
Detection of common single nucleotide polymorphisms synthesizing quantitative trait association of rarer causal variants
- 1Department of Gene Diagnostics and Therapeutics, Research Institute, National Center for Global Health and Medicine, Tokyo 162-8655, Japan;
- 2Pathogen Genomics Center, National Institute of Infectious Diseases, Tokyo 162-8640, Japan;
- 3Shimane University Hospital, Izumo 693-8501, Japan;
- 4Department of Geriatric Medicine and Nephrology, Osaka University Graduate School of Medicine, Suita 565-0871, Japan;
- 5Amagasaki Health Medical Foundation, Amagasaki 661-0012, Japan
Abstract
Genome-wide association (GWA) studies have identified hundreds of common (minor allele frequency ≥5%) single nucleotide polymorphisms (SNPs) associated with phenotype traits or diseases, yet causal variants accounting for the association signals have rarely been determined. A question then raised is whether a GWA signal represents an “indirect association” as a proxy of a strongly correlated causal variant with similar frequency, or a “synthetic association” of one or more rarer causal variants in linkage disequilibrium (D′ ≈ 1, but r2 not large); answering the question generally requires extensive resequencing and association analysis. Instead, we propose to test statistically whether a quantitative trait (QT) association of an SNP represents a synthetic association or not by inspecting the QT distribution at each genotype, not requiring the causal variant(s) to be known. We devised two test statistics and assessed the power by mathematical analysis and simulation. Testing the heterogeneity of variance was powerful when low-frequency causal alleles are linked mostly to one SNP allele, while testing the skewness outperformed when the causal alleles are linked evenly to either of the SNP alleles. By testing a statistic combining these two in 5000 individuals, we could detect synthetic association of a GWA signal when causal alleles sum up to 3% in frequency. Such signal only partially explains the heritability contributed by the whole locus. The proposed test is useful for designing fine mapping after studying association of common SNPs exhaustively; we can prioritize which GWA signal and which individuals to be resequenced, and identify the causal variants efficiently.
Genome-wide association (GWA) studies have identified hundreds of common (minor allele frequency [MAF] ≥5%) single nucleotide polymorphisms (SNPs) associated with a few hundred traits or diseases, yet the associated SNPs and their proxies mostly do not show evident function related to the target trait, and eventual identification of causal variants accounting for GWA signals has been challenging (Wellcome Trust Case Control Consortium 2007; McCarthy et al. 2008). A question that is then raised is whether a common SNP identified in a GWA study represents an “indirect association” as a proxy of a strongly correlated causal variant with similar frequency, or a “synthetic association” of one or more rarer causal variants that are in linkage disequilibrium (LD) (D′ ≈ 1, but r2 not large) with the common SNP (Cirulli and Goldstein 2010; Dickson et al. 2010).
Synthetic association accounted for GWA signals in several studies. In a GWA study for dose of anticoagulant drug warfarin, the strongest association signal in the CYP2C9 gene was observed at an SNP rs4917639, whose minor allele (frequency 18%) is a composite of two functional alleles CYP2C9*2 (rs1799853, frequency 11%) and CYP2C9*3 (rs1057910, frequency 7%) (Wadelius et al. 2007; Takeuchi et al. 2009). In a GWA study for anemia in patients treated for chronic hepatitis, the strongest signal was observed at an SNP rs6051702 in C20orf194, whose minor allele (frequency 19%) is almost exactly a composite of two causal variants in the neighboring ITPA gene (frequency 8% and 12%) (Fellay et al. 2010). When there are many rare causal variants, but no common one, as in the HBB gene for sickle cell anemia or the GJB2/GJB6 locus for hearing loss, the association of common SNPs detected in GWA studies were attributable to the rare variants (Dickson et al. 2010). Using simulations, Dickson and colleagues showed that synthetic association is likely to occur when there are multiple rare variants in a locus (Dickson et al. 2010).
In general, identification of the causal variants accounting for a synthetic association requires extensive resequencing and association analysis. Instead, here we propose to test statistically whether a quantitative trait (QT) association of an SNP represents a synthetic association or not by inspecting only the QT distribution at each genotype of the SNP, without a priori knowledge about rarer causal variants. We focus on two statistics of the QT distribution: the heterogeneity of variance (i.e., heteroscedasticity) among SNP genotypes and the skewness. The statistical tests were examined in real data of the apolipoprotein E (APOE) gene, and in simulated data for representative models of synthetic association. Moreover, we formulated a general mathematical model of synthetic association, and assessed the test statistics theoretically. The two statistics were suitable for complementary scenarios: Heteroscedasticity was more sensitive than skewness when low-frequency (<5%) causal alleles were linked mostly to one SNP allele, while skewness outperformed when the causal alleles were linked in balance to either of the two SNP alleles. We thus devised a test combining the two statistics, which was powerful for any of the assumed models.
Results
Test of heteroscedasticity
We first show a schematic example of synthetic association and illustrate how QT variance can differ among individuals classified by marker SNP genotypes. We assume a common marker SNP with alleles A and a, and a single causal variant with alleles B1 and b1. The allele B1 (5% in frequency) is always linked to allele A (20% in frequency); thus, existing haplotype classes are AB1, Ab1, and ab1. We assume the QT is normally distributed with the unit variance and the mean equal to 2, 1, and 0 within a subgroup of individuals having genotype B1/B1, B1/b1, and b1/b1, respectively. The QT distribution in the whole population becomes a mixture of the normal distributions combined according to the frequency of genotypes B1/B1, B1/b1, and b1/b1 (Fig. 1A). Individuals with A/A genotype at the marker SNP are enriched with the genotypes of B1/B1 and B1/b1 at the causal variant, thus their QT distribution widens (Fig. 1B). On the contrary, individuals with a/a genotype at the marker all have b1/b1 genotype at the causal variant, and their QT variance equals one (Fig. 1D). The QT variance is the largest in the subgroup with A/A genotype, which is linked more frequently to the low-frequency causal allele B1, and the smallest in the subgroup with a/a genotype. Indeed, QT variance among individuals of a specific marker genotype enlarges proportionally to two factors: the variance of the causal genotype within the subgroup, and the squared effect-size of the causal allele (equation M2). The low-frequency causal variant causes the synthetic association of the marker SNP, and the heteroscedasticity of QT distribution among the marker genotypes.

Probability distribution of the QT value within subgroups classified by marker SNP genotypes. (A) In the whole population, the total QT distribution (gray curve) comprises a mixture of normal distributions (black curves) with unit variance and the mean 0, 1, or 2, which correspond to genotypes b1/b1, B1/b1, and B1/B1 at the causal variant. As genotype B1/B1 is rare (0.25%), the corresponding curve appears flat. (B) QT distribution among individuals with A/A genotype at the marker. As B1/B1 and B1/b1 genotypes are enriched in this subgroup due to LD, the variance is enlarged, as noticeable from the lower peak and wider distribution of the gray curve. (C) Individuals with the A/a genotype have either genotypes b1/b1 or B1/b1, and the QT variance is moderately enlarged. (D) All individuals with a/a genotype at the marker have b1/b1 genotype at the causal variant. The QT variance is 1.10 in A, 1.38 in B, 1.19 in C, and 1 in D.
We could exemplify the detection of synthetic association using heteroscedasticity in the APOE gene, which is known to associate with LDL cholesterol (LDL-C) level through three classical isoforms coded by two functional (or causal) variants—rs7412 (Arg158Cys) and rs429358 (Cys112Arg). As compared with E3 (the most common isoform), E2 (coded by rs7412) and E4 (coded by rs429358) decreased and increased the LDL-C level, respectively (Weisgraber et al. 1981; Weisgraber 1994; Bennet et al. 2007). The two variants had MAF <10% (in Europeans and East Asians) and were not included in SNP chips of GWA scan (except for the recent ones containing >1 million SNPs). In a GWA study for lipids in 1210 Japanese (F Takeuchi, et al., in prep.), we initially found four SNPs near APOE to attain locus-wise significant P-values for LDL-C association, although any of these were not significant after adjustment for the two functional variants. When only the chip SNPs were analyzed, rs405509 and rs377702 showed statistically independent signals of association (Supplemental Fig. 1). In a larger panel of 4840 individuals, the association signals remained at the two chip SNPs, and heteroscedasticity was significant for rs405509 (P = 0.019) (Table 1). Indeed, the causal minor alleles of rs7412 (T) and rs429358 (C) were linked to alternate alleles of rs405509 (C and A, respectively), demonstrating synthetic association (Fig. 2). The two causal variants could simultaneously enlarge the QT variance at all three genotypes of rs405509, and consequently, diminish heteroscedasticity (equation M5). However, in this case, as the effect-size of rs7412 was much larger than that of rs429358, heteroscedasticity remained detectable; rs7412 enlarged the QT variance at C/C genotype of rs405509 to 1.182, whereas rs429358 kept the QT variance at A/A genotype at 0.978. On the other hand, the heteroscedasticity of rs377702 did not reach statistical significance due to its recombination with rs7412 (D′ = 0.34). Thus, even if we identified the association signals at rs405509 and rs377702 via the GWA scan, by detecting heteroscedasticity we could notice the presence of synthetic association and the necessity to search for variants not on the chip.
Testing heteroscedasticity of SNPs in the APOE locus associated with LDL-C
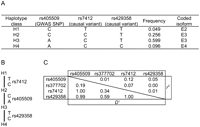
Haplotype classes (A), their phylogeny (B) for the marker SNP rs405509 showing synthetic association of functional variants rs7412 and rs429358 in the APOE locus. LD coefficients between the SNPs associated with LDL-C (C). Haplotype frequencies were calculated using the PLINK software (Purcell et al. 2007).
We next estimated the power to detect synthetic association at an SNP that could be identified in a GWA study. We assumed
that the marker SNP has MAF ≥5%, and that the proportion of QT variance explained by the marker is R2mrk = 0.00592, a borderline level to attain genome-wide significance (see Supplemental Notes). Figure 3 illustrates the statistical power for detecting heteroscedasticity in 5000 individuals. We examined four representative models
of synthetic association by simulation. Under Model 1, there are l causal variants with alleles B1 and b1, B2 and b2, up to Bl and bl, and the low-frequency causal alleles Bi have a uniform effect (e.g., increase QT) and are all linked to marker allele A. The QT variance enlarges for individuals with A/A genotype at the marker since they carry various numbers of the causal alleles, whereas individuals with a/a genotype at the marker carry none. Heteroscedasticity of the marker was detectable (power >0.8) in the region marked with
an asterisk: For example, when the A allele frequency, pA ≥ 45%, or alternatively when pA = 25% and the cumulative frequency of causal alleles is <3%. For a fixed value of pA, the power for detecting heteroscedasticity increases as the cumulative frequency of causal alleles decreases. When pA becomes small, the detectable range narrows; the highest cumulative frequency in the detectable range changes proportionally
to 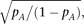 as estimated in equation M12.
as estimated in equation M12.
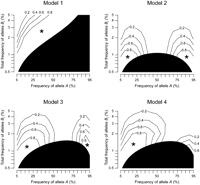
Power for detecting synthetic association by testing heteroscedasticity. The power was computed from simulation under four representative genetic models of synthetic association (see Methods), assuming the strength of marker association (R2mrk) of 0.00592. Horizontal and vertical axes represent the frequency of the marker allele A, and the cumulative frequency of causal alleles Bi (linked to allele A), respectively. The asterisk indicates the region where synthetic association is detectable with power >0.8. The black region of the parameter space should be neglected, as it does not include causal variants accounting for the marker association.
We next examine Models 2–4, where both of the marker alleles are loaded with low-frequency causal alleles. In addition to l causal variants with alleles B1 and b1, B2 and b2, up to Bl and bl, there are m other causal variants with alleles C1 and c1, C2 and c2, up to Cm and cm, and we designate the low-frequency alleles Bi and cj as causal. The two groups of causal alleles, Bi and cj, affect the QT in opposing directions and are linked to alternate alleles A and a of the marker, respectively, and thus synthetically generate the marker association. The QT variance at marker genotype A/A enlarges due to the causal alleles Bi, and the variance at marker genotype a/a enlarges due to the causal alleles cj (equation M3). Indeed, the variances for all marker genotypes increase and become less heterogeneous than under Model 1. Under Model 2, there is exact balance in effect-size and cumulative frequency between the two groups of causal alleles. The heteroscedasticity disappears if pA = 50% (equation M5), and became undetectably weak around the frequency (Fig. 3). The heteroscedasticity was detectable when pA is close to 5% or 95%: For example, when pA = 15% or 85% and the cumulative frequency of the causal alleles Bi, which equals the cumulative frequency of cj, is <1%. Under Models 3 and 4, where the causal alleles Bi and cj are not balanced, heteroscedasticity still disappeared, but around a different marker allele frequency. Under Model 3, the effect-size of the causal variants is uniform, yet the cumulative frequency of alleles cj is half that of alleles Bi, and under Model 4, the cumulative frequencies are identical, yet the effect-size of alleles Cj is half that of alleles Bi. Heteroscedasticity was undetectable around pA = 65% and 80% under Models 3 and 4, respectively. At pA = 25% heteroscedasticity was detectable when the cumulative frequency of Bi alleles was <2%.
Test of skewness
As the test of heteroscedasticity could not detect synthetic association at a certain marker allele frequency around pA = 50%, when both alleles of the marker were loaded with low-frequency causal alleles (Fig. 3, Models 2–4) we introduced the test of skewness to cope with such a case. We observed that synthetic association skews the
QT distribution at the marker genotypes A/A and a/a oppositely (equation M7): QT distribution among individuals with marker genotype A/A is skewed toward the effect direction of causal alleles Bi, and the QT distribution at genotype a/a is skewed toward the opposite direction, which is the effect direction of causal alleles cj. Thus, we added the skewness test statistics for the two genotypes, taking the direction into account (equation M8). Accordingly, under Model 2, the test of skewness could detect synthetic association around pA = 50% (Fig. 4). The detectable range with regard to the cumulative frequency of causal alleles Bi is wide (extends up to 2%) when pA is around 50% and is narrow when near 5% or 95%; as estimated in equation M14, the maximum detectable cumulative frequency is proportional to 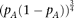 . On the other hand, the skewness test was less powerful than the heteroscedasticity test when all causal alleles are linked
to one marker allele (Model 1 in Fig. 4 vs. Fig. 3).
. On the other hand, the skewness test was less powerful than the heteroscedasticity test when all causal alleles are linked
to one marker allele (Model 1 in Fig. 4 vs. Fig. 3).
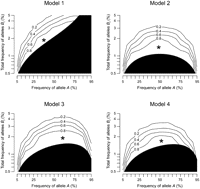
Power for detecting synthetic association by testing skewness. The power was computed from simulation under four representative genetic models, assuming the strength of marker association (R2mrk) of 0.00592. The format of the figure is the same as Figure 3.
Combined test
Between the two tests to detect synthetic association, the test of heteroscedasticity was more powerful when one marker allele was loaded with the causal alleles (Model 1), and the test of skewness was more powerful when both of the marker alleles were loaded with a balanced amount of causal alleles (Model 2). By combining the two tests (equation M10), we devised the third test that was powerful under all of the models (Fig. 5). The detectable range for the cumulative frequency of Bi alleles exceeds 1% when the causal variants are exactly balanced (Model 2), and is up to 2% otherwise. Overall, we could detect synthetic association if the cumulative frequency of all causal alleles, Bi and cj altogether, is <3%.
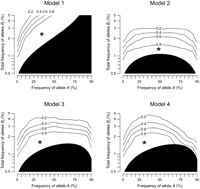
Power for detecting synthetic association by the combined test of heteroscedasticity and skewness. The power was computed from simulation under four representative genetic models, assuming the strength of marker association (R2mrk) of 0.00592. The format of the figure is the same as Figure 3.
The power to detect synthetic association is influenced by the strength of marker SNP association and sample size. So far, we studied association at a borderline level of genome-wide significance, which is much weaker than some reported SNPs, for example, of lipid traits (Chasman et al. 2009). When the strength of association is doubled to R2mrk = 0.0118, synthetic association could be detected if the cumulative frequency of all causal alleles is <6%, in a wider range (Supplemental Fig. 2). When the sample size is halved to 2500 individuals, the detectable region narrowed (Supplemental Fig. 3), because the χ2 statistics of the tests are proportional to the sample size (equations M5 and M9).
Discussion
As seen through mathematical analysis and simulation, we could detect an SNP representing synthetic association of rarer causal variant(s) by testing heteroscedasticity and skewness. The test only requires the genotype–phenotype data obtained in association studies, and the causal variants can be unknown. Whereas previous studies of synthetic association were based on empirical results and simulation (Dickson et al. 2010), we introduced a general mathematical formulation (see Methods) and estimated the variance and skewness of the marker SNP. We also performed computer simulations under representative models of synthetic association and obtained concordant results. The test of heteroscedasticity outperformed the test of skewness when low-frequency causal alleles were linked mostly to one SNP allele, while the test of skewness was better when the causal alleles were linked in balance to either of the two SNP alleles. The test combining the two could detect synthetic association if the cumulative frequency of causal alleles is <3% when tested in 5000 individuals for a marker SNP associated with QT at a borderline level of genome-wide significance (Fig. 5).
Genetic or environmental factors not correlated or interacting with the tested marker SNP do not skew the proposed test statistics. Thus, even when there is allelic heterogeneity, the variants not in LD with the marker SNP have no effects on the test. Although we modeled the causal variants to have an additive effect on QT, the mode of inheritance does not change the results, because homozygotes for a low-frequency allele are rare and negligible. In the power assessment by simulation, we modeled the causal variants to be in complete LD (D′ = 1) with the marker. When LD decays, the heteroscedasticity or skewness at the marker becomes weaker and less detectable. However, since the marker is associated with QT, causal variant(s) of the same directional effect should be loaded mostly to one allele of the marker, thus the decay of LD would be limited.
There are a few limitations in using heteroscedasticity and skewness to detect synthetic association. False positives arise if a causal variant itself shows heteroscedasticity. This can result from a strong gene–environment interaction. Indeed, the test of heteroscedasticity has been used for detecting such interaction (Pare et al. 2010). Another possible source of false positives is population stratification; if two subpopulations have a different mean QT at a specific causal genotype, the QT variance enlarges when the subpopulations are combined. Although a realistic level of population stratification is unlikely problematic (Supplemental Table 1), we recommend applying the test to each cohort separately.
Although the tests we proposed are limited to QTs, the idea of stratifying individuals by marker genotype leads to another test of synthetic association, which is applicable to quantitative as well as dichotomous traits. Here, we compare the association of neighboring common SNPs among the strata. If rare causal alleles are loaded onto the marker allele A, but not on the allele a, neighboring SNPs in LD with the causal variants will show association in the individuals with A/A or A/a genotypes, but not in the individuals with a/a genotype. As a result, the association P-value of neighboring SNPs would be distributed differently among the strata. Contrarily, if the marker SNP represents indirect association, no neighboring SNPs will be associated in any of the strata.
The proposed test can be helpful in understanding the “missing heritability” that GWA studies failed to account for (Maher 2008). If synthetic association is detected at a GWA signal SNP, the heritability of the SNP is likely an underestimate of the heritability of the whole locus (Dickson et al. 2010). Actually, in the APOE example, the explained variance was much smaller for the leading SNPs on a GWA chip (R2 = 0.006 and 0.005) than for the two causal variants (R2 = 0.040 and 0.008; see Table 1). In other words, our method can identify loci that contribute to a trait more than what we would expect from GWA study results.
It is unknown what proportion of GWA signals are due to synthetic association rather than indirect association. The situation is likely to differ by the function and molecular evolution of the genes. For example, several common causal variants are known for pharmacological traits that have not been under evolutionary selection (Cirulli and Goldstein 2010). On the contrary, only rare causal variants are found in genes for renal salt reabsorption, which have been under purifying selection; homozygotes of mutant alleles are susceptible to severe renal salt wasting and hypotension, although heterozygotes confer health benefits from lower blood pressure in postreproductive ages (Ji et al. 2008). Although the proposed test would help detecting synthetic association of a marker SNP, the discovery of the causal variants can require resequencing of a large number of individuals if the causal variants are rare. The aim of the proposed test is to assess potential synthetic association at a particular locus and then use the information to help design future resequencing studies.
Testing synthetic association is useful for designing fine mapping after exhaustively interrogating association of common SNPs at a locus. Exhaustive analysis of common SNPs (MAF ≥5%) is becoming accomplishable by genotyping with SNP chips of the GWA test and by imputing the unassayed SNPs using the HapMap or the 1000 genomes project data. The next focus is to explore rarer variants by resequencing and to identify the causal variants. Since resequencing is still expensive, we need to prioritize which GWA loci and which individuals are to be resequenced. Such information is obtainable by testing synthetic association of the common leading SNP(s), showing the strongest association in a locus. If synthetic association is detected for the leading SNP(s), rarer variants need to be examined in order to pinpoint the variants causing synthetic association. Moreover, if heteroscedasticity is detected, we can discover the causal variants efficiently by resequencing individuals having the homozygote genotype with larger QT variance, and especially those having extreme QT values, who are enriched with the rare causal alleles. Alternatively, if the test for synthetic association is not significant (in >5000 samples), the leading SNP(s) or their proxies are likely causal. Whereas conventional fine-mapping techniques aim to find the causal SNP(s) or haplotype(s) from a set of SNPs tested for association (McCarthy et al. 2008), our method is unique in suggesting that causal variants can be discovered if the study is extended to rarer variants.
Numerous SNP associations have been identified in recent GWA studies, yet our understanding of causal variants is very limited; it is not easy to prove functional changes, let alone the causality with the associated phenotype (Cirulli and Goldstein 2010). We proposed a simple statistical test, which helps to detect whether a common SNP associated with a QT is a noncausal marker in LD with rarer causal variant(s). The proposed test statistic can serve as a milepost in fine mapping and help understand the genetic structure of complex traits.
Methods
Modeling the probability distribution of genotype and QT
We model a QT-associated marker SNP with alleles (referred to as the marker alleles), A and a. We assume the low-frequency allele of each causal variant—which we call the causal allele—is linked exclusively to one of the marker alleles; l causal variants each have alleles B1 and b1, B2 and b2, up to Bl and bl, where the causal allele Bi is linked to marker allele A; m other causal variants each have alleles C1 and c1, C2 and c2, up to Cm and cm, where the causal allele cj is linked to marker allele a. We impose one assumption for mathematical convenience. Among the haplotype classes sharing a specific marker allele (A or a), we assume the probability distribution of causal variant alleles are independent among the variants; for example, among the haplotype classes carrying the A allele, the frequency (conditional on the marker allele being A) of the haplotype class carrying both B1 and B2 should equal the product of the frequencies of classes carrying B1 and B2, which is very small (e.g., 0.01%, if the frequency is 1% both for B1 and B2). The assumed frequency would differ only marginally from the actual frequency: The haplotype class carrying both B1 and B2 does not exist initially if the two causal variants arose separately in the phylogeny, and increases to the assumed frequency by recombination. This assumption enables us to rewrite the test statistics into simple forms (see Supplemental Notes). As we assume Hardy-Weinberg equilibrium, the frequencies of multivariant genotypes can be calculated from those of the haplotype classes.
Each individual's dose of the capital letter alleles, A, Bi, or Cj, is represented by random variables, x, yi, or zj, respectively, and the QT value is represented by a random variable q. The allele Bi (or Cj) is modeled to affect the QT by di (or ej, respectively); specifically, the QT value q of an individual with multivariant genotype (y1,…,yl,z1,…,zm) has the probability density of a normal distribution with the unit variance and the mean of 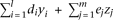 . Thus, the probability density function of the genotype and QT level (x,y1,…,yl,z1,…,zm,q) becomes
. Thus, the probability density function of the genotype and QT level (x,y1,…,yl,z1,…,zm,q) becomes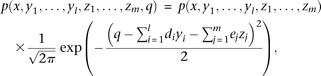 where p(x,y1,…,yl,z1,…,zm) represents the frequency of the multivariant genotype (x,y1,…,yl,z1,…,zm). The expectation of the QT value is
where p(x,y1,…,yl,z1,…,zm) represents the frequency of the multivariant genotype (x,y1,…,yl,z1,…,zm). The expectation of the QT value is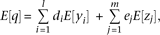 where E[·] represents the expectation (see equation S5 in the Supplemental Notes for derivation). Similarly, when conditioned on
a specific genotype x0 at the marker,
where E[·] represents the expectation (see equation S5 in the Supplemental Notes for derivation). Similarly, when conditioned on
a specific genotype x0 at the marker,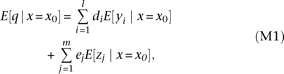 where E[·|x=x0] represents the conditional expectation.
where E[·|x=x0] represents the conditional expectation.
Variance
The variance of QT among individuals having a specific marker genotype x0 becomes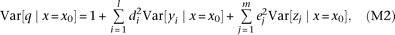 where Var[·|x=x0] represents the conditional variance (see Supplemental equation S6 for derivation). Equation M2 indicates that the inflation (above one) of QT variance decomposes into a sum of terms, each corresponding to one causal
variant and determined by the square of the effect-size, di or ej, and the conditional variance of genotype. Designating the frequencies of alleles A, Bi and cj as pA, pBi, and
where Var[·|x=x0] represents the conditional variance (see Supplemental equation S6 for derivation). Equation M2 indicates that the inflation (above one) of QT variance decomposes into a sum of terms, each corresponding to one causal
variant and determined by the square of the effect-size, di or ej, and the conditional variance of genotype. Designating the frequencies of alleles A, Bi and cj as pA, pBi, and  , respectively, the genotype variance becomes
, respectively, the genotype variance becomes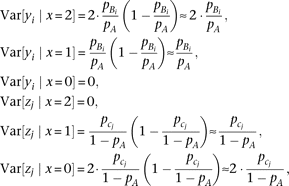 where the approximation is under
where the approximation is under 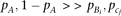 . By substituting the genotype variance into equation M2, we obtain
. By substituting the genotype variance into equation M2, we obtain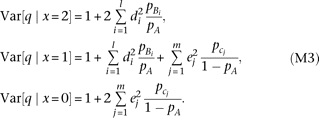 Thus, the QT variance at marker genotypes x = 2 (A/A) and x = 0 (a/a) is determined by the contribution of alleles Bi and cj, respectively, and the average of the two variances equals the variance at genotype x = 1 (A/a). The average of three variances weighted by marker genotype frequency becomes
Thus, the QT variance at marker genotypes x = 2 (A/A) and x = 0 (a/a) is determined by the contribution of alleles Bi and cj, respectively, and the average of the two variances equals the variance at genotype x = 1 (A/a). The average of three variances weighted by marker genotype frequency becomes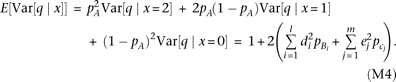 We test the heterogeneity of QT variance (i.e., heteroscedasticity) among marker genotypes using Bartlett's test (Bartlett 1937). The χ2 statistic (two degrees of freedom) is expected to become
We test the heterogeneity of QT variance (i.e., heteroscedasticity) among marker genotypes using Bartlett's test (Bartlett 1937). The χ2 statistic (two degrees of freedom) is expected to become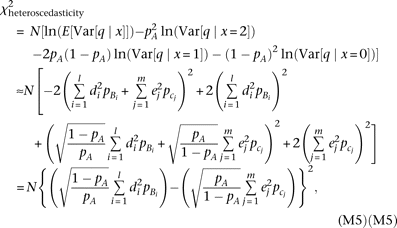 when the sample size N is large (thus, the constant for the Bartlett's test statistic equals one); for the second equality, equations M3 and M4 were substituted, and ln(1 + x) was approximated as x−x2/2. In the curly brackets of the final formula in equation M5, the contribution by causal alleles Bi (linked to marker allele A) is subtracted by the contribution of alleles cj (linked to allele a). Thus, the statistic for heteroscedasticity is maximized when all low-frequency causal alleles are linked to the same marker
allele, and diminishes when they are linked evenly to both of the marker alleles.
when the sample size N is large (thus, the constant for the Bartlett's test statistic equals one); for the second equality, equations M3 and M4 were substituted, and ln(1 + x) was approximated as x−x2/2. In the curly brackets of the final formula in equation M5, the contribution by causal alleles Bi (linked to marker allele A) is subtracted by the contribution of alleles cj (linked to allele a). Thus, the statistic for heteroscedasticity is maximized when all low-frequency causal alleles are linked to the same marker
allele, and diminishes when they are linked evenly to both of the marker alleles.
Skewness
The third central moment of the QT distribution among the individuals having a specific marker genotype x0 is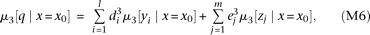 which decomposes into a sum of terms, each contributed by one causal variant (see Supplemental equation S7 for derivation);
μ3[·|x=x0] represents the conditional third central moment. The third central moment of the genotypes yi and zj conditional on a marker genotype becomes
which decomposes into a sum of terms, each contributed by one causal variant (see Supplemental equation S7 for derivation);
μ3[·|x=x0] represents the conditional third central moment. The third central moment of the genotypes yi and zj conditional on a marker genotype becomes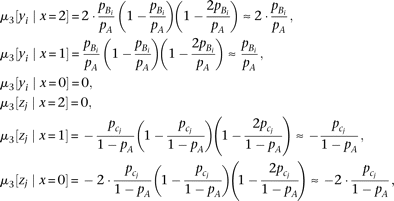 where the approximation is under
where the approximation is under 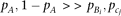 . By substituting the genotype moment into equation M6, we obtain
. By substituting the genotype moment into equation M6, we obtain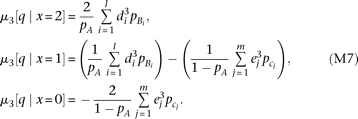 The z statistic (standard normal distribution) for testing skewness (Stuart et al. 1999) of QT among the individuals with genotype x0 becomes
The z statistic (standard normal distribution) for testing skewness (Stuart et al. 1999) of QT among the individuals with genotype x0 becomes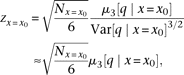 where
where 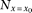 is the number of the individuals, and the variance in the denominator is approximated as one for this statistic. By substituting
equation M7, and converting
is the number of the individuals, and the variance in the denominator is approximated as one for this statistic. By substituting
equation M7, and converting 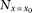 into N multiplied by the marker genotype frequency,
into N multiplied by the marker genotype frequency,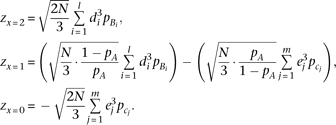 As the QT distributions at the two homozygote marker genotypes should be skewed to opposite directions under synthetic association,
zx = 2 and zx = 0 would have opposite signs. We take their difference and obtain a χ2 statistic with one degree of freedom,
As the QT distributions at the two homozygote marker genotypes should be skewed to opposite directions under synthetic association,
zx = 2 and zx = 0 would have opposite signs. We take their difference and obtain a χ2 statistic with one degree of freedom,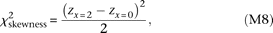 which we adopt as the test statistic for skewness. The test statistic is expected to become
which we adopt as the test statistic for skewness. The test statistic is expected to become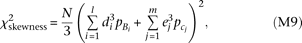 reflecting the contribution by causal alleles Bi and cj.
reflecting the contribution by causal alleles Bi and cj.
Statistical tests of synthetic association and type I error rate
We combine two types of statistics, heteroscedasticity and skewness, to devise the combined test. Using Fisher's method, the
P-values for testing heteroscedasticity and skewness, pheteroscedasticity and pskewness, respectively, are combined as a χ2 statistic (four degrees of freedom),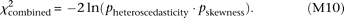 The significance level was set to 0.05 for all tests.
The significance level was set to 0.05 for all tests.
Before testing, we applied rank-based inverse normal transformation (Blom 1958) to the whole QT distribution. The transformation avoids detecting spurious signals when the QT distribution is skewed as
a whole. The transformed QT value qi of the i-th individual is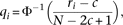 where ri is the rank of the individual, N is the total number of individuals, c = 3/8, and Φ−1 is the standard normal quantile. We strongly recommend applying the transformation, although it can cause false positives
when the marker association is extremely strong, as explained below.
where ri is the rank of the individual, N is the total number of individuals, c = 3/8, and Φ−1 is the standard normal quantile. We strongly recommend applying the transformation, although it can cause false positives
when the marker association is extremely strong, as explained below.
Type I error rate of the tests were assessed from simulated and empirical data. Under the “null hypothesis” of indirect association, we inspected the distribution of nominal P-value, and assessed the test as accurate, conservative, or anticonservative, if the actual type I error rate was equal, smaller, or larger than the nominal P-value, respectively; an anticonservative test cannot be used. For a marker showing association at a borderline level of genome-wide significance (R2mrk = 0.00592), the heteroscedasticity test was accurate, but the skewness test tended to be conservative, due to the inverse normal transformation (Supplemental Fig. 4); this was not calibrated. As the tests for heteroscedasticity and skewness were not correlated, they could be combined using Fisher's method. When the marker association was as large as R2mrk = 0.1, which is exceptional for GWA signals, the inverse normal transformation caused spurious heteroscedasticity and skewness, thus the proposed tests were not valid. For gene expression data (Stranger et al. 2007), the heteroscedasticity test was accurate, and the skewness test was slightly conservative (Supplemental Fig. 5; Supplemental Table 2).
Models for simulation
If the strength of the marker SNP association 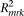 , and the frequency of variants
, and the frequency of variants 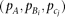 are specified, we can calculate the effect-size of the causal variants (di,ej) by solving Supplemental equation S3, and determine the genetic model. We systematically explore four representative models
of synthetic association by simulation. (Plots of di according to variant frequency are shown in Supplemental Fig. 6.)
are specified, we can calculate the effect-size of the causal variants (di,ej) by solving Supplemental equation S3, and determine the genetic model. We systematically explore four representative models
of synthetic association by simulation. (Plots of di according to variant frequency are shown in Supplemental Fig. 6.)
Model 1
All causal alleles linked to marker allele A have identical effect-size, and there are no causal alleles linked to allele a. By solving Supplemental equation S3 under 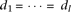 and m = 0,
and m = 0,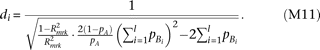 By substituting this to the test statistic of heteroscedasticity (equation M5), and solving for
By substituting this to the test statistic of heteroscedasticity (equation M5), and solving for 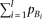 ,
,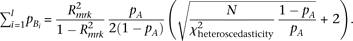 Thus, when R2mrk = 0.00592 and N = 5000, heteroscedasticity is detectable at a power >0.8 under a significance level of 0.05 (which requires a noncentrality
parameter of χ2 > 9.64 for the χ2 distribution with two degrees of freedom; “+2” in the parenthesis is negligible) if
Thus, when R2mrk = 0.00592 and N = 5000, heteroscedasticity is detectable at a power >0.8 under a significance level of 0.05 (which requires a noncentrality
parameter of χ2 > 9.64 for the χ2 distribution with two degrees of freedom; “+2” in the parenthesis is negligible) if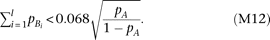 Heteroscedasticity is detectable if the cumulative frequency of causal alleles (left term) is smaller than a certain function
of the frequency of the marker allele A (right term); the detectable range with regard to
Heteroscedasticity is detectable if the cumulative frequency of causal alleles (left term) is smaller than a certain function
of the frequency of the marker allele A (right term); the detectable range with regard to 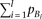 is wider when pA is larger.
is wider when pA is larger.
Model 2
Causal alleles are linked to the two marker alleles in a balanced way, such that the effect-size is uniform, as 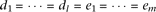 , and the cumulative frequencies equal between causal alleles Bi (linked to marker allele A) and causal alleles cj (linked to marker allele a), as
, and the cumulative frequencies equal between causal alleles Bi (linked to marker allele A) and causal alleles cj (linked to marker allele a), as 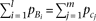 . By solving Supplemental equation S3 under the constraints,
. By solving Supplemental equation S3 under the constraints,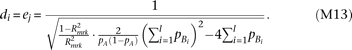 By substituting this (approximating the last term in the square-root as zero) to the test statistic of skewness (equation M9), and solving for
By substituting this (approximating the last term in the square-root as zero) to the test statistic of skewness (equation M9), and solving for 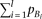 ,
,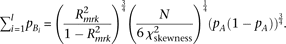 Thus, when R2mrk = 0.00592 and N = 5000, skewness is detectable at a power >0.8 under a significance level of 0.05 (which requires a noncentrality parameter
of χ2 > 7.85 for the χ2 distribution with one degree of freedom) if
Thus, when R2mrk = 0.00592 and N = 5000, skewness is detectable at a power >0.8 under a significance level of 0.05 (which requires a noncentrality parameter
of χ2 > 7.85 for the χ2 distribution with one degree of freedom) if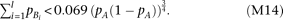 Skewness is detectable if the cumulative frequency of causal alleles Bi (left term) is smaller than a certain function of the frequency of the marker allele A (right term); the detectable range with regard to
Skewness is detectable if the cumulative frequency of causal alleles Bi (left term) is smaller than a certain function of the frequency of the marker allele A (right term); the detectable range with regard to 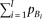 is widest when pA is around 0.5.
is widest when pA is around 0.5.
Model 3
The effect-size of causal alleles is uniform, as 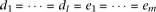 , yet the cumulative frequency of the causal alleles Bi is twice the cumulative frequency of causal alleles cj, as
, yet the cumulative frequency of the causal alleles Bi is twice the cumulative frequency of causal alleles cj, as 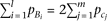 . Then,
. Then,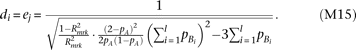
Model 4
The cumulative frequencies are equal between causal alleles linked to the two marker alleles, as 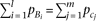 , yet the effect-size of causal alleles Bi is twice the effect-size of causal alleles cj, as
, yet the effect-size of causal alleles Bi is twice the effect-size of causal alleles cj, as 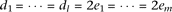 . Then,
. Then,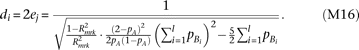
Power assessment by simulation
We assessed the power of the three tests under each of the four models by simulation. In any of the models, we assumed that
effect-size equals among the causal alleles linked to the same marker allele (i.e., 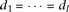 and
and 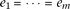 ). In such a case, the tests remain the same if instead there was one composite allele of Bi's and another composite of cj's. Using this property, we actually simulated the special case with one causal allele linked to each one of the marker alleles;
the simulation results apply to the general case with multiple causal alleles.
). In such a case, the tests remain the same if instead there was one composite allele of Bi's and another composite of cj's. Using this property, we actually simulated the special case with one causal allele linked to each one of the marker alleles;
the simulation results apply to the general case with multiple causal alleles.
Simulations were performed under the following parameter values; R2mrk = 0.00592, 0.0118; 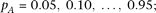
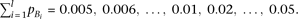 Other parameters—
Other parameters—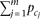 , di, and ej—were determined according to constraints. We randomly generated 5000 (or 2500) individuals using simulation and applied the
tests. The power was assessed from 1000 simulation trials. We used the R software for computation.
, di, and ej—were determined according to constraints. We randomly generated 5000 (or 2500) individuals using simulation and applied the
tests. The power was assessed from 1000 simulation trials. We used the R software for computation.
Acknowledgments
We thank the participants in the lipid study and Drs. Toru Nabika (Shimane University), Tomohiro Katsuya (Osaka University), and Yukio Yamori (Mukogawa Women's University). We also thank anonymous reviewers for their constructive comments. This work was supported by the Program for Promotion of Fundamental Studies in Health Sciences of the National Institute of Biomedical Innovation Organization; a Grant of the National Center for Global Health and Medicine; and the Ministry of Health, Labor and Welfare.
Footnotes
-
↵6 Corresponding author.
E-mail fumihiko{at}takeuchi.name.
-
[Supplemental material is available for this article. The synthetic association test software is freely available at http://www.fumihiko.takeuchi.name/PUBLICATIONS/synthetic.R.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.115832.110.
- Received September 24, 2010.
- Accepted March 9, 2011.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press








