
The presence of RNA polymerase II, active or stalled, predicts epigenetic fate of promoter CpG islands
- 1 Carcinogenesis Division, National Cancer Center Research Institute, 104-0045 Tokyo, Japan;
- 2 Epidemiology and Prevention Division, Research Center for Cancer Prevention and Screening, National Cancer Center, 104-0045 Tokyo, Japan
Abstract
Instructive mechanisms are present for induction of DNA methylation, as shown by methylation of specific CpG islands (CGIs) by specific inducers and in specific cancers. However, instructive factors involved are poorly understood, except for involvement of low transcription and trimethylation of histone H3 lysine 27 (H3K27me3). Here, we used methylated DNA immunoprecipitation (MeDIP) combined with a CGI oligonucleotide microarray analysis, and identified 5510 and 521 genes with promoter CGIs resistant and susceptible, respectively, to DNA methylation in prostate cancer cell lines. Expression analysis revealed that the susceptible genes had low transcription in a normal prostatic epithelial cell line. Chromatin immunoprecipitation with microarray hybridization (CHiP-chip) analysis of RNA polymerase II (Pol II) and histone modifications showed that, even among the genes with low transcription, the presence of Pol II was associated with marked resistance to DNA methylation (OR = 0.22; 95% CI = 0.12–0.38), and H3K27me3 was associated with increased susceptibility (OR = 11.20; 95% CI = 7.14–17.55). The same was true in normal human mammary epithelial cells for 5430 and 733 genes resistant and susceptible, respectively, to DNA methylation in breast cancer cell lines. These results showed that the presence of Pol II, active or stalled, and H3K27me3 can predict the epigenetic fate of promoter CGIs independently of transcription levels.
Epigenetic alterations, along with genetic alterations, are known to play critical roles in human carcinogenesis and other acquired diseases (Laird and Jaenisch 1996; Robertson 2005; Jones and Baylin 2007). Especially, DNA methylation of promoter CpG islands (CGIs) has been known to be involved in silencing of tumor-suppressor and other genes (Ushijima 2005; Eckhardt et al. 2006; Jones and Baylin 2007). In addition, a critical role of methylation of the nucleosome-free region (NFR) just upstream of a transcription start site (TSS) was recently demonstrated in nucleosome occupation and thus in gene silencing (Li et al. 2007; Lin et al. 2007).
Epigenetic alterations, different from genetic alterations, have unique natures, such as gene specificity (Costello et al. 2000; Esteller et al. 2001; Keshet et al. 2006; Nakajima et al. 2009; Oka et al. 2009), high levels of accumulation in normal-appearing tissues (Kondo et al. 2000; Maekita et al. 2006; Ushijima 2007), and deep involvement of inflammation in their induction (Issa et al. 2001; Ushijima and Okochi-Takada 2005; Maekita et al. 2006). Especially, the presence of gene specificity, originally suggested by the presence of tumor type-specific DNA methylation patterns (Costello et al. 2000; Esteller et al. 2001), is now confirmed by methylation of specific genes in non-cancerous tissues exposed to specific carcinogenic factors (Nakajima et al. 2009; Oka et al. 2009). Selection biases for genes with growth advantage can be avoided by analysis of non-cancerous, therefore polyclonal, tissues (Mihara et al. 2006). The gene specificity of DNA methylation induction depending on cell types and carcinogenic factors shows that there are instructive mechanisms for DNA methylation induction, in contrast to the random nature of mutation induction.
As mechanisms for instructive induction, limited information is available so far, including low transcription levels and some histone modifications. Exogenous and endogenous genes are likely to become methylated only when they have low transcription levels (Song et al. 2002; De Smet et al. 2004). Most genes methylated in cancer tissues had no or low transcription in their normal counterpart cells (Ushijima 2005; Keshet et al. 2006). Transcription factors, such as SP1/SP3 and MLL, protected CpG sites from becoming methylated, independent of and dependent on transcription levels, respectively (Boumber et al. 2008; Erfurth et al. 2008). In addition, trimethylation of histone H3 lysine 27 (H3K27me3), a target of Polycomb repressive complex (PRC) 2 (Hansen et al. 2008), was enriched in normal cells and embryonic stem (ES) cells at genes that can be methylated in cancers (Ohm et al. 2007; Schlesinger et al. 2007; Widschwendter et al. 2007; Hahn et al. 2008; Rodriguez et al. 2008). Nevertheless, at a genome level, many genes have low transcription levels and H3K27me3 but are still resistant to DNA methylation induction, indicating that some critical factors are likely to be still missing.
In this study, we hypothesized that RNA polymerase II (Pol II) binding around TSSs can function as a protective factor for DNA methylation induction. Accumulation of Pol II at genes with low transcription levels (stalled Pol II) was recently found in as high as 12% of protein-coding genes in Drosophila melanogaster (Muse et al. 2007; Zeitlinger et al. 2007) and in humans (Guenther et al. 2007). We demonstrate in a genome-wide manner that Pol II binding, active or stalled, and histone modifications in normal cells predict genes resistant and susceptible to DNA methylation in cancers.
Results
Identification of genes with promoter CGIs resistant and susceptible to DNA methylation
To identify genes with promoter CGIs resistant and susceptible to induction of DNA methylation in human prostate cancers, four prostate cancer cell lines (PC3, LNCaP, 22Rv1, and Du145), along with a normal prostatic epithelial cell line (RWPE1), were analyzed using methylated DNA immunoprecipitation (MeDIP) combined with a human CGI oligonucleotide microarray that covered 27,800 CGIs (MeDIP-CGI microarray analysis).
First, appropriate cutoff values of our original output values “DNA methylation values” (Me values) were determined using 145 samples (29 CGIs in five cell lines) (Supplemental Table S1). As cutoff values with high specificity and little compromise of sensitivity, cutoff values of 0.6 and 0.4 were selected for methylated and unmethylated CGIs, respectively (Supplemental Fig. S1). The specificity and sensitivity for methylated (unmethylated) CGIs with these values were 0.95 (0.96) and 0.85 (0.82), respectively. DNA methylation status of a CGI or putative NFR was judged as unmethylated (UM), moderately methylated (MM), and highly methylated (HM) when the average of Me values of the probes within the region was 0–0.4, 0.4–0.6, and 0.6–1.0, respectively. The validity of our methods was also supported by the fact that promoter CGIs were more likely to be unmethylated (68%–82%) than those in gene bodies (54%–63%), which conformed with previous observations (Supplemental Table S2; Ushijima et al. 2003; Eckhardt et al. 2006; Rakyan et al. 2008).
The susceptibility of genes was determined by methylation analysis of 8930 NFRs (Li et al. 2007). Genes with NFRs unmethylated (Me value, 0–0.4) in the normal cell line and all the four cancer cell lines were defined as DNA methylation-resistant genes. On the other hand, those unmethylated in the normal cell line but highly methylated (Me value, 0.6–1.0) in at least one of the four cancer cell lines were defined as DNA methylation-susceptible genes (Fig. 1A). Susceptible genes were further divided into S1, S2, S3, and S4 subclasses according to the DNA methylation frequency in cancer cell lines (highly methylated in one, two, three, and four, respectively, of the four cancer cell lines). In addition, genes unmethylated in the normal cell line but moderately methylated (Me value, 0.4–0.6) in at least one of the four cancer cell lines were defined as genes with intermediate susceptibility (intermediate genes). In prostate cancers, 5510, 1330, and 521 genes with promoter CGIs were classified as resistant, intermediate, and susceptible genes, respectively (Fig. 1B). DNA methylation levels of NFRs were largely consistent with those of further upstream regions up to −800 bp, and downstream regions up to +800 bp (Fig. 1C).
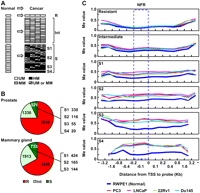
Identification of methylation-resistant and methylation-susceptible genes and their methylation profiles in various genomic regions against TSSs. (A) Definition of genes resistant and susceptible to induction of DNA methylation. Genes unmethylated (UM) (white) in the normal cell line (cells) and all cancer cell lines were defined as resistant genes (R). Genes unmethylated in the normal cell line (cells) but highly methylated (HM) (black) in at least one of the four cancer cell lines were defined as susceptible genes (S). Susceptible genes were further divided into four subclasses according to DNA methylation frequency in the cancer cell lines (S1–S4). Genes unmethylated in the normal cell line (cells) but moderately methylated (MM) (gray) in the cancer cell lines were defined as genes with intermediate susceptibility (intermediate genes: Int). (B) The fractions of resistant (red), intermediate (light green), and susceptible (green) genes in the prostate and the mammary gland. (Right side of the pie graph) Numbers of susceptible genes in each subclass (S1–S4). (C) DNA methylation levels at various positions against the TSSs in the normal prostatic cell line and four cancer cell lines. Average Me values of CGIs continuous from their NFRs are shown. (Blue dotted rectangle) The NFRs. Methylation levels of the NFRs were similar to those of upstream regions up to −800 bp and downstream regions up to +800 bp.
To avoid any tissue bias and statistical errors, we also analyzed three human breast cancer cell lines (MCF7, ZR-75-1, and MDA-MB-468), along with normal human mammary epithelial cells (HMEC). As in the prostate, the promoter CGIs were more likely to be unmethylated (68%–90%) than the CGIs located in gene bodies (52%–70%) (Supplemental Table S2). Using the same definition as in the prostate cancers, 5430, 1913, and 733 genes with promoter CGIs were classified as resistant, intermediate, and susceptible genes, respectively (Fig. 1B). As in prostate cancers, DNA methylation levels were also largely consistent among the NFRs, further upstream regions, and downstream regions in human breast cancers (Supplemental Fig. S2). Between breast and prostate cancers, only 261 genes, 36% of the susceptible genes in breast cancers and 50% of those in prostate cancers, were commonly susceptible, showing the presence of tissue specificity.
To explore possible selection bias for the resistant and susceptible genes due to gene functions, functional annotation analysis of resistant and susceptible genes was performed. In the prostate, 203 and 154 processes out of 16,621 biological processes were enriched among the resistant and susceptible genes, respectively. Among the resistant genes, processes involved in basic cellular processes such as metabolic process, RNA processing, and RNA splicing were enriched. In contrast, among the susceptible genes, biological processes involved in the developmental processes of specific cells or tissues, such as nervous system development, and embryonic development, were enriched (Table 1). Similar enrichment of genes involved in specific biological processes was also observed in the mammary glands.
Functional annotation analysis of genes with different DNA methylation susceptibility
Low transcription levels of DNA methylation-susceptible genes in normal cell lines
For a limited number of genes, the susceptibility of genes with low transcription to DNA methylation has been reported in cell lines (Song et al. 2002; De Smet et al. 2004) and in human tissue (Ushijima and Okochi-Takada 2005; Nakajima et al. 2009). To analyze this susceptibility in a genome-wide manner, we performed expression analysis in the normal prostatic cell line using a GeneChip oligonucleotide microarray. Owing to the difference of array platforms between the CGI oligonucleotide microarray and the GeneChip oligonucleotide expression microarray, we were able to measure transcription levels of the 7574 genes out of 8930 genes with promoter CGIs in the normal prostatic cell line. The accuracy of the transcription levels obtained by the GeneChip oligonucleotide microarray was validated by observing a strong correlation between the microarray data and mRNA levels obtained by quantitative RT-PCR (correlation coefficient = 0.95 and 0.97 in RWPE1 and HMEC, respectively) (Supplemental Fig. S3). When the transcription levels were analyzed according to the DNA methylation status in the normal prostatic cell line itself, as expected, highly methylated genes had remarkably low transcription levels (Supplemental Fig. S4).
Genes highly methylated in prostate cancer cell lines had low transcription levels in the normal prostatic cell line (Fig. 2A). When transcription levels of resistant, intermediate, and susceptible genes were compared, susceptible genes had lower transcription levels than resistant genes. Even among the susceptible genes, genes with frequent DNA methylation had lower transcription levels than those with infrequent DNA methylation (Fig. 2B). When fractions of genes with high, moderate, and low transcription levels were analyzed in the 7574 total, 4567 resistant, and 479 susceptible genes, the susceptible genes had a significantly larger fraction of genes with low transcription (63%) than the total genes (38%; P < 0.001, χ2 test) (Fig. 2C). Even among the susceptible genes, genes with more frequent DNA methylation had the larger fraction of genes with low transcription (Supplemental Fig. S5). These results showed that aberrant DNA methylation is preferentially induced in genes with low transcription, as previously reported (Song et al. 2002; De Smet et al. 2004; Ushijima 2005; Keshet et al. 2006; Nakajima et al. 2009), in a genome-wide manner.
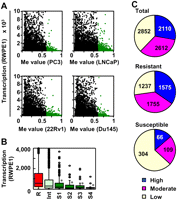
Low transcription levels of DNA methylation-susceptible genes in the normal prostatic cell line (RWPE1). (A) The association between DNA methylation levels (Me value of the NFRs) in each of the four prostate cancer cell lines (PC3, LNCaP, 22Rv1, and Du145) and transcription levels in RWPE1. (Green dots) Genes highly methylated in a cancer cell line. Genes highly methylated in a cancer cell line had low transcription levels in the normal cell line. (B) Transcription levels of resistant (R), intermediate (Int), and susceptible (S1–S4) genes in RWPE1. The boxes represent the 75th and 25th percentiles, and the line in the box represents the 50th percentile (the median). Whiskers represent the maximum data within (75th percentile + 1.5 × [75th percentile − 25th percentile]) and the minimum data within (25th percentile − 1.5 × [75th percentile − 25th percentile]). (Dots) The data not included between the whiskers. Transcription levels of Int, S1, S2, S3, and S4 were compared to that of R by the Mann-Whitney U-test (*P < 1 × 10−5). Susceptible genes had significantly lower expression levels than resistant genes. (C) The fraction of genes with high (blue) (signal intensity > 1000), moderate (pink) (250–1000), and low (yellow) (<250) transcription. Susceptible genes had a significantly larger fraction of genes with low transcription than the total genes.
In the mammary glands, the susceptible genes also had a significantly larger fraction of genes with low transcription (74%) than the total genes (37%; P < 0.001, χ2 test) (Supplemental Figs. S5, S6).
Levels of histone modifications and Pol II binding were associated with DNA methylation susceptibility
Although most genes susceptible to DNA methylation in cancers had low transcription in the normal cell line (cells), the converse was not true: 1237 of 2852 (prostate) and 1048 of 2750 (breast) genes with low transcription in the normal cell line (cells) were still resistant to DNA methylation in cancers (Fig. 2C; Supplemental Fig. S6). This indicated that factors besides low transcription are also involved in DNA methylation susceptibility. To address this issue, we analyzed both active (acetylation of histone H3 [H3Ac] and trimethylation of histone H3 lysine 4 [H3K4me3]) and inactive (trimethylation of histone H3 lysine 9 [H3K9me3] and H3K27me3) histone modifications and Pol II binding at and adjacent to the NFRs in a genome-wide manner. Since the length of sheared DNA used for chromatin immunoprecipitation (ChIP) analysis ranged mainly from 200 to 1000 bp, analysis of probes within the NFRs automatically reflected histone modifications adjacent to the NFRs even if nucleosomes were absent in the NFRs. The data obtained by the ChIP with microarray hybridization (ChIP-chip) analysis were validated by analyzing correlations between the signal ratio (immunoprecipitated DNA [IP]/whole cell extract [WCE]) obtained by ChIP-chip and those obtained by quantitative ChIP-PCR (Supplemental Fig. S7).
Using only genes with low transcription, we analyzed the association between the candidate instructive factors in the normal prostatic cell line and susceptibility to DNA methylation in prostate cancer cell lines. It was clear that H3Ac and H3K4me3 were elevated in resistant genes, and H3K27me3 was elevated in susceptible genes (Fig. 3A). In contrast, the H3K9me3 level was not different between resistant and susceptible genes. Notably, Pol II binding was remarkably higher in resistant genes (Fig. 3B). When further upstream regions and downstream regions were analyzed, resistant genes had elevated H3Ac and H3K4me3 mainly in their downstream regions, and susceptible genes had elevated H3K27me3 in their downstream regions and further upstream regions (Fig. 3C). Pol II binding was elevated mainly in the NFRs and then in downstream regions of resistant genes (Fig. 3C). In the mammary glands, exactly the same tendency was observed (Supplemental Fig. S8).
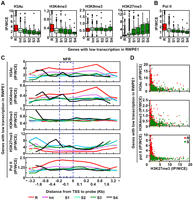
The association between the levels of candidate instructive factors in RWPE1 and DNA methylation susceptibility, among genes with low transcription in RWPE1. (A) Histone modification levels of genes with different susceptibilities to DNA methylation. For the box plot and statistical methods, refer to the legend to Figure 2B. Active histone modifications were elevated in resistant genes, and H3K27me3 was elevated in susceptible genes. (B) The association between Pol II binding and DNA methylation susceptibility. Pol II binding was associated with resistance even among genes with low transcription. (C) Levels of histone modifications and Pol II binding at various positions against the TSSs in RWPE1. Average levels of histone modifications and Pol II binding of CGIs continuous from their NFRs are shown. (Blue dotted rectangle) The NFRs. (D) The combination effect of one of the three active factors (H3Ac, H3K4me3, and Pol II binding) (y-axis) and H3K27me3 (x-axis) on resistance and susceptibility of genes with low transcription. (Red dots) DNA methylation-resistant genes; (green dots) DNA methylation-susceptible genes; they were separated by any of the three combinations.
Next, within the normal prostatic cell line, the association between histone modifications and transcription levels was analyzed. Conforming to previous reports (Barski et al. 2007; Wang et al. 2008), genes with high and low transcription had elevated active and inactive histone modifications (Supplemental Fig. S9). Notably, among genes with low transcription, those without DNA methylation had elevated H3K27me3, confirming a previous report that H3K27me3 is involved in gene silencing independent of DNA methylation (Kondo et al. 2008). Within the normal mammary epithelial cells, the same tendency was observed.
Strongest association of Pol II binding with resistance to DNA methylation
The combination effect of H3K27me3 and one of the three active factors (H3Ac, H3K4me3, and Pol II binding) on DNA methylation susceptibility was then examined (Fig. 3D). All the three combinations were informative in distinguishing the resistant and susceptible genes, while Pol II binding gave the clearest discrimination. Multivariate logistic regression analysis was then performed to compare precisely the independent effects of H3Ac, H3K4me3, H3K9me3, H3K27me3, and Pol II binding on DNA methylation susceptibility. The genes with low transcription in the normal cell line (cells) were divided into quintiles according to the amounts of H3Ac, H3K4me3, H3K9me3, H3K27me3, and Pol II binding at the NFRs. Compared with the genes in the lowest quintile, multivariate-adjusted odds ratios (ORs) of genes in the other quintiles to become moderately or highly methylated in cancers (Int, and S1–S4 for the prostates; Int, and S1–S3 for the mammary glands) were calculated (Table 2). In the prostates, Pol II binding had the strongest independent association with resistance, and H3K27me3 had a strong and significant association with susceptibility. In the mammary glands, similar associations were observed. If the analysis was performed for the multivariate-adjusted odds ratio of genes to become highly methylated (S1–S4 for the prostates; and S1–S3 for the mammary glands), the association of Pol II binding became even clearer (Supplemental Table S3).
The association between the levels of candidate instructive factors and susceptibility to DNA methylation (Int and S)
Finally, regardless of their transcription levels, all the genes were classified into genes with “active Pol II” (high/moderate transcription, high Pol II), those with “stalled Pol II” (low transcription, high Pol II), and those with “low Pol II” (low Pol II). The group of genes with low Pol II was further subdivided into those with and without H3K27me3. In the normal prostatic cell line, 47%, 13%, and 40% of genes had active, stalled, and low Pol II, respectively (Fig. 4A). Both genes with active Pol II and genes with stalled Pol II consisted mostly of resistant genes (Fig. 4B). In contrast, genes with low Pol II contained larger fractions of susceptible genes, and the presence of H3K27me3 remarkably increased the fraction. Similar results were obtained also in the mammary glands (Supplemental Fig. S10).
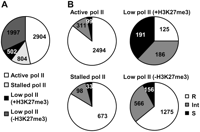
The association between Pol II binding and DNA methylation resistance in the total 6207 genes, regardless of transcription levels. (A) Classification of genes by Pol II status and H3K27me3 in the normal prostatic cell line. We were able to analyze transcription levels for 4567 of 5510 resistant, 1161 of 1330 intermediate, and 479 of 521 susceptible genes (total 6207 of 7361 genes) due to a difference in microarray platforms. Genes with high Pol II levels and high/moderate transcription levels were considered as those with “active Pol II.” Genes with high Pol II levels but low transcription levels were considered as those with “stalled Pol II.” Genes with low Pol II were further subdivided into those with and without H3K27me3. The numbers of genes with active, stalled, and low Pol II are shown. (B) The fractions of resistant, intermediate, and susceptible genes according to the Pol II and H3K27me3 statuses. Genes with either active or stalled Pol II had a larger fraction of resistant genes, and genes with low Pol II had a larger fraction of susceptible and intermediate genes.
Discussion
In this study, we showed that Pol II binding in the NFRs in normal cell lines (cells) was closely associated with resistance to DNA methylation in cancer cell lines (cells) for the first time. The association between Pol II binding and resistance to DNA methylation was independent of transcriptional levels. It was also independent from the promoting effect of H3K27me3, and the combination of Pol II binding and H3K27me3 could explain a large part of the instructive mechanisms for induction of DNA methylation. These data provided fundamental information on how the epigenetic fate of promoter CGIs is determined. The association between Pol II binding and resistance to DNA methylation can be potentially useful in the prediction of genes that will become silenced in cancer and other diseases.
Our multivariate analysis involving Pol II binding and histone modifications showed that the association between active histone modifications and resistance to DNA methylation was mostly overridden by that of Pol II binding, while the association between H3K27me3 and susceptibility to DNA methylation remained. It was reported that active histone modifications are involved in anchoring of the basal transcription factor TFIID (Vermeulen et al. 2007), which forms a transcription complex with Pol II. H3K4me3 is recognized by the PHD domain of TFIID, and acetylation of histone H3 lysine 9 and lysine 14 potentiates this interaction. It was therefore suggested that Pol II binding more directly works as a protection mechanism than active histone modifications, and that H3K27me3 has an independent mode of action.
Pol II forms a huge transcription complex of ∼3 MDa with general transcription factors and other proteins (Boeger et al. 2005), and such a huge complex around promoter CGIs is expected to compete with DNA methyltransferases and their associated proteins. On the other hand, H3K27me3 is recognized by PRC2/3 (Hansen et al. 2008), which contains EZH2. Since EZH2 interacts with DNMT3A and DNMT3B (Vire et al. 2006), H3K27me3 is expected to signal binding of DNMT3A and DNMT3B. Taken together, Pol II binding and H3K27me3 are likely to function by preventing and promoting, respectively, recruitment of DNA methylation complexes.
Functional annotation analysis revealed that most of the susceptible genes were involved in the developmental processes of specific cells or tissues. Genes in this category were considered unnecessary for normal cells that have already differentiated. This raised alternative possibilities: The lack of current need for a gene is one of the instructive factors, or an unnecessary gene has a low level of Pol II, which is associated with methylation susceptibility. To distinguish these two possibilities, we examined overrepresentation of susceptible genes among genes with low Pol II levels after classification of genes by their function (Supplemental Table S4). As a result, in any categories of genes, susceptible genes were overrepresented among the genes with low Pol II levels, showing that the presence of Pol II was an independent factor for resistance to DNA methylation from functions of genes.
Specific genome structures are also known to be involved in the specificity of genes methylated, in addition to the instructive factors analyzed here. The presence of a repetitive sequence has been reported to be capable of functioning as a source of aberrant DNA methylation (Yates et al. 1999). In addition to methylation induction of individual genes, a cluster of genes can be methylated simultaneously in a cancer (Frigola et al. 2006). In this study, 64% and 50% of the susceptible genes in breast and prostate cancers, respectively, were unique to individual tumors. The susceptibility specific to a tissue is more likely to be due to Pol II binding and H3K27me3 rather, while susceptibility common to different tissues can be due to specific genome structures.
Genes moderately methylated were considered to be methylated in a fraction of cancer cells and thus to have been methylated after clonal expansion started. Genes highly methylated were considered to be present in all the cancer cells, and thus to have been methylated before clonal expansion. Therefore, DNA methylation susceptibility in normal cell line (cells) might be more precisely measured using genes highly methylated (Supplemental Table S3) than using genes highly and moderately methylated (Table 2).
As materials, we used normal and cancer cell lines to perform efficient and precise ChIP experiments. It is known that cancer cell lines generally show a larger number of methylated genes than primary tumor cells when a single cancer cell line and a primary tumor sample are compared. However, when a large number of primary tumor samples are analyzed, most DNA methylation found in cancer cell lines is also observed in at least one of the primary tumor samples (Sato et al. 2003; Lodygin et al. 2005; Yamashita et al. 2006). Therefore, it is considered that DNA methylation susceptibility identified in cancer cell lines reflects that in the primary cancer cells as a whole.
In summary, Pol II binding and H3K27me3 in normal cell lines (cells) could predict the epigenetic fate of genes with promoter CGIs in cancer cell lines independently of transcription activity and are major components of instructive mechanisms of DNA methylation induction.
Methods
Cell culture
PC3, LNCaP, 22Rv1, Du145, MCF7, ZR-75-1, and MDA-MB468 (American Type Culture Collection) were maintained in RPMI1640. RWPE1 (American Type Culture Collection) was maintained in keratinocyte-SFM containing 5 ng/mL rEGF, 50 μg/mL bovine pituitary extract (Invitrogen). HMEC (Clonetics) was maintained in mammary epithelial cell serum-free growth medium containing 1% growth supplement (CELL Applications).
ChIP assay
About 1 × 107 cells were cross-linked with 1% formaldehyde for 10 min at room temperature, and washed with ice cold 1× PBS (−) twice. Cells were re-suspended in lysis buffer (50 mM Tris-HCl at pH 8.0, 1 mM EDTA, 1% [w/v] SDS), incubated for 10 min on ice, and then sonicated to shear DNA to an average length ranging from 200 to 1000 bp with a Bioruptor UCD-250 (Cosmo Bio). After DNA shearing, the lysate was centrifuged at 13,000 rpm for 10 min, and supernatant was recovered. The volume of supernatant containing 30 μg of sheared DNA was adjusted to 100 μL with lysis buffer, and then was diluted with 900 μL of dilution buffer (50 mM Tris-HCl at pH 8.0, 167 mM NaCl, 1.1% [w/v] Triton X-100, 0.11% [w/v] sodium deoxycholate [DOC]). Twenty microliters of sheared chromatin was recovered and was used as input DNA.
Diluted lysate was incubated with 2 μg of antibody against H3K4me3 (07-473; Millipore), H3K9me3 (07-442; Millipore), H3K27me3 (07-449; Millipore), H3Ac (06-599; Millipore), or Pol II (ab5095; Abcam), which was reported to be capable of detecting stalled Pol II (Muse et al. 2007) overnight at 4°C with rotation, and then immuno-complexes were collected with 25 μL of Dynabeads Protein A (Invitrogen Dynal AS). Collected beads were washed with 1× RIPA buffer (50 mM Tris-HCl at pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% [w/v] Triton X-100, 0.1% [w/v] SDS, 0.1% [w/v] DOC) containing 150 mM NaCl twice, 1× RIPA buffer containing 500 mM NaCl twice, LiCl wash buffer (10 mM Tris-HCl at pH 8.0, 0.25 M LiCl, 1 mM EDTA, 0.5% [w/v] NP-40, 0.5% [w/v] DOC), and 1× TE containing 50 mM NaCl. Beads were re-suspended with 1× TE, and the cross-links were reversed in the presence of 200 mM NaCl overnight at 65°C. DNA was recovered with RNase A and proteinase K treatment, followed by phenol extraction and ethanol precipitation, and dissolved in 100 μL of 1× TE. One microliter of DNA was used for quantitative ChIP-PCR to confirm the specificity of our ChIP technique (Supplemental Fig. S11) or to validate microarray results (Supplemental Fig. S7). Quantitative ChIP-PCR was performed using SYBR Green I (BioWhittaker Molecular Applications) and an iCycler Thermal Cycler (Bio-Rad Laboratories) as described previously (Nakajima et al. 2009). The primers used in quantitative ChIP-PCR are listed in Supplemental Table S5 (Kirmizis et al. 2004).
MeDIP
Five micrograms of genomic DNA was sheared by sonication using a VP-5s homogenizer (TAITEC) to a length of ∼300 bp (Supplemental Fig. S12). Generally, there are nine to 53 CpG sites in 300-bp regions of promoter CGI (Nakajima et al. 2009), and this number of CpG sites is sufficient for efficient immunoprecipitation by MeDIP (Keshet et al. 2006). After heat denaturation for 10 min at 95°C, DNA was incubated with 5 μg of antibody against 5-methyl cytidine (Diagnode) in 1× IP buffer (10 mM Na-phosphate at pH 7.0, 140 mM NaCl, 0.05% [w/v] Triton X-100) overnight at 4°C with rotation. Immuno-complexes were collected with 70 μL of Dynabeads Protein A, washed with 1× IP buffer four times, and were recovered by Proteinase K treatment, followed by phenol extraction and ethanol precipitation. DNA was dissolved in 26 μL of 1× TE.
CGI oligonucleotide microarray analysis
Genome-wide analysis of DNA methylation, histone modifications, and Pol II binding was carried out using a human CGI oligonucleotide microarray (Agilent technologies) that contained 237,220 probes in or within 95 bp of CGI covering 27,800 CGIs, with an average probe spacing of 100 bp.
For MeDIP-CGI microarray analysis, immunoprecipitated DNAs from 4.33 μg of sonicated DNA and 0.96 μg of input DNA, without any amplification, were labeled with Cy5 and Cy3, respectively, using an Agilent Genomic DNA Labeling kit PLUS (Agilent technologies). Labeled DNA was hybridized to the microarray for 40 h at 67°C with constant rotation (20 rpm), and then scanned with an Agilent G2565BA microarray scanner (Agilent Technologies). The scanned data were processed using Feature Extraction Ver.9.1 (Agilent Technologies), and the IP (Cy5) and WCE (Cy3) signal values were obtained. These two values were normalized using background subtraction, and signal log ratio [log2(IP/WCE)] and P[Xbar] were obtained using Agilent G4477AA ChIP Analytics 1.3 software (Agilent Technologies). Xbar is a signal value for a probe that takes account of signals for neighboring probes (within 1 kb), and P[Xbar] is a probability of how the Xbar value is deviated from a normal distribution of Xbar values of the entire genome of a sample.
For ChIP-chip analysis, 500 ng of immunoprecipitated and input DNA, without any amplification, was labeled with Cy5 and Cy3, respectively, and then hybridized with the microarray. A scan of the microarray and the data processing were performed as described above. The levels of each histone modification or Pol II binding were assessed by the signal ratio (IP/WCE). Genes were classified into those with high and low levels of each histone modification or Pol II binding when they had signal intensities higher and lower, respectively, than the average signal intensity of total probes. The microarray data (MeDIP-CGI microarray and ChIP-chip analyses) were submitted to the GEO database under accession no. GSE15154.
Calculation of Me value
The Me value of each probe was calculated as Me value = [signal log ratio × (1 − P[Xbar]) − 1.3]/2.6 + 0.5. The Me value was developed to give a value between 0 and 1 that linearly correlates with the amount of methylated DNA molecules at a specific locus and is not influenced by the genome-overall methylation levels. The Me value of a single probe is known to correlate well with an average DNA methylation level of CpG sites within 200 bp from the probe (Yamashita et al. 2009).
Definition of genomic regions
The position of each probe against a TSS was determined using UCSC hg18 (NCBI Build 36.1, March 2006). A CGI was defined as an assembly of probes with intervals <500 bp. CGIs were classified into four categories, promoter CGIs (within 10 kb upstream of the TSS), divergent CGIs (within 10 kb upstream of the TSSs of two genes that are transcribed in opposite directions), gene body CGIs, and downstream CGIs (within 10 kb downstream from genes). A CGI spanning both a promoter region and gene body was split into a promoter CGI and a gene body CGI. A putative NFR was defined as a region between a TSS, determined by UCSC hg18 (NCBI Build 36.1, March 2006), and its 200 bp upstream. Since TSSs are inherently variable for some genes (Suzuki et al. 2001), and the size of NFRs are different according to studies (Yuan et al. 2005; Gal-Yam et al. 2006), the locations are approximate, but expected to be correct as a whole. According to these definitions, 34,697 assemblies of probes were defined as CGIs, and 9624 assemblies were defined as NFRs. Genes with multiple NFRs because of their multiple TSSs were analyzed as different genes. DNA methylation status and histone modifications/Pol II binding in each CGI (or NFR) were assessed by an average Me value and signal ratio, respectively, of the probes located within each CGI (or NFR). A single CGI (or NFR) contains 6.8 (2.0) probes on average.
Gene expression analysis by oligonucleotide microarray
Expression microarray analysis was performed by a GeneChip Human Genome U133 Plus 2.0 expression microarray (Affymetrix) that contained 54,000 probe sets from 39,000 genes. From 8 μg of total RNA, the first-strand cDNA was synthesized with SuperScript III reverse transcriptase (Invitrogen) and a T7-(dT) 24 primer (Amersham Bioscience). Double-stranded cDNA was then synthesized, and biotin-labeled cRNA was synthesized using a BioArray HighYield RNA transcript labeling kit (Enzo). Twenty micrograms of labeled cRNA was fragmented and hybridized to the GeneChip oligonucleotide microarray. The microarray was stained and scanned according to the protocol from Affymetrix. The scanned data were processed using GeneChip operating software (ver. 1.4). The signal intensity of each probe was normalized so that the average signal intensity of all the probes on a microarray would be 500. Average signal intensity of all the probes for a gene was used as its transcription level. Genes were classified into those with high (>1000), moderate (250–1000), and low (<250) transcription according to their signal intensities.
Multivariate analysis and other statistical tests
To evaluate the independent contribution of each predictor variable (H3Ac, H3K4me3, Pol II binding, H3K9me3, or H3K27me3 level) in relation to the other four predictor variables on DNA methylation susceptibility (an outcome variable), multivariate logistic regression analysis was performed. Susceptible genes were defined as (1) those moderately and highly methylated in cancer cell lines (Int, and S1–S4 for the prostates; Int, and S1–S3 for the mammary glands), or (2) those highly methylated in cancer cell lines (S1–S4 for the prostates; and S1–S3 for the mammary glands). The predictor variables were classified into quintiles according to H3Ac, H3K4me3, Pol II binding, H3K9me3, or H3K27me3 levels of the NFRs to create dummy variables. This was done because a log linear relationship was unclear between the raw value (signal ratio of each gene) and DNA methylation susceptibility. Multivariate-adjusted ORs and 95% confidence intervals (CIs) of genes in each quintile for DNA methylation susceptibility were calculated, including all predictor variables simultaneously in the model using SAS software, ver. 9.1 (SAS Institute Inc, SAS/STAT 9.1 User's Guide, SAS Institute Inc., Cary, NC). Using the lowest quintile as a reference, we calculated multivariate-adjusted ORs of genes in each quintile, which reflect DNA methylation susceptibility relative to the reference while controlling for the simultaneous effect of all the other predictor variables included in the model.
The fractions of genes with low transcription were compared between different groups of genes by the χ2-test. The transcription, histone modification, and Pol II binding levels were compared between two groups of genes by the Mann-Whitney's U-test.
Functional annotation analysis
Functional annotation analysis was performed by DAVID bioinformatics resources (Dennis et al. 2003; Huang et al. 2009). The enrichment of genes in a biological process (a Gene Ontology criterion) was analyzed by comparing a fraction of genes with an ontology among the resistant (or susceptible) genes with that among all the genes.
Acknowledgments
We thank Hiroyuki Sasaki for his critical reading of this manuscript. H.T. is a recipient of Research Resident Fellowships from the Foundation for Promotion of Cancer Research. This study was supported by Grants-in-Aid for the Third-Term Comprehensive Cancer Control Strategy from the Ministry of Health, Labour and Welfare, Japan; for the Priority-area Research from the Ministry of Education, Science, Culture, and Sport, Japan; and a grant from Uehara Life Science Foundation.
Footnotes
-
↵3 Corresponding author.
E-mail tushijim{at}ncc.go.jp; fax 81-3-5565-1753.
-
[Supplemental material is available online at http://www.genome.org. The microarray data from this study have been submitted to Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) under accession no. GSE15154.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.093310.109.
-
- Received March 1, 2009.
- Accepted July 30, 2009.
- Copyright © 2009 by Cold Spring Harbor Laboratory Press








