
A novel mode of enhancer evolution: The Tal1 stem cell enhancer recruited a MIR element to specifically boost its activity
Abstract
Altered cis-regulation is thought to underpin much of metazoan evolution, yet the underlying mechanisms remain largely obscure. The stem cell leukemia TAL1 (also known as SCL) transcription factor is essential for the normal development of blood stem cells and we have previously shown that the Tal1 +19 enhancer directs expression to hematopoietic stem cells, hematopoietic progenitors, and to endothelium. Here we demonstrate that an adjacent region 1 kb upstream (+18 element) is in an open chromatin configuration and carries active histone marks but does not function as an enhancer in transgenic mice. Instead, it boosts activity of the +19 enhancer both in stable transfection assays and during differentiation of embryonic stem (ES) cells carrying single-copy reporter constructs targeted to the Hprt locus. The +18 element contains a mammalian interspersed repeat (MIR) which is essential for the +18 function and which was transposed to the Tal1 locus ∼160 million years ago at the time of the mammalian/marsupial branchpoint. Our data demonstrate a previously unrecognized mechanism whereby enhancer activity is modulated by a transposon exerting a “booster” function which would go undetected by conventional transgenic approaches.
Increased biological complexity during evolution does not correlate with gene number but rather with increasing complexity of gene regulation (Levine and Tjian 2003). However, the evolution of cis-regulatory mechanisms remains poorly understood. This is largely due to the fact that, compared with coding sequences, very little is known about the way in which regulatory information is encoded in the genome. Developmental programs are thought to be executed through transcriptional networks which provide the capability to propagate transcriptional changes through feed forward as well as positive and negative feedback loops (Davidson 2006). Individual cis-regulatory elements are key components of these transcription factor networks since they function as information-processing units by integrating the inputs of their respective upstream regulators into tightly controlled spatiotemporal expression patterns.
Repetitive DNA, largely derived from transposable elements, is known to make up ∼50% of the human genome (Lander et al. 2001) and has historically been thought of as “junk” DNA. Following the recent completion of the opossum genome project, it has become increasingly clear that transposable element-derived DNA may play a larger role in gene regulation than previously thought (Gentles et al. 2007). Several topical reports have highlighted that thousands of these elements are under strong purifying selection and are therefore likely to have gained a beneficial function that promotes host fitness (Kazazian 2004; Bejerano et al. 2006; Xie et al. 2006). Indeed, it has been suggested that at least 16% of eutherian-specific conserved noncoding elements are derived from transposons and that these are commonly found in the gene loci of key developmental genes, suggesting that they play a role in controlling expression of these genes (Mikkelsen et al. 2007). To date, very few of these elements have been functionally validated. However, there have been specific instances where repetitive elements have been shown to function as silencers (Donnelly et al. 1999) and polyadenylation or alternative splice sites (Medstrand et al. 2005), although experimental validation has so far been largely restricted to those elements derived from the more recent, primate-specific Alu family of repeats. However, a role for more ancient repeats in gene regulation is also now emerging with the discovery of two neuronal enhancers that are derived from ancient short interspersed nucleotide elements (SINE) retroposons (Bejerano et al. 2006; Santangelo et al. 2007).
Hematopoietic stem cell (HSC) specification and subsequent differentiation into the many mature blood lineages is one of the best-understood vertebrate developmental systems. The stem cell leukemia (SCL) transcription factor, also known as TAL1, is essential for the specification of HSC and the development of hematopoiesis (Porcher et al. 1996; Robb et al. 1996). As part of a systematic analysis of the transcriptional mechanisms regulating the Tal1 locus, we have identified multiple cell type-specific enhancers, each of which directs expression to a subdomain of the overall Tal1 expression pattern (Gottgens et al. 1997; Sanchez et al. 1999; Sinclair et al. 1999; Gottgens et al. 2000, 2001; Sanchez et al. 2001; Gottgens et al. 2002a, b, 2004; Delabesse et al. 2005; Silberstein et al. 2005; Ogilvy et al. 2007). The Tal1 +19 enhancer lies 19 kb downstream from the first exon of Tal1 and directs expression to hemangioblasts, HSCs, hematopoietic progenitors, and endothelium (Sanchez et al. 1999; Gottgens et al. 2002b). Activity of this element is dependent on two conserved Ets and one conserved GATA binding site. We have also developed bio informatic tools to identify functionally related regulatory enhancers situated in the Fli1, Hhex (also known as Hex), and Smad6 gene loci (Donaldson et al. 2005a, b; Donaldson and Gottgens 2006; Pimanda et al. 2007a), and are thus beginning to define key components of the transcriptional networks controlling HSCs (Pimanda et al. 2007a, b).
In this paper we demonstrate that activity of the +19 enhancer is modulated by a novel “booster” element created by the insertion of a mammalian interspersed repeat (MIR) transposon ∼160 million years ago.
Results
Chromatin structure of the mouse Tal1 stem cell enhancer
Our previous analysis of the regulation of the murine Tal1 gene identified a 5.5-kb fragment 3′ to the Tal1 coding region in which we mapped the approximate positions of two regions of DNase I hypersensitivity (+18 and +19 regions; nomenclature reflects the approximate distance in kilobases from the beginning of exon 1; Gottgens et al. 1997). Transgenic analysis showed that this fragment directed expression of a lacZ reporter gene to endothelium and blood progenitors throughout ontogeny (Sanchez et al. 1999) as well as to the vast majority of long term repopulating HSCs from fetal liver and adult bone marrow (Sanchez et al. 2001). Subsequent transgenic studies demonstrated that a 640-bp fragment containing the +19 hypersensitive site was sufficient to drive reporter gene expression in embryonic tissues (Gottgens et al. 2002a) but was prone to being silenced in adult tissues (Silberstein et al. 2005). The function of the +18 remained obscure.
To assess the potential significance of the +18 region we first performed comparative sequence analysis of the intergenic segment between Tal1 and Pdzk1ip1. This analysis demonstrated that, in addition to the previously described +19 and +23 enhancers (Gottgens et al. 2000), the region at +18 was also conserved between mouse and human (Fig. 1A). Real-time PCR mapping (Follows et al. 2007) was used to confirm the presence of a DNase I-hypersensitive site in the +18 region of the myeloid progenitor cell line 416B (Fig. 1B; Dexter et al. 1979). The location of the hypersensitive site was further refined using a restriction endonuclease accessibility assay with a resolution of ∼100 bp (Gottgens et al. 2001) and was found to correspond precisely with the +18 peak of sequence homology (Fig. 1C). Chromatin immunoprecipitation (ChIP) analysis of 416B cells demonstrated clear binding of GATA2 to the +19 region, confirming our previous results (Gottgens et al. 2002b), but little binding to the +18 region (Fig. 1B). In contrast, histone H3K9 acetylation was more prominent over the +18 region compared to the +19 region (Fig. 1B). Taken together these results demonstrate that the +18 region is conserved and contains a DNase I-hypersensitive site together with acetylation of H3K9, all features of a cis-regulatory element.

Identification of the + 18 region as a region of open chromatin. (A) Sequence homology plot between mouse and human highlighting the +19 and +23 elements with known regulatory functions and the conserved peak at +18. Numbering corresponds to distances in kilobases from the start of exon 1a with the exons shown as black boxes and the repeat regions marked in gray. (B) Chromatin status and transcription factor occupancy in the Tal1 3′ flanking region. DNase I/ChIP assays were performed in the 416B cell line with either DNase I (black bars), AcK9 (gray bars), or GATA2 (white bars) and analyzed by real-time PCR using primers across a 4-kb segment of the Tal1 locus from +17 to +20. The levels of enrichment were normalized to untreated DNase I control or to IgG and plotted as fold increase over the +20 control region. (C) Restriction endonuclease accessibility assay showing the mapping of the +18 hypersensitive site (arrowhead). Nuclei were incubated with increasing concentrations of PalI and the extracted DNA digested with HindIII. Southern blot analysis was performed with the indicated probe.
The +18 region does not function as a classical enhancer
While analysis of regulatory elements in cell lines is relatively high throughput and cost effective, only in vivo analysis using transgenic animals can identify true in vivo enhancer activity across different tissues. We generated lacZ reporter constructs containing the +18 region (460-bp fragment), the +19 region (640-bp fragment), or both (1.1-kb fragment) together with the SV40 minimal promoter (Fig. 2A). These constructs were used to create transgenic embryos which were analyzed at mid-gestation by staining for β-galactosidase activity. The +19 and the +18,19 constructs both gave rise to strong hematopoietic and endothelial staining in 5/7 and 2/2 transgenic embryos, respectively (Fig. 2B; Table 1). In contrast, this pattern was absent in all transgenic embryos carrying the +18 construct (n = 21) and in all those with the construct containing the SV40 minimal promoter alone (n = 9) (Fig. 2B; Table 1). Although all four Tal1 enhancers described so far are able to function with the SV40 promoter (Sanchez et al. 1999; Sinclair et al. 1999; Gottgens et al. 2000, 2002b, 2004; Delabesse et al. 2005), we considered the possibility that the +18 element may be unable to interact with this promoter and may require the endogenous Tal1 promoter. We therefore generated constructs carrying the Tal1 promoter region with and without the +18 element. As previously described, a 3.8-kb Tal1 promoter fragment directs expression to the midbrain and spinal cord (Sinclair et al. 1999). However, inclusion of the +18 element did not result in any additional activity (Fig. 2B; Table 1). Taken together these data demonstrate that the +18 region does not show any enhancer activity in transgenic mouse embryos at mid-gestation, a time point when all organ systems are already developing.
Activity of Tal1 regulatory elements in transgenic embryos
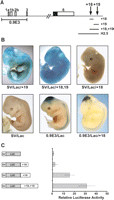
The +18 region does not behave as a classical enhancer. (A) Schematic of the Tal1 locus indicating the positions of the 0.9E3 Tal1 promoter, +18 and +19 regions. (B) Whole-mount staining of mid-gestation transgenic embryos expressing lacZ under the control of either the SV40 minimal promoter or the 0.9E3 Tal1 promoter with and without the +18, the +19, or the +18/+19 elements combined. (C) The +18 region boosts the activity of the Tal1 +19 enhancer in 416B stable transfection assays. Shown on the left are the reporter constructs of the +18 element, the 640-bp +19 element, and the 1.1-kb +18/+19 fragment inserted downstream from the SV40 luciferase reporter cassette. The results are shown on the right with the luciferase values presented as fold increase over the SV40 luciferase control, which was assigned a value of 1.
The +18 region can boost the activity of the +19 enhancer in stable transfection assays
Having established that the +18 region did not function as an independent enhancer, we next asked whether it might be able to modulate activity of the +19 enhancer. Consistent with our transgenic results a luciferase reporter construct containing the +18 element on its own was inactive in stable transfection experiments using 416B cells (Fig. 2C). In contrast, the + 19 enhancer generated a 15-fold increase in luciferase activity compared to a construct containing the SV40 promoter alone. Inclusion of the +18 element as well as the +19 enhancer reliably produced a further twofold increase in luciferase activity. These results therefore suggested that the +18 region may have a role in boosting the activity of the +19 enhancer, which is consistent with our observation that deletion of the +18 region from a 2.5-kb Tal1 3′ enhancer fragment results in a 50% reduction of enhancer activity in stable transfection assays (Supplemental Fig. 1). The activity of the +18 region was further assessed using a beta-geo reporter cassette in stable transfection assays using cotransfection of a puromycin marker for stable selection. Pools of stably transfected clones were analyzed after 3-wk selection in puromycin by flow cytometry. Cells containing the combined +18 and +19 elements showed a fourfold higher mean fluorescence than cells containing the +19 alone (Supplemental Fig. 2), demonstrating that the +18 region increases the transcriptional activity of the +19 enhancer when stably integrated into chromatin.
Single-copy analysis of +18 function during embryonic stem (ES) cell differentiation
The interpretation of stable transfection experiments is complicated by the fact that the reporter constructs are usually integrated as multiple concatenated copies and are also subject to position effects. Furthermore, transformed cell lines are unlikely to provide a normal transcriptional environment. To circumvent these issues, we used a gene targeting method that allows single-copy transgenes to be inserted upstream of the X-linked Hprt1 gene (Bronson et al. 1996; Misra et al. 2001) in HM-1 ES cells. This male ES cell line carries an inactivating deletion of the promoter and exons 1 and 2 of the Hprt1 gene. Upon successful homologous recombination with an exogenous Hprt1 targeting vector, the functional activity of Hprt1 is restored, permitting selection of correctly targeted clones in HAT media. Targeting vectors were generated using three different lacZ reporter constructs: SV40 minimal promoter alone; the promoter with the +19 enhancer; the promoter with the +19 enhancer and the +18 element (Fig. 3A). Each targeting vector was used to generate ES cell lines carrying a single copy of the appropriate reporter construct targeted to the Hprt locus. Activity of the reporter constructs was then assessed during ES cell differentiation.
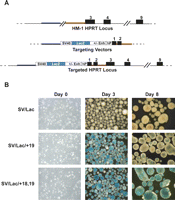
The +18 region can boost the activity of the Tal1 +19 stem cell enhancer. (A) Schematic representation of the Hprt locus in the HM-1 ES cell line, which carries a deletion of exons 1 and 2. SV40 lacZ reporter constructs with or without the 1.1-kb +18/+19 fragment or the +19 element were cloned into an Hprt targeting vector containing the promoter and exon 1 of the human HPRT locus and 2.9 kb of the mouse genomic locus including exon 2. The targeted Hprt locus contains the SV40 lacZ reporter cassettes integrated upstream of the promoter of the fully functional Hprt locus. (B) ES cells expressing either the SVLac control, SVLac/+19, or SVLac/+18,+19 were plated into differentiation assays and analyzed at days 0, 3, and 8 of differentiation. lacZ activity was assessed by staining with X-gal for 2 h.
Several groups have shown that Tal1 switches on around day 2.5 of ES cell differentiation and that this expression coincides with the onset of hemangioblast specification (Elefanty et al. 1997; Fehling et al. 2003). Prior to plating in differentiation assays, X-gal was used to stain each of the ES cell lines targeted with SV/Lac, SV/Lac/+19, and SV/Lac/+18,19. As would be expected no staining was observed in any of the targeted cell lines (Fig. 3B).
Embryoid bodies (EBs) were then generated from each of the targeted ES cell lines and lacZ activity assessed at day 3 and day 8 of differentiation (Fig. 3B). No staining was observed in EBs with the SV40 lacZ control vector at either day of differentiation. X-gal staining of EBs carrying the +19 enhancer alone (SV/Lac/+19) showed high levels of lacZ expression in day 3 EBs, which markedly decreased by day 8 of differentiation. In striking contrast, EBs containing both +18 and +19 regions (SV/Lac/+18,19) showed high levels of lacZ activity observed by X-gal staining at both days 3 and 8 of differentiation. lacZ activity in EBs was also quantified using a colorimetric assay. In cells carrying SV/Lac/+19 alone, the mean lacZ activity per 1 × 106 cells showed a twofold reduction in lacZ activity between days 3 and 8 of differentiation whereas cells containing SV/Lac/+18,19 showed a fourfold increase (Supplemental Fig. 3). These data demonstrate that, during hematopoietic differentiation of ES cells, a single copy of the +18 element boosts activity of an adjacent +19 stem cell enhancer by almost an order of magnitude.
Functional activity of the +18 region resides in a mammalian interspersed repeat
To investigate the +18 region more closely, we focused on the sequence conservation plot between mouse and human (Fig. 1A), which showed several blocks of sequence identity. To our surprise, these coincided with a stretch of 211 bp masked as repetitive sequence in the UCSC and Ensembl genome browsers. To determine the nature of the repeat, we screened the DNA sequence of the +18 region with the RepeatMasker program (A.F.A. Smit, R. Hubley, and P. Green, 1996–2004. RepeatMasker Open-3.0; http://www.repeatmasker.org), which identified the +18 repeat as a member of the mammalian interspersed repeat (MIR) family (Fig. 4A; Smit and Riggs 1995). In stable transfection assays using 416B cells (Fig. 4B), a 1.1-kb fragment containing both +18 and +19 elements produced a 90-fold increase in luciferase activity relative to a construct containing the SV40 promoter alone. Deletion of 460 bp containing the +18 element halved activity of the construct. A similar effect was observed following deletion of the 200 bp containing the MIR repeat.

A MIR repeat within the +18 region boosts the activity of the Tal1 +19 enhancer. (A) Mouse/human nucleotide sequence of the +18 region aligned to the MIR consensus sequence, in which the A and B boxes of the MIR tRNA region are underlined. The nucleotide sequence contained within the boxed region denotes the 200 bp of the +18 region that was deleted in the SV/Luc/+18,19ΔMIR construct, and the conserved sequence that was mutated to a BglII restriction site for SV/Luc/+18,19M1 is marked. (B) Mutation analysis of the +18 region. Shown on the left are the luciferase reporter constructs depicting the deletion in the +18 region and the mutation in the first block of sequence conservation highlighted in A. The mutation constructs were analyzed in 416B stable transfection assay and the luciferase values presented as fold increase over the SV40 luciferase control, which was assigned a value of 1. Data are presented as the mean luciferase activity of three biological replicates performed in quadruplicate plus or minus the standard error of the mean. (C) In vivo DMS footprinting of the +18 MIR repeat region (antisense strand). Protections (◦) were observed over the M1 region of the MIR repeat in two independent 416B samples compared to the naked genomic control. (D) Shown on the left are the luciferase reporter with constructs, either the heterologous SV40 enhancer or the tissue-specific Lyl1 promoter proximal enhancer, inserted downstream from the SV40 luciferase reporter cassette with and without the +18 region. The luciferase activities are shown as fold increase over SV40 luciferase with either the SV40 enhancer or the Lyl1 enhancer, each of which was assigned a value of 1.
In vivo DMS footprinting was performed to search for DNA sequence motifs that might mediate the boosting effect of the +18 region. No footprints were observed in the 400-bp +18 region apart from the protection of three bases (Fig. 4C, open circles) around the first block of sequence conservation (M1, Fig. 4A) situated between the MIR A and B box sequences. We therefore generated a construct whereby we mutated the M1 region to a BglII restriction site and assessed its activity in our 416B assay. Analysis of the mutant construct, SV/Luc/+18,19M1, also showed a decrease in luciferase activity comparable to the SV/Luc/+18,19ΔMIR construct (Fig. 4C). Bioinformatic analysis of the conserved motif M1 using the TRES web server (TRES: comparative promoter sequence analysis; Katti et al. 2000) showed that this conserved motif did not match any of the 222 TRANSFAC consensus binding site positional weight matrices or any of the 5919 experimentally characterized sites present in the TRES database. We also used the recently described software tool STAMP, which is specifically designed to predict the identity or structural class of proteins binding to a given sequence motif (Mahony et al. 2007). Using the JASPAR and TRANSFAC data sets, STAMP also failed to identify any match to the motif M1. Taken together these results indicate that the boosting effect of the +18 region is mediated by sequence derived from a MIR repeat and that much of this activity depends on the conserved sequence that lies within the tRNA promoter-like domain of this repeat element.
To determine whether the boosting activity of the +18 region is nonspecific or restricted to a subset of enhancers, we inserted the 460-bp +18 element upstream of an SV40 enhancer and also a Lyl1 promoter/enhancer element (Lyl1) that we have previously shown to con tain an Ets/Ets/GATA motif related to the Tal1 +19 enhancer (Chan et al. 2007). Both the SV/luc/SVEnh and SV/luc/Lyl1P constructs are highly active in stable transfection assays in 416B cells compared to the SV40 promoter alone (data not shown). When inserted into these two constructs, the +18 region failed to increase activity of the SV40 enhancer any further but dramatically boosted the activity of the Lyl1 element (Fig. 4D). These results demonstrate that the +18 element is able to increase the activity of two functionally related hematopoietic/endothelial enhancers (Tal1 +19 and Lyl1), but that it does not act nonspecifically on all enhancers. The question as to whether the activity of other enhancers may be modulated in a similar way to the Tal1 +18/+19 interaction will require comprehensive analysis using the multiple assays carried out here. However, we have noted that another hematopoietic enhancer (Fli+12; Donaldson et al. 2005b) is situated <1 kb downstream from an exapted MER repeat. Moreover, stable transfection assays suggest that this repeat region is also capable of functioning as a positive modulator enhancer activity (see Supplemental Fig. 4).
This led us to examine if the presence of MIR repeats within close proximity to cis-regulatory regions might be a more widespread phenomenon. To this end, we considered 2768 lymphoid DNase I-hypersensitive sites that have been mapped as part of the ENCODE project (Sabo et al. 2004). As a control, we performed the same analysis on 1000 sets of 2768 randomly chosen genomic coordinates to determine a normal distribution, allowing us to assign z-scores to any subsequent results. On average, we found 20% of randomly chosen coordinates have MIR repeats within 500 bp. In contrast, when we analyzed the 2768 lymphoid DNase I-hypersensitive sites, we found that 26% have a MIR repeat within 500 bp (Z-score 7.65 when compared to random genomic coordinates). MIR repeats had previously been reported to be evenly distributed with respect to the location of coding genes (Medstrand et al. 2002). Given the recent availability of data sets containing genome-wide patterns of histone modifications (Barski et al. 2007), we chose to further investigate the distribution pattern of MIR repeats (see Methods). We found that 36% of H3K4me1 sites, 23% of H3K4me3 sites, and 28% of H3K27me3 sites contain a MIR within 500 bp, suggesting that MIR repeat distribution throughout the genome is not random with the prevalence of MIR repeats next to H3K4me1 sites being particularly striking. Finally, we interrogated a recent genome-wide data set of DNase I-hypersensitive sites in human CD4 T-cells which reported fairly precise regions of hypersensitivity (Boyle et al. 2008). Out of the 95,723 DNase I-hypersensitive sites reported in the paper, 19% overlap with sequence annotated as MIR repeat compared to 12% of 95,723 randomly chosen regions of the same length as the 95,723 DNase I-hypersensitive regions. Taken together, the above analyses therefore indicated that MIR repeats may be enriched near regulatory regions consistent with a more widespread role in cis-regulation.
Sequential evolution of the Tal1 stem cell enhancer
Our observation that the +18 region was derived from a MIR repeat suggested that it evolved from a transposition event sometime during early mammalian evolution. To investigate the timing of this transposition, we obtained genomic sequences of the Tal1 locus for 10 mammals and also chicken as an outgroup for comparative analysis. Mouse, human, dog, platypus, and chicken sequences were obtained from BAC clones isolated by us previously and sequenced by the Wellcome Trust Sanger Institute (Gottgens et al. 2000, 2002a), while the remaining sequences were downloaded from the Ensembl genome browser. The 11 sequences were aligned using seven different alignment programs (ClustalW, MLagan, MAVID, Dialign, DialignT, TCoffee, MUSCLE) to minimize the likelihood of missing significant alignment blocks.
Our comparative sequence analysis demonstrated that both the +18 and +19 elements were present in all eight eutherian mammals that were analyzed (Fig. 5A; data not shown), but only the +19 could be detected in the more distantly related opossum and platypus. To confirm that our failure to detect homology with the +18 region in platypus and opossum was not a shortcoming of all seven alignment programs, we made use of the fact that the eutherian +18 sequence is recognized as MIR-derived by the RepeatMasker program. No MIR repeats were identified within 10 kb of the platypus +19 sequence, and a MIR-derived sequence 600 bp upstream of the opossum +19 element was inserted in the opposite orientation to the MIR-derived eutherian +18 region. Our results are therefore consistent with a model where the +18 MIR element integrated next to the +19 enhancer at some point between the marsupial branchpoint and the base of extant eutherians (∼160 to 120 million years ago), and was subsequently conserved throughout the radiation of placental mammals (Fig. 5B).
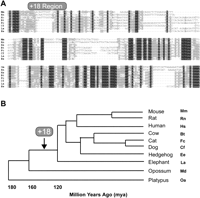
Evolution of the Tal1 stem cell enhancer region. (A) Multiple species sequence alignment of the +18 region detected in eight of the mammals analyzed. Mm, Mus musculus; Rn, Rattus norvegicus; Hs, Homo sapiens; Bt, Bos taurus; Fc, Felis catus; Cf, Canis familiaris; La, Loxodonta africana; Ee, Erinaceus europaeus. (B) Phylogentic tree depicting the divergence of the species listed on the right (adapted from Murphy et al. 2007). Time window for the insertion of the Tal1 +18 region is indicated. Md, Monodelphis domestica; Oa, Ornitorhynchus anatinus.
Discussion
Transcriptional control is a key mechanism controlling the formation and subsequent behavior of blood stem cells. Many transcription factors such as TAL1, GATA2, ETV6 (also known as TEL), FLI1, or RUNX1 are critical for the specifica tion and/or subsequent development of HSCs. It is the combinatorial activity of these (and additional as yet unknown) transcription factors that characterizes the transcriptional environment of HSCs, also termed the “regulatory state” (Davidson 2006). The Tal1 +19 enhancer represents the best-characterized regulatory element that specifically responds to the HSC regulatory state. Using reporter genes, we have shown that the +19 enhancer directs expression to HSCs in transgenic mice. The Tal1 enhancer has also been used to generate mouse models for inducible transgene expression, retroviral gene transfer, and Cre-mediated recombination as well as the development of acute myeloid leukemia mouse models (Murphy et al. 2003; Eguchi et al. 2005; Gothert et al. 2005; Koschmieder et al. 2005). At the molecular level, activity of the +19 element critically depends on binding sites for the Ets and GATA families of transcription factors and this combination of transcription factors constitutes a regulatory code employed by several other enhancers (Landry et al. 2005; Pimanda et al. 2006, 2007a, b; Chan et al. 2007; Landry et al. 2008) that display similar expression patterns in transgenic assays.
Much less well understood are the mechanisms that control quantitative aspects of gene expression, even though expression levels of key regulators are critical both to the initial development and maintenance of HSCs as well as their transformation into malignant leukemia cells. For example, reducing the level of key regulators can cause leukemia development, as illustrated by the deletion of an upstream regulatory element of the mouse PU.1 (also known as SFPI1) transcription factor, which results in an 80% reduction of PU.1 expression without any gross alterations to the overall expression pattern (Rosenbauer et al. 2004). Interestingly, a single nucleotide polymorphism in the corresponding human element reduces enhancer activity and is associated with specific subtypes of human acute myeloid leukemia (Steidl et al. 2007). Several possible mechanisms for this so-called “dosing effect” of gene expression can be proposed: Firstly, within a given regulatory element there may be binding sites that mediate the quantitative effect. One such example seems to be the PU.1 enhancer where the human SNP appears to affect binding of SATB1, which apparently plays no part in setting expression patterns but instead boosts the overall activity of the enhancer (Steidl et al. 2007). Alternatively, multiple tissue-specific enhancers with overlapping expression patterns may be required to act in an additive fashion, thus ensuring appropriate levels of gene expression as seen for example in the β-globin LCR (for review, see Grosveld 1999). Here, we propose a third and novel mechanism whereby domestication of transposable element-derived DNA sequence functions in a quantitative manner to boost the activity of a tissue-specific enhancer.
The new element (+18 region) is located 1 kb upstream of the +19 Tal1 enhancer and can boost its activity, suggesting that the Tal1 stem cell enhancer may encompass a larger gene regulatory domain. From our ES cell differentiation assays it appears that the +18 region may contribute to the function of the Tal1 stem cell enhancer by maintaining expression of the +19 enhancer, and this may explain the lack of transgene expression in adult mice when the +19 enhancer is used alone (Silberstein et al. 2005). Given that the +18 and +19 regions show the same tissue-specific pattern of DNase I hypersensitivity, their physical proximity, and specificity of enhancement, it is likely that they constitute a regulatory unit within the Tal1 gene locus. The DNA sequence in the +18 region is derived from an ancient MIR repeat. This family of repeats can be found in all mammalian orders and is thought to have arisen more than 130 million years ago (Jurka et al. 1995; Smit and Riggs 1995). MIR repeats constitute ∼1%–2% of our DNA, initially characterized by a 70-bp core sequence (Donehower et al. 1989) and more recently were shown to be part of a longer 260-bp tRNA-derived SINE (Jurka et al. 1995; Smit and Riggs 1995; Gilbert and Labuda 1999).
When the MIR-derived +18 region was assayed in standard classical enhancer assays, it failed to show any activity and therefore may easily have been disregarded when screening for regulatory elements. However, when tested upstream of hematopoietic enhancers, it behaved in a quantitative manner to boost the activity of these elements. We considered the possibility that the function of the +18 region was related to boundary elements or insulators. These latter elements are thought to play key roles in organizing higher order chromatin structure and can serve to protect a given gene locus from repressing influences of neighboring regions of heterochromatin. At the molecular level, insulator/boundary elements are characterized by the presence of ubiquitous DNase I-hypersensitive sites and binding by the multi-zinc finger protein CTCF (Xi et al. 2007). In contrast, the Tal1 +18 region is characterized by a tissue-specific hypersensitive site and not bound by CTCF (see Supplemental Fig. 5). Moreover, within our single-copy Hprt targeting constructs, the +18 region is located between the SV promoter and the Tal1 +19 enhancer and therefore not in the right position to protect the +19 enhancer from encroaching inhibitory chromatin. We also demonstrate that MIR elements are commonly found in the vicinity of DNase I-hypersensitive sites, raising the possibility of a more widespread regulatory role for this family of repeats. The precise mechanism whereby the +18 element boosts the activity of an associated enhancer remains unclear. Nevertheless, it is clear from the data presented here that we have defined a novel role for transposon-derived DNA sequence and thus added to the growing list of possible functions of repetitive DNA, which until fairly recently was considered to be “junk” DNA.
Regulatory variation is fundamental to evolution, and transposable repeat sequences (which make up at least 50% of the human genome; Kazazian 2004) may make a significant contribution (Britten and Davidson 1971). With the advent of whole genome sequencing projects and the availability of comprehensive EST data sets, it has indeed been possible to verify that up to 25% of genes incorporate transposable elements into their promoters and/or untranslated regions (Jordan et al. 2003; van de Lagemaat et al. 2003). Moreover, in the few cases studied in detail, transposable element-derived DNA was found to play a functional role in promoter activity (Landry et al. 2002; Dunn et al. 2003). Promoters only represent a relatively small proportion of active gene regulatory elements as evidenced by a recent genome-wide survey of DNase I-hypersensitive sites, which demonstrate that only 16%–21% of DNase I-hypersensitive sites were found in promoters or first exons of known genes (Boyle et al. 2008). The vast majority of gene regulatory activity therefore appears to reside in distal regulatory elements. However, very little is known about a possible role for transposable elements in the evolution of distal regulatory elements, a situation most likely explained by the relative ease of identifying promoters compared to distal regulatory elements. Despite this, a few examples of repetitive DNA gaining specific functions as distal regulatory elements have emerged where transposable elements have been shown to act as enhancers (Hambor et al. 1993; Bejerano et al. 2006; Santangelo et al. 2007) and silencers (Donnelly et al. 1999). Indeed, a recent report suggests that many exapted nonexonic elements may be preferentially involved in distal cis-regulation (Lowe et al. 2007)
Given the recent reports that 16% of eutherian-specific conserved noncoding sequence is derived from transposable elements (Mikkelsen et al. 2007) and that a proportion of these exapted noncoding elements are under strong purifying selection (Lowe et al. 2007), it is becoming increasingly clear that repetitive elements have been a driving force in carving the landscape of the human genome. Here we describe a novel function, whereby a repeat-derived sequence that would generally have been discounted in many of the techniques used to delineate gene regulatory pathways has become domesticated and acts to boost the activity of an adjacent tissue-specific enhancer.
Methods
Preparation of DNase I-treated template, chromatin immunoprecipitation, and real-time PCR analysis
DNase I-digested material was prepared from the 416B cell line as previously described (Follows et al. 2006, 2007). ChIP assays were performed as previously described (Landry et al. 2005). Briefly 416B cells were treated with 0.4% formaldehyde and the cross-linked chromatin was retrieved by nuclei isolation and lysis. The chromatin was sonicated to yield an average fragment size of ∼300 bp and immunoprecipitated with anti-GATA2 (sc-9008x, Santa Cruz) to recover DNA-bound transcription factors and an anti-acetyl H3K9 antibody (06-599, Upstate Biotechnology) to recover acetylated histones. Enrichment was measured by real-time PCR using Sybr green (Stratagene). The levels of enrichment were normalized to those obtained with a control rabbit antibody and were calculated as a fold increase over that measured at a control region. The Tal1 +20 region was used as a control for both the enrichment of GATA2 and acetyl H3K9. The following pairs of primers were used: 17PW1f: CCGAG-GATTCTGATCACTCC; 17PW1r: GGCACTTTTCTTCCCTTGTG; 18PW1f: AGGAAGG ACAGT-TGGTGTGG; 18PW1R: CTCAAAGTCACAAGCCAGCA; 19PW1f: GCAAGGTTGGGAGGTAT-CTG; 19PW1R: GAGCTG GAAGGCAAAGTGAG; 20PW1f: AAGAATGGGATGTGCTTTGG; 20PW1R: GCAGCTGTTCTCCACTACGG.
Restriction endonuclease accessibility assay and transgenic mouse analysis
416B cells were maintained as described previously (Bockamp et al. 1997). The restriction endonuclease assay was performed as described (Gottgens et al. 2001) using PalI digestion and a 210-bp HindIII/StuI fragment ∼2 kb 3′ of exon 6 as a probe. Transgenic lacZ reporter constructs were driven by the 3.8-kb 0.9E3 fragment of Tal1 (Sanchez et al. 1999; Sinclair et al. 1999) or the SV40 minimal promoter. DNA was linearized using BamHI and SalI, and F0 transgenic embryos were generated by pronuclear injection as described (Sanchez et al. 1999). Embryos were harvested at mid-gestation (embryonic day 11–12.5) and analyzed as described (Sanchez et al. 1999).
Reporter constructs and transfection assays
Reporter constructs were generated by cloning the following fragments of the Tal1 locus (numbering refers to the Tal1 locus database entry AJ131017) downstream from the luciferase gene in pGL2 (Promega) or downstream from an SV40 minimal promoter containing a lacZ reporter cassette. The +18/+19 regions of Tal1 were cloned as a 1.1-kb NcoI/HindIII fragment from 78,359 to 79,480 and an SspI site in the middle of this region was used to generate the shorter 480-bp NcoI/SspI fragment from 78,359 to 78,840 and the 640-bp SspI/HindIII fragment from 78,840 to 79,480 for the +18 and +19 constructs, respectively. The mutation constructs were made as previously described (Gottgens et al. 2002b) and verified by sequencing. Stable transfections and luciferase assays were performed as described (Gottgens et al. 1997). Data are presented as a representative plot of three biological replicates performed in quadruplicate plus/minus standard deviation unless otherwise stated.
ES cell culture and targeting
The HM-1 ES cell line was a kind gift from David Melton (Magin et al. 1992). ES cells were cultured on mitotically inactivated primary embryonic fibroblasts in standard ES cell media supplemented with LIF. For the introduction of transgenes at the Hprt locus ∼1 × 108 ES cells were electroporated with 100 μg of Pme I linearized DNA (0.8Kv,3uF Gene Pulser, Bio-Rad) followed by selection in HAT media (Sigma). HAT-resistant colonies were picked after 7–10-d growth in HAT-containing media. Targeted ES cells were differentiated in 90-mm petri dishes in IMDM supplemented with 15% FCS, 2 mM L-glutamine, 300 μg/mL transferrin, 4 × 10−4 M MTG, 50 μg/mL ascorbic acid, and 5% PFHM-II. Harvested EBs were either fixed and stained for 2 h with X-gal as for the transgenic embryos or disrupted with trypsin for 3 min, and 1 × 106 cells lysed and assayed for lacZ activity using the synthetic β-galactosidase substrate ONPG as described (Bockamp et al. 1995).
In vivo DMS footprinting
Footprinting was performed as previously described (Follows et al. 2003). Briefly cells or genomic DNA were incubated at room temperature in 0.2% DMS solution in PBS for 5 min. The reaction was stopped with multiple washes with ice-cold PBS. Following cell lysis and DNA extraction, DNA was cleaved with 0.1 M piperidine at 90°C for 10 min and analyzed by ligation-mediated PCR. PCR products were labeled by primer extension using 32P-labeled nested primers and analyzed on 6% denaturing polyacrylamide gels. Linker and primer sequences are available on request. Experiments were performed on three separate occasions with material from two independent DMS-treated cell and genomic DNA preparations.
Comparison of MIR repeats with genomic annotation
Five sets of genome annotation corresponding to the human genome (NCBI v35/hg17/May 2004) were obtained for this study, including a genome-wide set of coordinates for MIR family repeats to be compared against four other data sets, namely: two collections of DNase I-hypersensitive sites, histone methylation sites, and random genomic regions. All MIR family repeats were obtained using the UCSC genome table browser (http://genome.ucsc.edu). Genome coordinates were output in the BED format, selecting the group: “Variation and Repeats”, track: “RepeatMasker”, and table: “rmsk” (n = 591,703). Two sets of DNase I hypersensitivity site annotation were extracted using the table browser. The first ENCODE-based set, by Sabo et al. (2004), was retrieved by selecting the group: “ENCODE Chromosome, Chromatin and DNA Structure”, track: “UW Dnase-array”, and table: “DNase I HSs” (n = 2768). The second genome-wide set, by Boyle et al. (2008), was retrieved by selecting the group: “Expression and Regulation”, track: “Duke DNase Sites”, and table: “dukeDnaseCd4Sites” (n = 95,723). Histone methylation sites were extracted from the supplementary Web site relating to the work of Barski et al. (2007). “Summary BED files” for H3K4me1, H3K4me3, and H3K27me3 modifications were downloaded. A Perl script was used to convert the files from “variable step” to BED format and extract all regions containing a minimum number of tags, resulting in the most significant ∼2000 regions per histone modification. The minimum numbers of tags for H3K4me1, H3K4me3, and H3K27me3 modifications represented in the extracted data where 42, 87, and 9 respectively. The genome coordinates were converted from human version hg18 to hg17 using the “liftOver” conversion function of Galaxy (Giardine et al. 2005; http://g2.trac.bx.psu.edu/).
To allow the calculation of Z-scores (to evaluate the chance of randomly finding an overlap between the ENCODE DNase I-hypersensitive sites and MIR repeats), 1000 sets of 2768 random genome coordinates, each of 250 nucleotides in length, were generated using a Perl script. A second Perl script was used to calculate the percentage overlap of each random genome coordinate set (extended by 500 flanking nucleotides each side, creating a set of extended coordinates) with the MIR repeat coordinates. The mean and standard deviation of the percentage overlaps for the 1000 comparisons were then calculated.
The comparison of the MIR repeat coordinates to the other data sets was performed using Galaxy. The coordinates of the two collections of DNase I-hypersensitive sites and histone methylation sites were extended by 500 flanking nucleotides each side, creating two sets of extended coordinates. The “intersection” (by at least one nucleotide) between the extended coordinates and the MIR family coordinates was determined.
Acknowledgments
This work was supported by the Cambridge MIT institute, Medical Research Council (MRC), Leukemia Research Fund (LRF), Biotechnology and Biological Sciences Research Fund (BBSRC), and the Wellcome Trust. We thank David Melton (University of Edinburgh) for the HM-1 cell line and Stephen Duncan (Medical College of Wisconsin) for the Hprt targeting vector. We thank Mike Chapman, Darren Grafham, and Team 47 at the Wellcome Trust Sanger Institute for the platypus sequences, and Sandie Piltz and Michelle Hammett for generating the transgenic embryos. We also thank Josette-Renée Landry and John Pimanda for the critical reading of this manuscript.
Footnotes
-
↵3 Present address: Centro Andaluz de Biologia del Desarrollo, Universidad Pablo de Olavide, Carretera de Utrera Km1, Sevilla 41013, Spain.
-
↵4 Joint senior authors.
-
↵5 Corresponding author.
↵5 E-mail bg200{at}cam.ac.uk; fax +44-1223-762670.
-
[Supplemental material is available online at www.genome.org.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.077008.108.
-
- Received February 4, 2008.
- Accepted June 5, 2008.
-
Freely available online through the Genome Research Open Access option.
- Copyright © 2008, Cold Spring Harbor Laboratory Press








