
Identification of active transcriptional regulatory modules by the functional assay of DNA from nucleosome-free regions
Abstract
The identification of transcriptional regulatory modules within mammalian genomes is a prerequisite to understanding the mechanisms controlling regulated gene expression. While high-throughput microarray- and sequencing-based approaches have been used to map the genomic locations of sites of nuclease hypersensitivity or target DNA sequences bound by specific protein factors, the identification of regulatory elements using functional assays, which would provide important complementary data, has been relatively rare. Here we present a method that permits the functional identification of active transcriptional regulatory modules using a simple procedure for the isolation and analysis of DNA derived from nucleosome-free regions (NFRs), the 2% of the cellular genome that contains these elements. The more than 100 new active regulatory DNAs identified in this manner from F9 cells correspond to both promoter-proximal and distal elements, and display several features predicted for endogenous transcriptional regulators, including localization within DNase-accessible chromatin and CpG islands, and proximity to expressed genes. Furthermore, comparison with published ChIP-seq data of ES-cell chromatin shows that the functional elements we identified correspond with genomic regions enriched for H3K4me3, a histone modification associated with active transcriptional regulatory elements, and that the correspondence of H3K4me3 with our promoter-distal elements is largely ES-cell specific. The majority of the distal elements exhibit enhancer activity. Importantly, these functional DNA fragments are an average 149 bp in length, greatly facilitating future applications to identify transcription factor binding sites mediating their activity. Thus, this approach provides a tool for the high-resolution identification of the functional components of active promoters and enhancers.
The precise regulation of gene expression is fundamental to all cellular processes and is largely mediated by the concerted activities of regulatory elements such as promoters and enhancers, which determine the level, timing, and cell-type specificity of gene transcription. The functional units within promoters or enhancers are one or more “cis regulatory modules,” which contain clusters of binding sites for multiple transcription factors (Istrail and Davidson 2005). Deciphering the components of mammalian transcriptional regulatory networks therefore requires identification of these functional elements and elucidation of both the genes that they control and the cellular context(s) in which they operate. To this end, high-throughput methods such as ChIP-chip and ChIP-seq have been developed to map genomic regions associated with specifically modified histones or transcriptional regulatory factors (Ren and Dynlacht 2004; Kim and Ren 2006; Barski et al. 2007; Johnson et al. 2007), and computational methods have also been used to identify evolutionarily conserved sequences that are likely to be functionally relevant (Liu et al. 2004). Complementary approaches have focused on the genome-wide mapping of nucleosome-free regions (NFRs) within chromatin, as it has been well established that these regions coincide with active regulatory DNA elements (Wu 1980; Elgin 1984; Gross and Garrard 1988; Felsenfeld 1996; Felsenfeld and Groudine 2003). However, several challenges inherent to these approaches remain. For example, NFRs are associated with active promoters, enhancers, insulators, silencers, and locus control regions, and it is often difficult to ascribe a specific regulatory function(s) to an individual NFR (Sabo et al. 2004; Crawford et al. 2006; Follows et al. 2006; Giresi et al. 2007; Xi et al. 2007). Furthermore, the genomic regions identified by these methods are relatively broad and often cannot precisely indicate the active regulatory modules within these loci.
A step toward resolution of these issues would be to integrate data derived from functional approaches designed for the direct identification of active transcriptional regulatory modules. Functional analyses have most often been employed as a validation tool for the evaluation of genomic regions that had been predicted by other means to function as promoters or enhancers. For example, Trinklein et al. (2003) first aligned cDNA sequences to the human genome sequence to identify the locations of the transcription start sites (TSSs) for several thousand genes and then tested the surrounding DNA sequences for promoter function using transient transfection assays. Another study functionally tested computationally defined, ultra-conserved human DNA fragments using an in vivo, mouse transgenic assay to identify enhancers (Pennacchio et al. 2006). However, the application of functional analyses as a tool for the de novo discovery of transcriptional regulatory modules has been hampered by the enormity of mammalian genomes, and consequently, there is only one report in which this approach was attempted. In this study (Khambata-Ford et al. 2003), DNA fragments generated by enzymatic digestion of total human genomic DNA were tested for their ability to drive expression of a promoterless GFP retroviral reporter plasmid, thereby identifying several hundred promoters.
Although assays such as those outlined above have been invaluable to furthering an understanding of promoter structure, identifying the genomic locations of elements that can activate transcription, and developing models for tissue-specific gene activation, they are by their nature unable to determine whether the endogenous counterparts of these modules are active within the chromatin environment of a given cell type. It would therefore be useful to develop a functional assay that not only identifies DNA segments that activate transcription but also closely reflects the chromatin status of the transcriptionally active element.
In this report, we describe a functional assay that aims to identify, with high resolution, the genomic locations of transcriptional regulatory modules that are active regulators of endogenous genes in the cell type under examination. To this end, we have developed a simple method to isolate DNA fragments from NFRs within F9 cell chromatin and to identify transcriptional regulatory modules within this population using a functional assay. As proof of principle, we show that the genomic positions of the functional elements identified in this manner correlate with multiple features, including association with genes that are expressed in F9 cells, and colocalization with modified histones characteristic of active promoters or enhancers, that support a predicted role for these sequences in the transcriptional activation of endogenous genes in F9 cells. We also demonstrate that we can isolate both promoters and enhancers, some of which function in a cell type-restricted manner. Extensions of this approach have the potential to identify an array of regulatory modules that are active in different cell types, and contribute complementary functional information to the data sets of ChIP- and DNase-chip studies.
Results
Preparation of DNA fragments from NFRs
The enzymatic susceptibility of NFRs was exploited to isolate a population of DNA fragments that are enriched for transcriptional regulatory elements. Protein–DNA complexes within F9 embryonal carcinoma (EC) cells were formaldehyde cross-linked, and permeabilized nuclei prepared from these cells were incubated in the presence of the restriction enzyme HaeIII. HaeIII recognizes the sequence GGCC that occurs, on average, every 300 bp in the mouse genome, and should preferentially digest chromatin regions that are depleted of nucleosomes, allowing the release of DNA fragments small enough to diffuse out of the nuclei into the surrounding buffer. The nucleosome-dense, uncut chromatin was removed by centrifugation of the nuclei, and the HaeIII-released fragments were recovered from the supernatant (Fig. 1A).
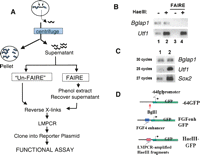
The HaeIII supernatants contain DNA derived from NFRs. (A) Method overview. Permeabilized nuclei are incubated with HaeIII. Silenced genes exhibit a densely packed nucleosome array (blue circles) that prohibits access of HaeIII to the DNA. This uncut DNA remains in the nuclei and is removed by centrifugation. In contrast, the relative absence of nucleosomes at the regulatory regions of actively transcribed genes renders the DNA accessible to HaeIII cleavage (small arrows). The released fragments diffuse out of the nucleus and can be recovered from the supernatant after centrifugation. (B) PCR analysis of HaeIII supernatants. F9 nuclei were incubated in the presence (+) or absence (−) of HaeIII. After centrifugation, DNAs in the supernatants were processed either with or without FAIRE treatment and analyzed for the relative levels of Bglap1 gene or Utf1 enhancer sequences using PCR. “Un-FAIRE” samples, lanes 1,2; FAIRE samples, lanes 3,4. (C) LMPCR enriches for DNA derived from NFRs. The relative levels of Utf1 enhancer, Sox2 enhancer, and Bglap1 DNA were compared before (lane 1) and after (lane 2) LMPCR amplification of DNAs in the HaeIII supernatants of the UnFAIRE sample. Note that after LMPCR, fewer cycles were required to visualize the Utf1 and Sox2 enhancer PCR products. (D) Reporter plasmids. The starting plasmid, −64GFP, contains the TATA box and transcription initiation site from the Fgf4 promoter cloned upstream of GFP coding sequences. The indicated BglII site was used for insertion of the Fgf4 enhancer to create the positive control plasmid FGFenhGFP, or LMPCR-amplified HaeIII fragments to create the HaeIII-GFP plasmid libraries.
The ability of this approach to enrich for active transcriptional regulatory elements was monitored using PCR. The formaldehyde cross-links were reversed in a portion of the supernatants, and the DNAs were purified (Fig. 1A, Un-FAIRE). Since the Utf1 enhancer is active in F9 cells (Nishimoto et al. 1999), it should reside in a NFR and be accessible to HaeIII cleavage. In addition, if these fragments efficiently diffuse out of the nuclei, we expect to observe more Utf1 enhancer DNA in the supernatant of HaeIII-digested nuclei than in that from undigested nuclei. As shown in Figure 1B, lanes 1 and 2, more Utf1 enhancer DNA is indeed present in the supernatant of the HaeIII-digested nuclei. In contrast, the amount of DNA sequences derived from the Bglap1 gene, which is inactive in F9 cells, was not changed by HaeIII cleavage (Bglap1, Fig. 1B, lanes 1,2).
FAIRE treatment
Figure 1B shows that some nonspecific DNA (e.g., Bglap1 sequences) is also present in the HaeIII supernatant. To eliminate this background, we employed a method denoted FAIRE (Nagy et al. 2003). FAIRE is based on the observation that sonicated nucleosome-bound, formaldehyde-cross-linked chromatin partitions in the interphase during phenol extraction, while nucleosome-free DNA remains in the supernatant. Supernatants from HaeIII-treated or -untreated nuclei were subjected to phenol extraction prior to cross-link reversal (FAIRE, Fig. 1A). PCR analysis showed that all Bglap1 sequences, in both HaeIII-treated and HaeIII-untreated samples, were removed by FAIRE (Fig. 1B, lanes 3,4). Utf1 DNA was also eliminated by FAIRE in the sample NOT digested with HaeIII (lane 3), presumably because it is embedded in a large segment of nucleosome-bound background DNA in this sample. In contrast, Utf1 DNA remained in the HaeIII-digested FAIRE supernatant, indicating that it had been released from the nuclei by HaeIII digestion and that it is nucleosome-free (Fig. 1B, lane 4).
LMPCR amplification of the HaeIII fragments
To prepare the HaeIII fragments for cloning into a reporter plasmid, ligation-mediated PCR (LMPCR) was employed. The double-stranded linker utilized (Supplemental Table 1) has a blunt end compatible for ligation to the HaeIII-digested DNA fragments but is unlikely to ligate to nonspecific DNA in the HaeIII supernatant. The linker was ligated to HaeIII DNAs in the FAIRE and UnFAIRE supernatants, followed by limited PCR amplification using primers complementary to the linker. PCR analysis showed that LMPCR specifically amplified DNA sequences derived from enhancers known to be active in F9 cells (i.e., Utf1, Sox2) but not Bglap1 DNA sequences (Fig. 1C). Thus, LMPCR provides a further specific enrichment of NFR-derived HaeIII fragments.
Functional analysis of DNA fragments derived from NFRs
To identify NFR-derived HaeIII DNA fragments that act as promoters or enhancers, they were tested for their ability to activate transcription of a GFP reporter plasmid in transfected F9 cells. The reporter plasmid –64GFP (Fig. 1D) contains the TATA box and transcription initiation site of the Fgf4 promoter (Ambrosetti et al. 1997) cloned upstream of the GFP gene in EGFP-1 (Clontech). A positive control plasmid, FGFenh-GFP was constructed by inserting a 250-bp fragment containing the murine Fgf4 enhancer (Curatola and Basilico 1990) at a BglII site located immediately upstream of the TATA box in the –64GFP plasmid (Fig. 1D).
The linker flanking the LMPCR-amplified HaeIII fragments contains a BglII site that was used to insert these DNAs into the –64GFP plasmid. Separate plasmid libraries, composed of either FAIRE-treated HaeIII DNAs or Un-FAIRE-treated HaeIII DNAs were prepared. Two hundred forty-three plasmids of FAIRE-treated HaeIII DNAs and 189 plasmids of Un-FAIRE-treated HaeIII fragments were individually transfected into F9 cells in 96-well plates, and the level of GFP expression was determined using an Envision microplate reader (Perkin Elmer). HaeIII-GFP test plasmids exhibiting reproducible GFP activation at least twice that of the −64GFP plasmid were scored positive. Using these criteria, ∼20% of the HaeIII fragments in each library activated GFP expression (Table 1). Enrichment for these elements was not significantly affected by FAIRE treatment, suggesting that the majority of the DNA in the HaeIII supernatants do in fact derive from NFRs and that background DNA is not a significant factor in the preparation of these libraries, presumably due to the specificity imparted by LMPCR.
Summary of the ability of different preparation methods to isolate activating transcriptional elements
To assess the significance of this result, we created a third plasmid library of HaeIII fragments derived from the digestion of naked, total genomic mouse DNA (random HaeIII DNAs). Analyses of 100 of these plasmids showed that, in contrast to the libraries of NFR-derived HaeIII fragments, only one random HaeIII DNA (clone R28) activated GFP expression. Thus DNA fragments derived from NFRs display a significantly greater ability to activate transcription of the GFP reporter plasmid than randomly isolated DNA fragments.
The functional HaeIII DNAs are bona fide transcriptional elements in F9 cells
NFR-derived HaeIII DNA fragments that are active in the functional assay reside in NFRs in situ
HaeIII fragments that activated GFP expression were sequenced and mapped to their location in the mouse genome using the BLAT algorithm (http://genome.ucsc.edu/cgi-bin/hgBlat). Representative DNAs are shown in Table 2. A list of all 88 elements can be found in Supplemental Table 2. Notably, all of the HaeIII DNAs were short, with an average length of 149 bp. All clones were unique and 97.6% mapped to a single position in the mouse genome. If the functionally selected HaeIII DNAs represent active regulatory elements in F9 cells, they should lie within NFRs. To test this, permeabilized F9 cell nuclei were treated with DNase I for increasing lengths of time, and the genomic DNAs were assayed using PCR and specific primers complementary to several of the active HaeIII DNAs. Figure 2 shows that sequences corresponding to the Bglap1 gene or R28, the single clone of the “random HaeIII library” that was able to activate GFP expression, are refractory to DNase I digestion, consistent with their location in silenced chromosomal regions in F9 cells. In contrast, sequences spanning the Utf1 enhancer, and all of the NFR-derived HaeIII fragments tested were exquisitely sensitive to DNase cleavage (Fig. 2). This observation confirms that NFR-derived fragments that are active in the GFP assay reside in NFRs within F9 cell chromatin.
Active HaeIII fragments display features characteristic of transcriptional regulatory elements

Functional HaeIII DNAs reside in NFRs in situ. F9 cell nuclei were treated for 0,1, 3, or 5 min with DNase I as indicated above each lane. The genomic DNAs were analyzed using PCR and primers spanning the sequences indicated on top.
NFR-derived fragments that activate GFP expression map to genomic regions with features of enhancers or promoters regulating endogenous genes
We classified the 56.8% of the functional NFR-derived DNA fragments that mapped within 2 kb of an annotated RefSeq transcription start site (TSS) as proximal elements. These included several bidirectional promoters. The remaining HaeIII DNAs were defined as distal elements (Table 2; Supplemental Table 2; Supplemental Fig. 2). Fifty-nine percent of the HaeIII DNAs overlapped a CpG island (Table 3), and 86.5% of these were proximal elements. If the elements that we have identified participate in the activation of endogenous genes, we expect those genes to be expressed in F9 cells. Candidate target genes, i.e., “associated genes,” were arbitrarily defined as RefSeq or known genes whose TSS is within 100 kb of the mapped HaeIII element, and their expression status in F9 cells was determined using Affymetrix expression microarrays. Notably, 100% of the proximal elements are associated with genes that are expressed in F9 cells. Twenty-six percent of the distal elements did not lie within 100 kb of a known TSS. However, 75% of the associated genes for the remaining distal elements are expressed in F9 cells. Thus, the majority of our functional elements are associated with actively transcribed genes.
Genomic characteristics of functionally selected, transcriptionally active HaeIII DNAs (active HaeIII elements) compared with random HaeIII fragments generated by the in silico digestion of mouse genomic DNA (random HaeIII DNAs)
To assess the significance of these correlations, we devised an algorithm that permits in silico HaeIII digestion of the total mouse genome and selection of fragments of the same average length as our functionally selected HaeIII DNAs to create a “background” model of random HaeIII fragments (Supplemental Table 3). Nearly half of the random HaeIII fragments corresponded to repeat elements, in contrast to only 2.4% of the active HaeIII DNAs, and were far less frequently associated with CpG islands (1.2%), proximity to an expressed gene (4%), or evolutionary conservation (Table 3; Supplemental Figs. 1 and 2). Thus, the functionally selected HaeIII DNAs map more significantly to genomic positions that are consistent with a role in transcriptional regulation.
Proximal elements are functional components of their natural promoters
DNA fragments spanning the 5′ end of proximal element 2-27b or 2-52 to ∼50 bp downstream of the TSS of each of their respective associated genes were cloned upstream of the luciferase gene within the pGL3 plasmid, as were similar fragments containing a deletion of the HaeIII elements (D27b, D2-52) (Fig. 3A). The levels of luciferase expression produced by each of the “full length” and deletion mutant constructs were compared after transfection of F9 cells. In both cases, promoter activity was compromised by deletion of the HaeIII DNA fragment (Fig. 3A), supporting the notion that they each represent functional components of these promoters.
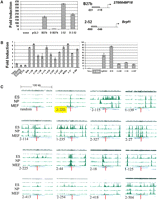
Functional HaeIII DNAs are bona fide transcriptional regulatory elements in F9 cells. (A) Proximal HaeIII fragments represent activating components of endogenous promoters in F9 cells. (Left panel) Luciferase expression exhibited by each of the plasmid constructs (right) relative to the pGL3 vector is indicated on the Y-axis. (Right panel) Plasmid constructs. The gray rectangles indicate the locations of the NFR-derived HaeIII fragments 27b and 2-52 relative to the associated genes’ TSS. These regions are deleted in constructs D27b and D2-53. (B) Luciferase assays to assess the ability of the HaeIII DNAs to act as enhancers. Seventeen distal elements identified in the GFP screen were cloned 2 kb downstream of the TK promoter in the pTK-luciferase reporter plasmid and transfected into F9 cells. Control plasmid 2-320 (highlighted by the gray box) contains a proximal element insert; FgfENH contains the Fgf4 enhancer to provide a positive control. Values were averaged and normalized to the luciferase activity of pTK-luciferase (assigned the value 1) and are expressed as fold induction relative to pTK-luciferase. Results for enhancers exhibiting moderate (left panel) or high (right panel) activity are shown. (C) Active HaeIII DNAs localize to genomic regions enriched for H3K4me3 in ES cells. The genomic coordinates (mm8 assembly) for each of the HaeIII DNAs were submitted to http://www.broad.mit.edu/seq_platform/chip/ to visualize their position relative to nucleosomes containing modifications of Histone H3. The HaeIII element analyzed is indicated on the lower left of each panel. “Random” indicates a representative HaeIII DNA sequence derived from the in silico HaeIII digestion of genomic mouse DNA. 2-320 is representative of the result observed for analysis of proximal elements and is highlighted in yellow. The remaining HaeIII elements are distal elements (Supplemental Table 2). Each panel shows an ∼100-kb segment containing the HaeIII element, and genomic locations of H3K4me3 are indicated in green. The red line indicates the position of the each of the HaeIII elements. The width of this line is not drawn to scale (the average length of these elements is 149 bp). The complete relationship between H3K4me3 and all of the elements is shown in Supplemental Figure 3.
The distal elements can function as enhancers
Since the distal elements do not lie in close proximity to an annotated TSS, we tested whether they could function as enhancers. Seventeen distal elements were each inserted 2 kb downstream of the herpes simplex virus thymidine kinase (TK) promoter within pTKLuc (Miyagi et al. 2004) and tested, along with control plasmids containing the Fgf4 enhancer or the proximal element 2-330 cloned at the same position, for their ability to activate luciferase expression after transfection in F9 cells. Sixteen (94.1%) of the distal elements tested showed enhancer activity, while 2-320 did not (Fig. 3B). Thus, functional targeting of NFRs also permits isolation of active enhancer elements.
Elements identified using the functional assay of NFRs correspond to genomic regions of trimethylated H3K4
A recent report by Mikkelsen et al. (2007) used ChIP-seq to assess the genomic distributions of trimethylated H3K4 (H3K4me3), which is associated with active transcriptional regulatory elements, and other modifications of H3 associated with repression or elongation, in ES, MEFs, and NP cells. Since F9 EC cells display largely overlapping gene expression profiles (Sperger et al. 2003) and shared mechanisms of transcriptional regulation with ES cells, we assessed whether any of these histone marks colocalized with our functional elements. This analysis showed that 100% of the proximal elements lay within broad peaks of H3K4me3 in ES cells and, with one exception, colocalized with H3K4me3 in all three cell lines (e.g., element 2-320) (Fig. 3C). Similar analysis of the distal elements showed that the vast majority localized precisely to distinct peaks of H3K4me3 (Fig. 3C) but not H3K27me3 or H3K9me3 marks associated with repression (data not shown). In contrast to the general correspondence of H3K4me3 with the proximal elements in all three cell lines, most of the distal elements aligned with H3K4me3 only in ES cells (Fig. 3C). The complete relationship between H3K4me3 and the proximal and distal elements in all three cell lines is depicted in Supplemental Figure 3. Thus, the colocalization of our functionally identified elements with H3K4me3 in ES cells is consistent with the possibility that these sequences define transcriptionally activating modules that contribute to the generation of this histone modification and the activation of endogenous gene transcription in ES and EC cells.
ChIP of NFR-derived DNAs
Functional NFRs harbor the binding sites for transcription factors that should remain associated with them as these DNAs diffuse out of the HaeIII-treated nuclei and therefore should be amenable to ChIP. Thus, we applied the functional assay to NFR-derived DNAs that had been immunoprecipitated by antibodies against the ES/EC cell transcription factor SOX2 (Yuan et al. 1995). Of the 75 plasmids tested, 50.6% displayed activated GFP expression (Table 1). The functional SOX2 elements showed a comparable distribution of proximal and distal elements as the “general” elements (Supplemental Fig. 2; Supplemental Table 2), and most of the associated genes are expressed in F9 cells but were less frequently associated with CpG islands (26.3% vs. 59% of the clones of Table 2). This may be related to the observation that tissue-specific genes tend to display CpG-poor promoters (Saxonov et al. 2006).
The transcription factors SOX2, POU5F1, and NANOG are important regulators of gene expression in ES and EC cells (Niwa 2007), and previous ChIP-chip and ChIP-PET analysis showed that these proteins bind many common target DNA regions (Boyer et al. 2005; Loh et al. 2006). Several of our active clones corresponded to subregions of these previously defined target sequences (Fig. 4A). The majority of the SOX2-IP’d NFRs activated GFP expression in F9 cells but not in transfected NIH3T3 mouse fibroblasts, which do not express SOX2, POU5F1, or NANOG (Fig. 4B). Furthermore, while most of the SOX2-IP’d DNAs aligned with genomic regions enriched for H3K4me3, the distal elements tended to do so only in ES cells, but not in NP cells or MEFs (Fig. 4C; Supplemental Fig. 3). These observations suggest that these subregions represent the critical functional core of the larger, previously identified target sequences, and support the notion that functional assay of NFR-derived DNA can provide high-resolution identification of endogenous transcriptional regulatory modules.
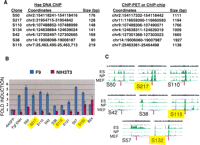
ChIP of NFRs followed by functional assays allows the high-resolution identification of functional target DNA sequences. (A) Overlap between functional SOX2-ChIP elements and previously reported target sequences. The genomic coordinates (assembly mm8) and the length of the DNA fragments for each of the target DNAs identified in each data set are indicated. (B) Transcriptional activation by the SOX2-ChIP’d NFRs listed in A, as well as several novel SOX2-ChIP HaeIII DNAs, were assessed after transfection of F9 or NIH3T3 cells. −64GFP and FgfENH are the negative and positive control plasmids, respectively (Fig. 1D), and GFP plasmids harboring each of the SOX2-ChIP’d NFRs are labeled as S. The control plasmid B24 (Supplemental Table 2) harbors a ubiquitously active HaeIII DNA. GFP expression elicited by each plasmid in F9 cells (blue bars) or NIH3T3 fibroblasts (red bars) were normalized to −64GFP and expressed as “fold induction.” (C) The genomic localization of each of the SOX2-ChIP elements (Supplemental Table 2) relative to H3K4me3 was determined as in Figure 3C. The elements highlighted in yellow correspond to proximal elements. The complete relationship of these elements with H3K4me3 is shown in Supplemental Figure 3.
Discussion
The identification of transcriptional regulatory modules by the functional analysis of DNA derived from NFRs presented here provides two major advantages over assays using total genomic DNA. First, the search space is limited to genomic regions that harbor active regulatory elements. NFRs represent only 2%–3% of the genome in a given cell (Xi et al. 2007), and focusing the functional assay on NFRs achieves a 20-fold enrichment for activating transcriptional elements compared with DNAs of HaeIII-digested total naked genomic DNA and affords a 50-fold enrichment if the NFRs are first subjected to ChIP prior to functional analysis. Second, information regarding the cell type in which these elements are active is inherent in our approach. Since the DNA fragments that activate transcription of the reporter gene derive from NFRs in F9 cell chromatin, it is likely that they also function in endogenous transcriptional regulation in these cells, in contrast to the random clone, R28, derived from HaeIII digestion of naked DNA. Although R28 activates GFP expression in the transient assay, and therefore may harbor a transcriptional regulatory element, this element does not appear to be functional for endogenous gene regulation in F9 cells since the DNase I assay (Fig. 2) showed that it exists in a region of condensed chromatin. The method presented here for the preparation of NFRs is simple and fast and results in a highly purified collection of these DNAs due to several key steps: (1) The nuclei act as a natural barrier that allows outward diffusion of the HaeIII-digested DNAs while retaining the bulk of the chromatin, thus affording easy fractionation of released NFRs by centrifugation; and (2) FAIRE treatment and/or LMPCR minimize background DNA not derived from NFRs.
Individual transcriptional regulatory modules are generally encompassed within 250 bp and the DNA fragments released by HaeIII digestion approximate this size, thereby allowing a high degree of resolution. A potential concern is that the functional assay of individual modules, which have been extricated from other regulatory influences in their natural genomic context, may not accurately reflect the role of these elements in transcriptional regulation of endogenous genes. While we cannot definitively eliminate this possibility, the strong correlation of our selected functional elements with parameters predictive of transcriptional regulatory elements, and the relative inability of random DNAs to activate the reporter gene, suggest that the majority of the elements identified by our assays correspond to activating components of endogenous promoters or enhancers. Although ∼15% of the distal elements that we have isolated do not align with any of the histone marks included in the Mikkelsen study (Mikkelsen et al. 2007) and may therefore represent false positives, it is also possible that these elements have distinct functions in ES and EC cells or correspond with histone modifications other than those analyzed. In this regard, it should be noted that some discrepancy remains concerning whether H3K4me3/H3K4me1 or H3K4me1 alone preferentially marks the site of enhancers (Barski et al. 2007; Heintzman et al. 2007).
Emerging evidence from genome-wide studies indicates that transcriptional activity in mammalian genomes is far more complex than was originally appreciated, involving multiple TSSs, anti-sense transcripts, and noncoding RNAs, and that the actual number of TSSs may exceed the current RefSeq annotations by 10-fold (The ENCODE Project Consortium 2007). Thus genes associated with the proximal elements can be assigned with a reasonable measure of confidence, but we are less certain whether the distal elements play a role in regulating their assigned associated genes or are functionally linked to another transcript. More definitive identification of target genes for these elements will require the application of additional methods such as 5C (chromosome conformation capture carbon copy) (Dostie et al. 2006).
Nearly all of the distal elements can act as enhancers and tend to correlate with ES cell-restricted peaks of H3K4me3 (Supplemental Fig. 3), suggesting that they may represent cell-specific elements. This correlation was observed for both the general elements of Table 2 as well as the SOX2 ChIP’d elements, and is consistent with a recent comparison of DNase accessible sites found in six cell lines that indicated that promoter-proximal NFRs tend to display “ubiquitous” peaks of H3K4me3, whereas distal NFRs colocalizing with H3K4me3 tended to be cell-type restricted (Xi et al. 2007). However, the functional analysis of the SOX2 IP’d elements indicates that some proximal elements activate transcription in an F9-specific manner even though H3K4me3 marks at these promoters are ubiquitous. If SOX2 participates in transcriptional activation at these promoters in ES and EC cells, this result suggests that the ubiquitous presence of an NFR or of H3K4me3 at a given promoter does not necessarily arise from the identical constellation of transcription factors or regulatory elements in all cell lines, further underscoring the importance of functional assays to define the active regulatory modules contributing to promoter function.
The full potential of this technique will be realized by its transformation into a truly high-throughput format and by increasing the probability of obtaining a full representation of active modules from NFRs. In regard to the latter point, it is likely that HaeIII digestion of some percentage of modules will have caused their inactivation. Thus, the recovery of all active modules within a cell may be enhanced by creating two different sets of NFRs, each prepared by the digestion of cell chromatin using restriction enzymes with distinct DNA recognition properties. Libraries of these NFR DNA fragments can be created using retroviral-GFP reporter constructs and active clones selected en masse using FACS. The active NFR-derived modules could then be recovered from the GFP-positive cells using PCR and identified using high-throughput techniques such as Solexa sequencing. Furthermore, although we have focused on the identification of functional regulatory components of promoters and enhancers, other regulatory elements, such as insulators and silencers, could also be functionally identified from NFRs using reporter constructs designed for revealing these activities. Eventual annotation of different regulatory elements by the integration of data generated by ChIP-chip/seq, DNase-chip/seq, and functional assays not only will provide information on the genomic location of these elements but could lead to new insights as to how their interplay results in specific transcriptional outcomes.
Methods
Preparation of NFR-derived or random HaeIII DNAs
Permeabilized nuclei were prepared from 108 F9 cells that had been formaldehyde-cross-linked for 10 min at room temperature. The pelleted nuclei were resuspended to a concentration of 20 × 106 nuclei/mL NEB2 (New England Biolabs) and divided into 500 μL aliquots. HaeIII was added to a final concentration of 0.2 units/μL, and the samples were incubated for 1 h at 30°C. After addition of EDTA (20 mM final), the nuclei were pelleted by centrifugation, and the supernatants were collected. Formaldehyde cross-links were reversed for half of the supernatants by the addition of SDS and NaCl, and overnight incubation at 65°C (UnFAIRE samples). The remainder of the supernatants were subjected to two phenol extractions prior to cross-link reversal (FAIRE samples). The supernatants were digested with Proteinase K and RNaseA, and the DNAs were purified using Qiaquick columns (Qiagen). For preparation of random HaeIII DNAs, 2 μg of F9 cell genomic DNA was digested with HaeIII, phenol extracted, and EtOH precipitated.
LMPCR amplification and cloning into the –64GFP reporter plasmid
UnFAIRE, FAIRE, or random HaeIII DNAs were ligated overnight to a double-stranded oligonucleotide linker (Supplemental Table 1), purified using Qiaquick columns, and PCR amplified using the HaeIII AMP primer (Supplemental Table 1). After purification, the amplified DNAs were digested with BglII, which cuts within the flanking linker sequences, and inserted at the BglII site within plasmid −64GFP (Fig. 1D). The ligated DNAs were used to transform DH5α, and individual miniprep DNAs were prepared from colonies transformed by the UnFAIRE-GFP, FAIRE-GFP, or random HaeIII–GFP plasmids.
Transfections
F9 cells were seeded on 96-well Viewplates (Perkin Elmer) at a concentration of 40 × 103 cells/100 μL/well. The following day, the cells were transfected with 0.5 μg of individual UnFAIRE-GFP, FAIRE-GFP, or random HaeIII-GFP plasmids using Lipofectamine2000 (Invitrogen). The level of GFP expression produced from each transfected plasmid was quantified using the FITC, bottom cell reader protocol of the EnVision microplate reader (Perkin Elmer).
Additional methods
Detailed protocols for the preparation and analysis of NFR-derived DNA plasmids and additional plasmids, oligonucleotide primers, creation of the in silico HaeIII digestion of genomic DNA, luciferase assays, SOX2 ChIP, and H3K4me3 and DNaseI analyses can be found in the Supplemental materials online. The microarray data discussed in this report have been deposited in NCBIs Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO Series accession number GSE10606.
Acknowledgments
We thank Aki Okuda for the pTkLuc plasmid, Derya Unutmaz for access to the Envision microplate reader, Brian Dynlacht and Laurent Pascual-Le Tallec for help in setting up ChIP, Beth Moorefield for helpful comments on the manuscript, and Tom Tao for excellent technical assistance. This work was supported by grant DE013745 from the NIDCR and is dedicated to the memory of FDNY Captain Eric Dailey.
Footnotes
-
↵1 Corresponding author.
↵1 E-mail dailel01{at}med.nyu.edu; fax (212) 263-8714.
-
[Supplemental material is available online at www.genome.org. The microarray data presented here have been submitted to the GEO database under accession no. GSE10606.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.073460.107.
-
- Received October 23, 2007.
- Accepted March 5, 2008.
- Copyright © 2008, Cold Spring Harbor Laboratory Press








