abundance after the Htg competition. A shift toward the origin (shorter lag-phase, hence more viable cells) signifies better Htg ability, while a shift away from the origin (longer lag-phase, hence fewer viable cells) signifies the opposite. The abundance ratio for any two growth curves was estimated as 2t/1.5, where t is the x-axis time shift in hours (divided by 1.5 to reflect the ∼1.5-h doubling time at 30°C). For example, to compare the wild-type to RH strain fitness, the abundance ratio of an RH strain before and after Htg was divided by the wild-type hybrid strain's abundance ratio for these same time points. The log2 of this value, representing the relative Htg ability of each RH strain compared with the wild type, is shown in Supplemental Figure 3. Similarly, abundance log2 ratios for pairs of RH strains are shown in Figure 3C and Table 1.
To test if the drug-resistance markers used for each strain are selectively neutral in our growth assays, we created eight hemizygous strains in which four alleles (the Y and S copies of BZZ1 and END3) were replaced by either of two different drug-resistance genes (hphMX4 or natMX4) and then measured the growth of these strains at both 30°C and 37°C, in five replicate experiments performed identically to our other in vitro growth assays. In all cases, we found no significant difference between the two drug-resistance genes when replacing the same allele (Supplemental Fig. 4). Our results are consistent with previous work showing a negligible effect of these genes on fitness, both in vitro and in vivo (Goldstein and McCusker 1999, 2001).
We estimated that fewer than ∼16.7% of yeast genes have diverged in their effects on Htg between Y and S as follows. Previously 72 genes have been tested for effects on Htg between Y and S (Steinmetz et al. 2002; Sinha et al. 2008; H Sinha and JH McCusker, unpubl.), and 12 have been found to be involved in Htg (eight Y+, four S+). However, 12/72 = 16.7% is likely to be an overestimate of genome-wide frequency of Htg-related genes in Y/S, since the 72 include genes from four separate Htg QTL (Steinmetz et al. 2002; Sinha et al. 2008), and at least one gene per QTL is guaranteed to be involved in Htg (each QTL was dissected until the causal gene(s) were identified). In addition, many of the 68 remaining genes were selected for maximal likelihood of affecting Htg (by several criteria, e.g., physical interaction with known Htg genes) (Sinha et al. 2008). However an important caveat is that previous studies have performed Htg assays on solid media, whereas here we have used liquid media. Our result of 11/11 genes showing S/Y divergence in Htg would be significantly (P < 0.05) enriched for Htg divergence as long as the genome-wide frequency of Htg-divergent genes is <76%.
Genetic association analysis
For this analysis, we utilized a data set of 88 densely genotyped strains, consisting of 44 clinical and 44 nonclinical strains (Muller et al. 2011). Clinical origin was thus treated as a categorical (binary) trait, much like disease status in a typical human GWAS. To calculate the strength of association, we performed a Fisher's exact test on all genetic markers within 5 kb of the start of each ORF. This window size was chosen based on previous estimates of the extent of linkage disequilibrium (LD) in S. cerevisiae (Schacherer et al. 2009). Because of this LD and also the fact that not all genetic variants are genotyped with this approach (Schacherer et al. 2009; Muller et al. 2011), we were not able to fine-map the associated variants, or determine if causal variants were protein-coding or noncoding, etc.
Muller et al. (2011) noted the presence of population structure among the 88 strains and estimated a correction factor of λ = 1.67 for the Fisher's exact test results (i.e., the corrected P-value = P1/1.67). While this adjusts our permissive cutoff from 5 × 10−4 to 0.01, it does not affect our enrichment calculations.
Morphological analysis
Frozen cell stocks were streaked out on YPD agar and grown for ∼48 h at 30°C. A single colony was grown to saturation in 3 mL YPD at 30°C with shaking. Saturated cultures were diluted 10-fold to a total volume of 180 μL in YPD in a 96-well plate and grown 4 h with shaking at 30°C. All subsequent fixation and staining was performed in the 96-well plate. Plates were spun at ∼400g to create cell pellets, and media was aspirated. Fixation was performed by adding 200 μL 3.7% formaldehyde, 100 mM potassium phosphate for 45 m; removing the fix; and adding fresh fix for another 45 m. Cells were washed 2× in 200 μL PBS and stained for 2 d in PBS, 100 ng/mL fluorescein isothiocyanate-Con A (Sigma), 2.8 μ Alexa Fluor 594 phalloidin (Molecular Probes). Cells were washed twice in PBS and diluted into Vectashield (Vector Labs), 70 ng/mL DAPI (Sigma) in a glass-bottomed 96-well plate (Matrical MGB096-1-2-LG). Sixty fields per well (∼300 cells) were captured on a Nikon Plan Apo 60x air objective. To prevent photobleaching of the rhodamine-phalloidin stain, a maximum of eight wells were plated and imaged at one time. Image files were prepared using custom Matlab scripts and analyzed using the CalMorph software (http://scmd.gi.k.u-tokyo.ac.jp/datamine/calmorph/) (Ohya et al. 2005). CalMorph divides cells into three cell types (unbudded, budded with one nucleus, and budded with two nuclei) and calculates values for 220 quantitative traits per cell.
To characterize morphological differences between genotypes, we first needed to remove phenotypic redundancy to assure, e.g., that phenotypes represented by many quantitative morphological traits are not overrepresented in our analysis. A common procedure to remove redundancy, principle components analysis, would proceed by creating then transforming a data matrix of cells (across all genotypes) by morphological traits and assumes no or few missing values in this matrix. Because cells are first classified into one of three cell types, however, each cell is missing values for approximately two-thirds of the available morphological phenotypes. Filling these values (with, for example, the mean value for each phenotype across all genotypes) could lead to statistical anomalies. Therefore we assembled three matrices containing cells from only one cell class; Box-Cox transformed the phenotypes to make them normally distributed, replaced rare missing values for each phenotype with the overall phenotype mean, and performed principle component analysis on each matrix. We compared the eigenvalues of this transformation to those resulting from principle component analysis when the data are first randomized to find “significant” components, operationally defined as those components with an eigenvalue greater than that of the randomized matrix. We found nine, 13, and 21 significant principle components in the unbudded, budded with one nucleus, and budded with two nuclei cell classes, respectively. Significant phenotypes from the three cell classes were ranked by their eigenvalues and assigned a phenotype number based on this ranking. Thus, phenotype 1 explains the most variance and phenotype 43 explains the least (Fig. 4D). Four experimental replicates of each genotype were performed resulting in extremely similar mean phenotypic values across all replicates. Thus, we pooled cells across all experimental replicates and reported the mean phenotype values of each genotype. The full data set of each phenotype mean for each strain is provided in Supplemental Table 3.
To compare RH strain pair differences to Y versus S parent strain differences, the fraction of the 43 components differing at Wilcoxon P < 0.05 was counted for each RH strain pair and also for Y versus S. Y versus S differed in 22/43 components; only two RH strain pairs differed by fewer (LAS17 with 20 components differing at P < 0.05, and ECM25 with 21).
Acknowledgments
We thank Marc Feldman, Ryosuke Kita, John McCusker, and Lars Steinmetz for helpful discussions and advice; Sangeeta Kowli for assistance making RH strains; and Alexander Johnson for ideas, advice, and making his laboratory available for the mouse experiments. H.B.F. is supported by the Alfred P. Sloan Foundation, the Pew Scholars in the Biomedical Sciences, and NIH grant 1R01GM097171-01A1. M.L.S. is supported by NSF grant IOS-0642999. H.S. is supported by intramural funds from the Tata Institute of Fundamental Research.
Footnotes
-
↵7 Corresponding author
E-mail hbfraser{at}stanford.edu
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.134080.111.
- Received October 27, 2011.
- Accepted May 22, 2012.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 3.0 Unported License), as described at http://creativecommons.org/licenses/by-nc/3.0/.








