
The making of a new pathogen: Insights from comparative population genomics of the domesticated wheat pathogen Mycosphaerella graminicola and its wild sister species
- Eva H. Stukenbrock1,5,6,
- Thomas Bataillon1,
- Julien Y. Dutheil1,
- Troels T. Hansen1,
- Ruiqiang Li2,
- Marcello Zala3,
- Bruce A. McDonald3,
- Jun Wang2,4 and
- Mikkel H. Schierup1
- 1Bioinformatics Research Center, Aarhus University, C.F. Moellers Alle, DK-8000 Aarhus C, Denmark;
- 2BGI-Shenzhen, Shenzhen 518083, China;
- 3ETH Zurich, Institute of Integrative Biology, 9082 Zurich, Switzerland;
- 4Department of Biology, University of Copenhagen, DK-2200 Copenhagen, Denmark
Abstract
The fungus Mycosphaerella graminicola emerged as a new pathogen of cultivated wheat during its domestication ∼11,000 yr ago. We assembled 12 high-quality full genome sequences to investigate the genetic footprints of selection in this wheat pathogen and closely related sister species that infect wild grasses. We demonstrate a strong effect of natural selection in shaping the pathogen genomes with only ∼3% of nonsynonymous mutations being effectively neutral. Forty percent of all fixed nonsynonymous substitutions, on the other hand, are driven by positive selection. Adaptive evolution has affected M. graminicola to the highest extent, consistent with recent host specialization. Positive selection has prominently altered genes encoding secreted proteins and putative pathogen effectors supporting the premise that molecular host–pathogen interaction is a strong driver of pathogen evolution. Recent divergence between pathogen sister species is attested by the high degree of incomplete lineage sorting (ILS) in their genomes. We exploit ILS to generate a genetic map of the species without any crossing data, document recent times of species divergence relative to genome divergence, and show that gene-rich regions or regions with low recombination experience stronger effects of natural selection on neutral diversity. Emergence of a new agricultural host selected a highly specialized and fast-evolving pathogen with unique evolutionary patterns compared with its wild relatives. The strong impact of natural selection, we document, is at odds with the small effective population sizes estimated and suggest that population sizes were historically large but likely unstable.
The ascomycete fungus Mycosphaerella graminicola is a major pathogen of wheat with a global distribution. M. graminicola originated in the Middle East ∼11,000 yr ago from wild species infecting other grasses (Stukenbrock et al. 2007). Speciation was associated with adaptation to domesticated wheat and its associated agro-ecosystem. In a previous study, we characterized the two progenitor pathogen species Mycosphaerella S1 and Mycosphaerella S2 (hereafter called S1 and S2) endemic to the Middle East. S1 and S2 were isolated from three distantly related wild grass species: Elymus repens, Dactylis glomerata, and Lolium multiflorum. M. graminicola is mainly found on cultivated wheat but is able to infect other grass species (Seifbarghi et al. 2009). The recent divergence of M. graminicola, S1, and S2 implies that only a limited number of evolutionary changes are likely to have occurred. Here we use a population genomics approach to investigate the extent of genome divergence between M. graminicola, S1, and S2. We identify footprints of natural selection and study patterns of genome evolution. We compare the evolution of a pathogen species on a domesticated host with evolution on “wild” host species. In plants and animals, domestication is often associated with genome-wide increased rates of nonsynonymous mutations resulting from a relaxation of purifying selection (Wang et al. 1999; Dobney and Larson 2006; Lu et al. 2006; Cruz et al. 2008). In studies of domesticated fungal species the trend is less clear. A comparison of laboratory and wild strains of the yeast fungus Saccharomyces cerevisiae showed an increase in nonsynonymous substitution rate in the domesticated species, indicating a relaxation of purifying selection (Gu et al. 2005). But in the fungus Aspergillus oryzae, also domesticated for use in fermentation of food products, no visible effects of domestication on the proteome were found compared with its wild sister lineage Aspergillus flavus (Rokas 2009). Besides the environmental change associated with domestication itself, other parameters such as population size, clonal versus sexual reproduction, and demography may explain the different outcomes from evolutionary studies of domesticated species. The effect of “domestication” of a parasite, i.e., coevolution with its host during domestication, has so far never been studied. A parasite on a domesticated host is also expected to experience strong directional selection generated by changes in the host. The closely related Mycosphaerella species allowed us to investigate the evolutionary consequence of coevolution and “domestication” of M. graminicola by comparing evolutionary rates in the agricultural and the wild species.
We hypothesize that sustained selection on wild hosts in S1 and S2 and adaptation to a new domesticated host in M. graminicola have been the primary drivers of the speciation process in these pathogens. Genes encoding secreted proteins potentially interact with host molecules and are therefore subjected to heterogeneous selection pressures imposed by hosts. These genes may therefore evolve more rapidly than other genes during host specialization. Positive selection in secreted proteins has been found in several plant pathogens, supporting the hypothesis that the evolution of these proteins is driven by coevolutionary arms races with host molecules (Rohmer et al. 2004; Stukenbrock and McDonald 2007; Win et al. 2007; Barrett et al. 2009; Stukenbrock et al. 2010). Our genome data set allows us to assess the extent of adaptive evolution in proteins targeted for secretion and to compare this with the extent of adaptive evolution in protein-coding genes throughout the genome. We furthermore aimed at identifying positively selected genes as candidates playing a major role in speciation or host specialization.
In a previous study we investigated the evolutionary history of the Mycosphaerella pathogens by applying an “Isolation with Migration” coalescence model (Nielsen and Wakeley 2001) to a small sequence data set from a population sample of the species (Stukenbrock et al. 2007). We estimated recent speciation times and ancient migration rates between the different species. The evolutionary genomic approach we apply here also includes a coalescence analysis that infers parameters of the ancestral species and the process of species divergence (Hobolth et al. 2007; Dutheil et al. 2009). It has previously been applied to the great apes to estimate ancestral population sizes and speciation times as well as to infer the amount of incomplete lineage sorting that results in incongruence between the genealogy of some regions of the genome and the species phylogeny (Dutheil et al. 2009).
Results and Discussion
Host specialization among closely related pathogen species
A detached leaf assay was used to measure virulence on cultivated and wild hosts through the percentage of leaf area covered by fungal fruiting structures called pycnidia (Supplemental Fig. S1). Detached leaves of Triticum aestivum (cultivar Drifter), E. repens, D. glomerata, L. perenne, and L. mulitiflorum, corresponding to the original host genera of the three fungal species, were inoculated with a standardized spore suspension for each pathogen strain. We also included the more distantly related Mycosphaerella pathogen Septoria passerinii in the experiment. The four species are related as outlined in Figure 1. M. graminicola is more virulent on the wheat host (P = 0.001) than the other species (Fig. 1, lower panel) but retained the ability to infect and propagate on other hosts (Table 1). S1 and S2 showed limited ability to infect wheat compared with M. graminicola. S1 is more virulent (P = 0.001) on L. multiflorum, while S2 is more virulent on D. glomerata (P = 0.03) (Fig. 1). We found a high variability in susceptibility among different plant individuals for each of the wild grasses, which likely is due to the fact that the seeds we used for testing originated from a genetically heterogeneous population. The variation for susceptibility among plants of the outcrossing wild grass hosts was considerably higher than among the wheat plants that all originated from one inbred cultivar. We conclude that these pathogens can show significant host adaptation even though they may coexist on the same host.
Percentage of leaf area covered by pycnidia on five different host species inoculated by isolates of M. graminicola, S1, S2, and S. passerinii
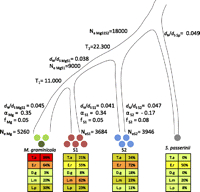
Species tree of M. graminicola, S1, S2, and S. passerinii. Circles indicate the number of genomes sequenced in each species. In our analyses we used the reference sequence of M. graminicola marked as a dark green circle. The lower panels of the figure summarize the ability of each species to infect the five grass species (T.a.) Triticum aestivum, (E.r.) Elymus repens, (D.g.) Dactylis glomerata, (L.m.) Lolium multiflorum, and (L.p.) Lolium perenne. The extent of host adaptation was estimated as percentage of leaf area covered by pycnidia among the different host species. Colors corresponding to the degree of infection are shown as 0%–20% (light green), 21%–40% (brownish green), 41%–60% (yellow), 61%–80% (orange), 81%–100% (red). We used a coalescence approach to estimate the split between M. graminicola and S1 (T1) and the more ancient split between S2 and the M. graminicola-S1 branch (T2). The population sizes of the ancestral species are indicated as NeMgS1 and NeMgS1S2 parameters. The evolutionary rate is estimated as the ratio of nonsynonymous to synonymous mutation rates dN/dS for each branch. Shown are also estimates of the parameter α quantifying the extent of adaptive evolution and the parameter f as a measure of the strength of purifying selection in each of the branches of M. graminicola, S1, and S2 using S. passerinii as the outgroup species. Contemporary effective population sizes were estimated from polymorphism data for each species and are given as NeMg, NeS1, and NeS2.
Generation of a population genomic data set
To infer patterns of genome evolution in M. graminicola, S1, S2, and the barley pathogen S. passerinii, we sequenced the genomes of 12 fungal individuals (two M. graminicola, five S1, four S2, and one S. passerinii) (Fig. 1). The isolates of M. graminicola, S1, and S2, were all collected at the center of origin of the pathogen in the Middle East and were selected according to their host species, mating type locus, and previously determined genomic fingerprint (Supplemental Table S1; Stukenbrock et al. 2007). De novo assembly of paired-end Illumina reads resulted in >40× coverage genome sequences (Supplemental Table S2). We compared contigs using the annotated reference genome of M. graminicola (Goodwin et al. 2011) as the backbone to construct a 12 × 28 Mb alignment (Supplemental Table S3). The genome alignment was compared with electrophoretic karyotypes of four of the isolates to assess collinearity of chromosomes. The genomes show a high degree of synteny for the 13 essential chromosomes but a considerably lower degree of synteny among the smaller dispensable chromosomes (chromosomes 14–21 in the sequenced reference isolate) (Mehrabi et al. 2007; Wittenberg et al. 2009; Goodwin et al. 2011). Interestingly, we find that six and four of the small chromosomes are also dispensable in S1 and S2, respectively, as revealed by whole chromosome presence/absence polymorphisms (Supplemental Table S4). We conclude that presence/absence polymorphism of whole chromosomes is a trait shared among the three species, consistent with the hypothesis that dispensable chromosomes were already present in the ancestors of M. graminicola.
Polymorphism and divergence in the M. graminicola species complex
The genomes of M. graminicola and S1 are on average 6% divergent at the nucleotide level, while M. graminicola and S2 are diverged by 10% (Supplemental Table S5). The amount of divergence (Dxy) and within-species nucleotide diversity (π) increases with decreasing chromosome size (Supplemental Fig. S2). Average divergence between M. graminicola and S1 is only three to four times higher than the polymorphism within each species. Given this modest amount of divergence, we expect some polymorphisms to predate speciation and thus to be shared between the two species. Between M. graminicola and S2 we do not expect polymorphism to be shared by descent to the same extent. Alternatively, shared polymorphisms may result from recurrent mutations, introgression, and/or balancing selection. We investigated shared polymorphisms by counting the proportion of shared polymorphic sites in 10-kb windows and distinguishing between sites segregating for the same polymorphism (type 1) and sites that actually contain different polymorphisms (type 2) (Supplemental Fig. S2; Supplemental Table S5).
We found higher amounts of shared polymorphisms between the two most closely related species (Supplemental Table S5), supporting the view that all shared polymorphisms cannot be derived by recurrent mutation. Furthermore, M. graminicola and S1 exhibit relatively more type 1 than type 2 shared sites, suggesting that a sizeable fraction of type 1 sites indeed are shared by descent (Fig. 2). The proportion of type 2 sites is similar between the M. graminicola and S1 and M. graminicola and S2 comparisons, suggesting that these most likely are caused by recurrent mutation. The increase in type 2 polymorphisms with decreasing chromosome size is again likely due to the higher amounts of polymorphisms in all species on smaller chromosomes. However, the excess of type 1 polymorphisms in M. graminicola and S1 shows a less clear correlation with chromosome sizes (Fig. 2). We hypothesize that changes in effective population size along the genomes can be important in determining how much polymorphism is shared between species (see also coalescent analysis below).
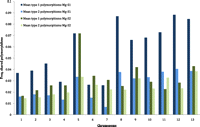
Proportion of polymorphic sites in M. graminicola that are also polymorphic in S1. Shared polymorphisms are distinguished as either type 1: same nucleotide variation between species; or type 2: different nucleotide polymorphisms between species.
Finally, some windows exhibit extremely high proportions of shared polymorphisms, which are best explained by balancing selection or recent introgression (Supplemental Fig. S3). Haplotype comparisons in these windows do not show increased similarity between species (data not shown) as expected if shared polymorphisms were originating from introgression. Balancing selection therefore remains as a likely hypothesis to be tested by functional studies of candidate regions.
Footprints of natural selection during pathogen domestication
We investigated patterns of evolution and natural selection in the domesticated and wild species in two ways: (1) by a phylogenetic analysis of patterns of nonsynonymous and synonymous substitution rates between species (Yang 2007) from all aligned genes and (2) by contrasting amounts of synonymous and nonsynonymous substitutions between species with patterns of within-species polymorphism using a modified McDonald-Kreitman approach (Welch 2006). When referring to estimation of evolutionary rates obtained from branch-specific models according to Yang (Yang and Nielsen 2000) implemented in the CODEML program, we use the term dN/dS. For analyses based on the Nei and Gojobori method (Nei and Gojobori 1986; Nei and Kumar 2000) and the McDonald-Kreitman approach (McDonald and Kreitman 1991; Eyre-Walker 2006; Welch 2006), we use the terms Ka/Ks for the between-species ratio of nonsynonymous to synonymous substitution and PN/PS for the within-species ratio of nonsynonymous to synonymous mutations (for parameter definitions, see Supplemental Table S6).
We first estimated dN/dS on the four species phylogeny of M. graminicola, S1, S2, and S. passerinii. We find that purifying selection—as measured through the dN/dS ratio—is strong in the Mycosphaerella species complex with an average dN/dS of only 0.045. This value is considerably lower than values obtained from whole genome analyses of two other ascomycetes. In the domesticated A. oryzae species, Rokas estimated a dN/dS ratio of 0.45 and in the wild sister species A. flavus 0.51 (Rokas 2009). In laboratory and wild strains of S. cerevisiae, dN/dS ratios of 0.20 and 0.10, respectively, were reported (Gu et al. 2005). The branch-specific dN/dS is significantly larger on the terminal M. graminicola branch compared with its closest relative S1 (dN/dS = 0.041; paired Wilcoxon test of 88,495 aligned codons on the 13 essential chromosomes, p = 3.7 × 10−5) (Fig. 3, dark blue circles). Interestingly, when comparing the “wild” pathogen species S1 and S2 and using S. passerinii as outgroup, we find that S2 has a higher dN/dS ratio than both M. graminicola and S1 (Fig. 1). To interpret the meaning of these differences, we note that dN/dS can increase either due to relaxed purifying selection or increased positive selection as both processes inflate the rate of nonsynonymous mutations. The relative roles of positive and purifying selection can be disentangled by analyzing jointly the counts of polymorphisms and divergence in coding regions with an approach developed by Eyre-Walker (2006) and extended by Welch (2006). We pooled the McDonald-Kreitman tables (McDonald and Kreitman 1991) of aligned genes for each chromosome and found that purifying selection in M. graminicola and S1 was very strong: ∼95% of nonsynonymous mutations were selected against, while in S2 the proportion was 92% (Fig. 1). S. passerinii was consistently used as outgroup in the analyses of adaptive and purifying selection to account for the high amount of incomplete lineage sorting between M. graminicola, S1, and S2 (see below). For individual chromosomes, there is no clear trend for differential amounts of purifying selection between M. graminicola and S1, suggesting that deleterious mutations were purged with the same efficacy on the different chromosomes in these two species (Fig. 3, light blue circles).
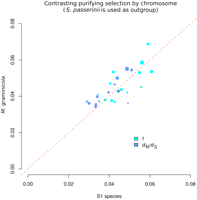
Contrasting evolutionary rates on the M. graminicola and S1 branches of the phylogeny. Plotted is dN/dS in M. graminicola versus S1 (blue dots) and the parameter “f” (green dots) showing the fraction of nonsynonymous mutations experiencing strong purifying selection as a measure of purifying selection in the two species. Each dot represents the average measure of a chromosome, and the size of the dot corresponds to the relative sizes of the different chromosomes (on a log scale). While dN/dS is significantly higher in M. graminicola, the strength of purifying selection is highly similar in the two species. S. passerinii is used as the outgroup in both analyses.
The proportion, alpha (α), of the remaining amino acid changing mutations that were fixed by positive selection is overall marginally higher in M. graminicola compared with S1 and negative (although not significantly different from zero) in S2. In M. graminicola ∼35% of the nonsynonymous mutations were fixed by positive selection, while signals of adaptive evolution were absent in S2. This also further argues that the increased dN/dS of the M. graminicola branch is driven at least partly by adaptive evolution.
Next, we correlated the ratio of amounts of nonsynonymous to synonymous polymorphism (PN/PS) in M. graminicola to recombination rates across chromosomes. Recombination rate estimates were computed in adjacent 100-kb windows using a coalescence analysis described in detail below. We found a weak but significant negative correlation between PN/PS and recombination rate in M. graminicola (Kendall's rank correlation, tau = −0.186, P = 0.0012) (Fig. 4A). The negative correlation may indicate a greater efficacy of selection in the M. graminicola genome compared with the other two species and again argues against a relaxation of purifying selection during domestication in M. graminicola. Taken together, the different analyses all support that the increase in dN/dS on the M. graminicola branch compared with its wild sister species results from higher amounts of adaptive evolution rather than mere accumulation of deleterious mutations.
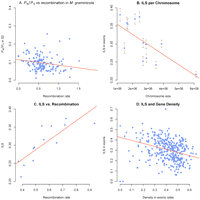
(A) Plot showing the negative correlation between PN/PS in M. graminicola and recombination rate. Both parameters were estimated as averages in windows of 100 kb (160 windows). One outlier window with an average value of PN/PS = 0.5 was excluded from the plot and regression analyses. (B) The frequency of incomplete lineage sorting (ILS) in exons plotted against chromosome size (chromosome 1–13) measured in base pairs. The strong negative correlation demonstrates a higher extent of ILS on smaller chromosomes reflecting the higher recombination rate on the smaller chromosomes. (C) Plot illustrating the positive correlation between the frequency of ILS on each essential chromosome (1–13) and average recombination rates for the same chromosomes. (D) The frequency of ILS in exonic sites plotted against exon density in 100-kb windows. The negative correlation illustrates that regions with a higher density of coding sites show less ILS compared to regions with fewer coding sites.
Positive selection in signal peptides
The genome alignment of M. graminicola–S1–S2 included more than 9000 protein-coding genes out of the 10,952 genes predicted in M. graminicola. We measured Ka and Ks to identify genes under positive selection. Only 27 genes show significantly higher Ka over Ks (P < 0.05, Z-test). None of these genes encode proteins with known function, but 11 of them were found to be positively selected in more than one species, supporting a putative role in divergence and/or host specialization (Fig. 5A).
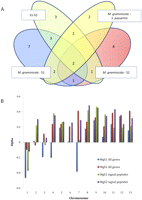
(A) Venn diagram demonstrating the number of genes showing evidence of positive selection between four different combinations of species. As illustrated, several genes showed a significantly higher Ka value in more than one pairwise species combination. (B) The parameter α estimated for all genes and for a subset of genes encoding signal peptides. The figure contrasts estimates of the two groups of genes on chromosomes 1–13 in the two combinations of species M. graminicola and S1 (MgS1) and M. graminicola and S2 (MgS2). Although the chromosomes show a high extent of heterogeneity, the signal peptide encoding genes overall have a significantly higher extent of adaptive evolution.
Pathogen effectors are secreted proteins that act to modify the plant immune system and thereby allow further access of the fungus inside the host (Kamoun 2007). We asked whether putative effectors exhibit more positively selected nonsynonymous mutations driven by adaptation to different host targets. Eight hundred two proteins targeted for secretion by the presence of a signal peptide were computationally identified (Kamoun 2007), and the proportion of nonsynonymous divergence attributable to positive selection, α, was assessed (Welch 2006). Overall, genes encoding secreted proteins exhibited a significantly higher amount of adaptive evolution (Fig. 5B; Supplemental Fig. S4), supporting the hypothesis of a coevolutionary arms race between host and pathogen molecules. This difference remained significant after correcting for difference in base composition between the two sets of genes (comparing M. graminicola and S1: P-value < 0.001 and comparing M. graminicola and S2: P-value < 0.01) (Supplemental Table S7). We additionally observed strong differences in the magnitude of α between chromosomes that likely reflect the differences in recombination rates and effective population sizes of the chromosomes (Fig. 6).
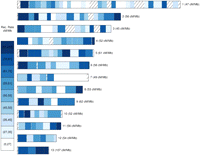
Recombination map of chromosomes 1–13 derived from coalHMM estimates of the parameter rho measured in units of substitutions. rho was estimated in windows of 100 kb along aligned chromosomes of four isolates: the M. graminicola reference genome, one genome of S1 and S2, and the outgroup sequence of S. passerinii. We converted rho to recombination rate measured in centimorgans per megabase (cM/Mb). Color differences indicate differences in recombination rate across chromosomes. The nine categories correspond to the nine quartiles of the observed distribution of recombination rate. Chromosome-wise values shown in parentheses are the median of all corresponding regions. Stripped regions show regions where rho could not be estimated due to insufficient alignment data.
We classified aligned genes using the Gene Ontology (GO) browser on the JGI M. graminicola genome portal. GO categories could be assigned to 1406 (15%) of the genes aligned between M. graminicola, S1, S2, and S. passerinii (Supplemental Table S8). We compared α and f values for the 27 most abundant GO categories and found significant support for heterogeneity of f and α among GO categories (permutation test; see Methods). The highest value of α was found between M. graminicola and S1 in the GO categories “amino acid transport” (α = 0.61, 65 genes assigned), “response to antibiotics” (α = 0.22, 22 genes), and “carbohydrate transport” (α = 0.20, 27 genes). Between M. graminicola and S2 the highest α values were likewise the GO categories “amino acid transport” (α = 0.59) and “response to antibiotics” (α = 0.32). Another GO category significantly affected by selection in M. graminicola but not in S1 and S2 is “signal transduction” (α = 0.16 for M. graminicola and S1 and α = −0.04 for S1 and S2), suggesting that changes in regulatory pathways have driven the divergence and specialization of M. graminicola.
Reproductive isolation through significant alteration of a mating-type locus
Although the detached leaf assay indicated that coinfection of the same host species can occur, we previously found no evidence of contemporary gene exchange between species based on gene flow analyses of large-scale population samples of the pathogens (Stukenbrock et al. 2007). Experimental crosses of M. graminicola and S1 were unsuccessful (G Kema, pers. comm.), further supporting the assumption that these species are reproductively isolated even though they exist in sympatry. However, the genome data reveal islands of shared polymorphisms that between M. graminicola and S1 could result from introgression. Reproductive barriers in ascomycetes can evolve by divergent selection on genes involved in the recognition of self and non-self. Several genes are involved in mating, but we focused on the two major idiomorphs of M. graminicola MAT1-1 and MAT1-2 (Waalwijk et al. 2002). We assessed the level of polymorphisms and substitutions in MAT1-1 and MAT1-2 of M. graminicola, S1, and S2 (Supplemental Table S9). The two idiomorph genes are highly conserved within species and contained no polymorphism in a global population sample including more than 70 isolates (data not shown). However, between species, the MAT1-2 locus has undergone several amino acid changes between M. graminicola and S1 and a change in start position between M. graminicola and S2. The MAT1-1 idiomorph, on the other hand, has accumulated fewer (5 vs. 22 between M. graminicola and S1 and 30 vs. 50 between M. graminicola and S2) amino acid changes. Altogether this suggests that reproductive isolation between the three species could have been mediated in part through selection on one of the major genes involved in mating.
Recent species divergence and extensive incomplete lineage sorting in pathogen genomes
We applied a coalescent hidden Markov model (coalHMM) (Hobolth et al. 2007; Dutheil et al. 2009) estimating and exploiting incomplete lineage sorting (ILS) between M. graminicola, S1, and S2 to assess speciation times, ancestral population sizes, and recombination rates. All parameters were estimated in units of substitutions, and in order to convert them to years and actual population sizes, we used the previously estimated speciation time of 11,000 yr as calibration (Stukenbrock et al. 2007). This assumption corresponds to a mutation rate per site per cell cycle of 3.3 × 10−8. This assumed mutation rate matches well the direct estimate of mutation rate from yeast (Lynch et al. 2008). It is, however, important to acknowledge the possible variation in cell cycles per year of the grass pathogens existing in highly different environments, which, in addition, have been changing since species divergence.
The average recombination rate in the ancestral species of M. graminicola and S1 was estimated to be on average 46 cM/Mb (Fig. 6; Supplemental Table S10), Using 100-kb stretches of aligned sequence, we constructed a recombination map of the ancestral species that shows great heterogeneity in recombination rates (Fig. 6) but with higher recombination rates near the telomeres and higher recombination rates on smaller chromosomes, matching previous estimates from a genetic map of M. graminicola (Wittenberg 2007). We note that this is the first broad-scale recombination map reconstructed without any crossing data.
The coalHMM analysis provided a divergence time for the S2 species of ∼23,000 yr. We estimate the effective population size of the S2 ancestor to be ∼18,000, while the population size of the ancestor of M. graminicola and S1 was only 9000. Using the same mutation rate, we estimate from contemporary amounts of polymorphism (SNPs in the three genomes of M. graminicola, in the five of S1, and in the four S2 genomes) that the current effective population size is ∼5000 for M. graminicola and ∼3500–4000 for S1 and S2 if one sexual cycle per year occurs (Fig. 1). If there are more sexual cycles per year, then these estimates should be multiplied by the number of sexual cycles per year. We place most confidence in the relative sizes and differences between the three species because the actual sizes as explained above rely on several assumptions for calibration. Our previous work also showed that M. graminicola has increased in population size while spreading around the world together with its wheat host (Banke and McDonald 2005; Stukenbrock et al. 2007). In M. graminicola Zhan and McDonald (2004) previously estimated an Ne of 25,000 using information from RFLP markers. This number is within the same range as the Ne values estimated here for the ancestral Mycosphaerella species (Fig. 1).
Interestingly, the small effective size estimates appear incompatible with the very strong selection observed, which predicts that the selection coefficient |Ne*s| ≫ 10 for 96%–97% of the genome. In Drosophila melanogaster, which exhibits a similar strength of purifying selection, effective sizes based on contemporary polymorphisms are estimated in the millions. Yet it has recently been proposed that even these estimates of Ne are smaller than the N relevant for natural selection due to fluctuations in population size (Karasov et al. 2010). We propose that fluctuations in population sizes must be much more dramatic in the M. graminicola species complex.
The pathogen genomes consist of a mosaic of sequences with different evolutionary histories. Thirty percent of the sites in the genome alignment are not concordant with the species tree and instead show either closer genomic relationship between M. graminicola and S2 or S1 and S2 as an effect of incomplete lineage sorting (ILS). The distribution of sites with ILS is nonrandom (Fig. 4B): We find a strong positive correlation between recombination rate and ILS (Kendall's rank correlation, tau = 0.7, P < 4 × 105) (Fig. 4C). We interpret this as evidence for less genetic hitch-hiking and less background selection on the more highly recombining chromosomes. This also provides a partial explanation for the higher rate of adaptive evolution observed on the smaller chromosomes (Fig. 5B) since the efficacy of selection is expected to be larger where the recombination rate is higher. A more direct way to investigate the effect of selection on patterns of ILS is to correlate the density of genes (as proxies for intensity of selection) with ILS for each 100-kb segment. Figure 4D shows that the amount of ILS observed is strongly negatively correlated with exon density (Kendall's tau = −0.22, P < 2.2 × 1016). It is not possible from this observation to determine whether this is due to more background selection or more selective sweeps in more gene-rich regions, but the selection analysis suggests that it might well be a mixture of selection against 95% of amino acid changing mutations and selection for (sweeps of) 2% of new amino acid–changing mutations (the remainder being effectively neutral).
The ILS correlations all suggest that natural selection (positive and negative) affects linked variation in these species despite recombination rates about 50 times larger than for the human genome. We can roughly estimate that selective sweeps constantly occur throughout the pathogen genomes: We estimate fixation of one new amino acid substitution by positive selection happening approximately once a year since M. graminicola and S1 diverged in the following way. The number of amino acid differences between the species is 188,000, of which ∼40% or 75,200 have been fixed by positive selection. The average genomic divergence time is 29,000 yr (11,000 yr of speciation plus coalescence in the common ancestor of expected 2 × 9000 [Ne ancestor of M. graminicola and S1] = 18,000 yr). So 75,200 selective sweeps have occurred over 58,000 yr. This rate of adaptive evolution in the Mycosphaerella pathogens exceeds estimates from whole genome studies in Drosophila by a factor of 20 (Andolfatto 2005). However, a recent study also quantifying adaptive evolution by α has found that adaptive protein divergence on the X chromosome reaches 85%–90% in Drosophila species (Andolfatto et al. 2011).
Conclusion
Using a population genomics approach, we demonstrate increased adaptive evolution in M. graminicola compared with S1 and S2. Differences in patterns of evolution between S1 and S2 demonstrate that natural populations of plant pathogens become specialized on different hosts by very different processes.
Positive selection has in particular affected genes encoding secreted proteins that likely interact with host molecules and potentially act as pathogen effectors. The pattern of overall strong positive selection on these genes is likely mediated by a coevolutionary arms race with host molecules.
The amount of incomplete lineage sorting in the genomes of M. graminicola, S1, and S2 provides evidence that genome divergence dates much further back than species divergence. Our analyses—based both on contemporary levels of polymorphism and patterns of ILS—are consistent with rather small present and ancestral effective population sizes of the species (Fig. 1). However, estimates of selective constraints point to fairly intense purifying selection and thus rather large (long-term effective) population sizes. Our study strongly supports the idea that the actual population size of M. graminicola must have been very high during the process of adaptation to a new domesticated host. Specialization on domesticated wheat did not entail any visible “domestication cost” that is otherwise often found in crop or livestock lineages (Lu et al. 2006; Haudry et al. 2007).
Methods
Host inoculation
Plants were obtained from seeds of the bread wheat variety Drifter that is highly susceptible to M. graminicola and from seeds of E. repens and D. glomerata provided by Otto Hauenstein Samen AG, Rafz Switzerland; D. glomerata, L. perenne, and L. multiflorum were provided by Fenaco Saemeraienzentrum, Winterthur, Switzerland. To study host adaptation, we used a detached leaf assay (Browne and Cooke 2004) with some modifications. Host leaf material was produced in a greenhouse. Each fungal strain was inoculated onto detached leaves taken from individual plants using a spore concentration of 107 per milliliter. Cotton swabs saturated with the spore solution were wiped across 5.5-cm leaf segments. Inoculated leaves were air-dried for 10 min and placed into a plastic box containing agar amended with benzimidazole. Sealed boxes were placed in the dark for 24 h at 18°C and subsequently placed in growth chambers at 70% humidity under alternating light/dark cycles with 16 h under white fluorescent lights (15 kLux) at 18°C followed by 8 h of darkness at 15°C. The percentage of leaf area covered by pycnidia (asexual fruiting bodies produced by the Mycosphaerella pathogens on leaf surfaces) at 22 d after inoculation was used as a measure of pathogenicity. The assay was repeated four times for each fungal isolate on each of the plant hosts. The significance of differences among means was evaluated using t-tests.
Electrophoretic karyotypes
Pulsed field gel electrophoresis was used to determine chromosome numbers and sizes in four of the resequenced isolates. DNA plugs were prepared by embedding intact spores in agarose and treating with EDTA, SDS, and protease XIV (Sigma-Aldrich) for 48 h at 60°C. PFGE was conducted with a contour-clamped homogeneous electric field (CHEF) Bio-Rad DR-II apparatus in 120 mL of 1× TAE, 0.8% to 1% agarose (Invitrogen) with different running conditions for large, medium, and small chromosomes (Stukenbrock et al. 2010). The standard chromosome size markers used were from S. cerevisiae (small chromosomes), Hansenula wingei (medium chromosomes), and Schizosaccharomyces pombe (large chromosomes). Gels were stained with ethidium bromide for 20 min, and karyotypes were made visible using a UV transilluminator. Chromosome bands were identified with visual inspection, and gel pictures were further analyzed using the software QuantityOne (Bio-Rad).
Genome assembly and alignment
Fungal genomes were sequenced by Illumina sequencing. Paired-end reads of 35–70 bp with insert size 150–500 nt were assembled using the SOAP de novo assembler (Li et al. 2008). Assembled contigs were aligned to the 21 annotated chromosomes of the JGI reference sequence of M. graminicola using LastZ included in the genome alignment software package BLASTZ (Schwartz et al. 2003). Alignment parameters were set as previously described in Stukenbrock et al. (2010). We merged the aligned chromosomes into a multiple genome alignment of 12 sequences using the reference sequence and coordinates as backbone. The 12 genome sequences can be accessed at the NCBI database (see “Data Access” below).
Population genomic analyses
We developed a Java-based genome browser to extract counts of the chromosome alignments (available from http://cs.au.dk/∼tth/viewer/). We recorded total counts of substitutions, polymorphisms, and shared polymorphisms in windows of 50 kb, 25 kb, and 10 kb according to the parameter of interest and the chromosome sizes. For inferences of nonsynonymous and synonymous substitutions, we used coordinate information from the JGI gene prediction in the reference sequence.
Evolutionary rates of the M. graminicola, S1, and S2 lineages were estimated by CODEML in the PAML software package using a branch model that allows for different dN/dS ratios (Yang and Nielsen 2000; Yang 2007). The parameters α and f were estimated from counts of polymorphism and divergence in synonymous and nonsynonymous categories using the likelihood framework implemented in the MK-Test software (Welch 2006; Obbard et al. 2009). We built a series of models, ranging from models assuming strict neutrality, where f and α are constrained to, respectively, 1 and 0, to models containing purifying or/and positive selection with varying intensities according to gene categories (or chromosomes). Given the sampling variance associated with these parameters (in particular α), we pooled MK tables by categories (gene categories or chromosomes). S. passerinii was used as outgroup for all analyses due to the high estimates of incomplete lineage sorting between M. graminicola, S1, and S2 obtained by the coalHMM approach. To compare the fit of these alternative models, we computed Akaike's Information Criterion (AIC) of each model Mi as AIC(Mi) = −2 ln L(Mi) + 2p(Mi), where ln L(Mi) is the (natural) log likelihood of Model Mi and p(Mi) the number of free parameters fitted. Previous work based on simulations suggests that AIC is a sensible choice for model selection in that context (Welch 2006). We evaluated 200 distinct models obtained by permutation of genes between categories. The resulting distribution of AIC allowed us to evaluate the significance of the partition of genes based on GO categories.
We estimated both parameters for pairs of species for the whole set of aligned genes and for a subset of genes encoding signal peptides. Signal peptides were identified using the software SignalP (Emanuelsson et al. 2007). Only genes with a hidden Markov model score >90 were assigned as signal peptides. We compared α and f estimates for signal peptides and nonsignal peptide genes. As genes with signal peptides appeared to have a significantly higher GC content, we performed an additional comparison by creating a subset of genes without signal peptides in which the GC content was within the range of “meanSP ± 2 stderr” (meanSP is the average GC and stderr the standard deviation of genes with signal peptide).
To identify single genes with evidence of positive selection, we used the Nei and Gojobori approach (Nei and Gojobori 1986) and assessed Ka and Ks rates for all genes with more than 100 aligned codons and used a Z test to identify genes with Ka > Ks.
Mating-type sequences
Partial mating-type genes were amplified in a population sample of more than 70 isolates including M. graminicola collected from different geographical regions and S1 and S2 isolates collected in Iran. PCR conditions and sequencing were as previously described (Zhan et al. 2002; Banke et al. 2004). Primers are listed in Supplemental Table S11. The program DnaSP v.5 was used to estimate nucleotide differences between the sequences (Rozas et al. 2003).
Coalescence analyses
The coalHMM approach used to infer coalescence parameters is described in Hobolth et al. (2007) and Dutheil et al. (2009) (Supplemental Fig. S5). We used four genome sequences for the analyses including the M. graminicola reference sequence, the S1 sequence of STIR04.3.11.1, the S2 sequence of STIR04.1.1.1, and the S. passerinii sequence of P63 (Supplemental Table S2). Analyses were initially performed on full chromosomes, on 200-kb aligned fragments, and on 100-kb fragments. All runs gave consistent parameter estimates.
We initially generated non-overlapping 1-kb windows along each chromosome. Windows in which at least one sequence had >40% gaps were discarded. The contiguous remaining windows were concatenated into alignment chunks and given as input to the coalHMM software. One set of parameters was estimated per alignment, but the Markov chain was reset after each alignment chunk. Next, we used the coalhmm program with the ILS model (2009 version) (Dutheil et al. 2009) to estimate the speciation times, ancestral population sizes, and recombination rate for each chromosome. Convergence was reached for all 13 essential chromosomes. We used a posterior decoding procedure to reconstruct the most likely genealogy for each position in the genome alignment. The amount of ILS for a given region was then computed as the proportion of sites in the region for which states Mg-S2 or S1-S2 (the states are illustrated in Supplemental Fig. S5) had the maximum posterior probability.
Data access
The genome project has been deposited at DDBJ/EMBL GenBank under the following accession numbers: STIR04 A48b: AEYQ00000000, STIR04 A26b: AFIP00000000, STIR04 2.2.1: AFIQ00000000, STIR04 3.1.1: AFI000000000, STIR04 4.3.1: AFIR00000000, STIR04 5.3: AFIS00000000, STIR04 5.9.1: AFIT00000000, STIR04 1.1.1: AFIU00000000, STIR04 1.1.2: AFIV00000000, STIR04 3.13.1: AFIW00000000, STIR04 3.3.2: AFIX00000000, S. passerinii P63: AFIY00000000. The versions described in this paper are the first versions of the assembled genome data.
Acknowledgments
We thank Freddy Bugge Christiansen and three anonymous reviewers for helpful comments to the manuscript, Gert Kema for testing the ability of M. graminicola and S1 to cross, and Tryggvi Stefansson for analyzing virulence data. The genome sequencing was financed by a research grant from the Faculty of Sciences, University of Copenhagen, Denmark and by a “L'Oreal Denmark for Women in Science Fellowship,” both to E.H.S. The research was additionally funded by the Danish Research Council through a postdoctoral grant to E.H.S. and an ERA-PG grant to M.H.S. The ETH Genetic Diversity Center was used to collect the MAT1-1 and MAT1-2 sequence data. The M. graminicola assembly used as reference in our study was produced by the U.S. Department of Energy Joint Genome Institute (http://www.jgi.doe.gov/) in collaboration with the user community.
Footnotes
-
↵6 Corresponding author.
E-mail eva.stukenbrock{at}mpi-marburg.mpg.de.
-
[Supplemental material is available for this article.]
-
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.118851.110.
- Received December 6, 2010.
- Accepted August 10, 2011.
- Copyright © 2011 by Cold Spring Harbor Laboratory Press








