
Transcription of foreign DNA in Escherichia coli
Abstract
Propagation of heterologous DNA in E. coli host cells is central to molecular biology. DNA constructs are often engineered for expression of recombinant protein in E. coli, but the extent of incidental transcription arising from natural regulatory sequences in cloned DNA remains underexplored. Here, we have used programmable microarrays and RT-PCR to measure, comprehensively, the transcription of H. influenzae, P. aeruginosa, and human DNA propagating in E. coli as bacterial artificial chromosomes. We find evidence that at least half of all H. influenzae genes are transcribed in E. coli. Highly transcribed genes are principally involved in energy metabolism, and their proximal promoter regions are significantly enriched with E. coli σ70 (also known as RpoD) binding sites. H. influenzae genes acquired from an ancient bacteriophage Mu insertion are also highly transcribed. Compared with H. influenzae, a smaller proportion of P. aeruginosa genes are transcribed in E. coli, and in E. coli there is punctuated transcription of human DNA. The presence of foreign DNA in E. coli disturbs the host transcriptional profile, with expression of the E. coli phage shock protein operon and the flagellar gene cluster being particularly strongly up-regulated. While cross-species transcriptional activation is expected to be enabling for horizontal gene transfer in bacteria, incidental expression of toxic genes can be problematic for DNA cloning. Ongoing characterization of cross-expression will help inform the design of biosynthetic gene clusters and synthetic microbial genomes.
Lateral transfer of genetic material is an important mechanism by which naturally competent bacteria acquire traits that increase fitness in a particular environment, such as antibiotic resistance or adaptation to a new energy source. Lateral transfer has been exploited in the laboratory to domesticate bacteria for the purposes of sorting and amplifying recombinant DNA and expressing and harvesting high value protein products. We are now shifting to a paradigm where not just individual genes, but entire biosynthetic pathways, and indeed complete microbial genomes, can conceivably be engineered and propagated in microbial hosts (Kodumal et al. 2004; Itaya et al. 2005, 2008; Ro et al. 2006; Holt et al. 2007; Gibson et al. 2008)
However, now and for the foreseeable future, engineering microbial cells requires propagation of large DNA constructs in a host cell, integrated either in the host genome or as episomal elements. For whole genome engineering, the final product may require segregation of donor and host genomes such that a novel organism is obtained that is encoded solely by the engineered genetic material. In other cases, a desired end point may be a hybrid organism that carries both host DNA and extra DNA that encodes novel biosynthetic capabilities. In either situation, there is a requirement for coresidence and cotranscription, in a single microbial cell, of both host and foreign genetic material. Further, when dealing with large constructs encoding genes or with complete genomes, expression of donor genetic material may be desirable because it activates the genetic program of interest, but it may be problematic if some of the gene products are incompatible or otherwise detrimental to the host cell. Indeed, it is well established that foreign genes propagated in Escherichia coli can have undesirable toxic effects, and this has been a long-standing problem for clone-based whole genome sequencing approaches, where toxic genes cause gaps in sequence assemblies (Holt et al. 2007; Sorek et al. 2007).
At the genome scale, the scope of expression of heterologous DNA in a bacterial host cell remains largely uncharacterized. Here we begin to address this issue. We have used programmable oligonucleotide microarrays and RT-PCR to explore the transcription of large segments of foreign DNA inserted into E. coli. We designed a custom nimblegen array containing highly redundant and substantially complete oligonucleotide probe sets for E. coli, Haemophilus influenzae Rd KW20, and Pseudomonas aeruginosa PAO1 annotated genes. We transformed E. coli, in separate experiments, with individual BAC clones from these organisms and probed arrays with cDNA derived from DNase-treated RNA isolated from the overnight culture of the transformants (Supplemental Fig. S1). We observe widespread transcription of genes from these organisms driven by their native promoter elements, and also changes in the transcriptional profile of the host cell in response to the insertion of heterologous DNA.
Results
To test probe specificity, arrays were hybridized, separately, with labeled cDNA derived from DNase-treated RNA from overnight cultures of each of the three bacterial species (E. coli, H. influenzae, and P. aeruginosa). Probes were overwhelmingly species specific (Fig. 1), and any probe that gave higher than background signal when hybridized with cDNA from other than its target species was excluded from further analysis. Interestingly, we observed that at stationary phase H. influenzae gene transcription is widespread, with 81% of the genes showing transcription signals at least one order of magnitude above background levels (Fig. 1). This contrasts with E. coli and P. aeruginosa, where under identical conditions 30% and 15% of genes, respectively, were transcribed. Under these growth conditions, we observe a strong negative correlation between genome size and transcription levels (Pearson correlation = −0.988).
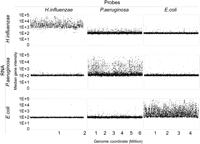
The profile of transcript abundance from cultures of E. coli, H. influenzae, and P. aeruginosa as measured by hybridization of labeled cDNA to a custom oligonuclotide microarray containing probes for most transcripts from each organism. Basal signal is ∼100 units in all hybridizations. A very small minority of probes shows cross-reactivity with other species, and these probes were excluded from analysis.
To assess the degree of transcription in E. coli of heterologous DNA from related gamma-proteobacteria, we transformed E. coli, in discrete experiments, with two different H. influenzae and three different P. aeruginosa bacterial artificial chromosomes (BACs). RNA was collected from an overnight culture of each bacterial clone, DNase treated, converted to cDNA, and assayed on our custom oligonucleotide array. For each gene, the median signal from all probes covering the locus was calculated and divided by the background noise (the median signal for that locus when probed with E. coli RNA alone) to obtain a signal-to-noise ratio (SNR). Arrays probed with cDNA derived from H. influenzae– and P. aeruginosa–transformed clones clearly showed transcription arising from the heterologous DNA (Table 1; Supplemental Fig. S2). While nearly all heterologous genes were transcribed at levels detectable above background, a much higher proportion of H. influenzae genes showed strong transcription compared with genes from P. aeruginosa. For example, the proportion of genes showing a SNR > 100 was 6.8% and 9.4% from the two H. influenzae BACs, respectively, but only a single P. aeruginosa BAC had any genes (1.5%) that gave signal in this range. Transcription of P. aeruginosa DNA was similar among the three BACs from this species, but one of the two H. influenzae BAC clones, clone Q, exhibited transcription levels substantially higher than the other H. influenzae BAC clone, clone S. For clone Q, 40 genes showed SNRs > 10, whereas for clone S, only 19 genes had SNRs > 10. Further investigation revealed that the highly expressing H. influenzae clone Q carried a previously identified ancient bacteriophage insertion, with bacteriophage Mu being the closest extant relative (Morgan et al. 2002), and the highly transcribed genes we observed were localized to this region (Supplemental Fig. S3A). The 34.5-kbp prophage segment (coordinates 1,559,963–1,594,474 of the H. influenzae genome) contains 48 genes, only 13 of which have predicted functions and only two (ymfP and ymfQ) of which have an E. coli ortholog. When H. influenzae clone Q was propagated in E. coli, these prophage genes exhibited 6.4-fold higher transcription (Student’s t-test, P < 1.0 × 10−5) than the non-prophage genes present on the same BAC (Supplemental Fig. S3A).
Distribution of signal-to-noise ratios for H. influenzae and P. aeruginosa genes on individual BAC experiments
To explore the whole genome profile of transcription of H. influenzae DNA in E. coli, a minimum tiling set of 18 large H. influenzae BAC clones was selected that encompassed the majority (90.5% of the sequence and 90.6% of the genes) of the H. influenzae genome (Holt et al. 2007). Clones were transformed separately into E. coli and grown overnight in 18 separate liquid cultures. RNA was isolated from each culture, quantified, and pooled in equal amounts. Again, labeled cDNA derived from the DNase-treated RNA pool was used to probe the custom oligonucleotide array. We observe widespread transcription of H. influenzae genes transformed in E. coli (Fig. 2A) with ∼50% of all H. influenzae genes assayed generating signals above background (SNR > 1). When averaged by clone, there is a distribution of transcription levels that ranges from 180 ± 3 (mean SNR ± SD) to 1624 ± 76. Mean transcription levels among clones are not significantly different, with the exception of the difference between the clone with lowest mean expression, E, and clone L, where genes are highly and more uniformly expressed (P < 0.05, Newman-Keuls multiple comparison test). There is a difference in gene content between clones E and L, which have 87 and 53 annotated genes, respectively. Six genes from clone L show high SNRs > 100, five of which are organized consecutively on the H. influenzae genome. Clone E does not show any evidence for clusters of coexpressed genes, and there are no genes at all that show SNR > 100. Interestingly, there is a small but significant inverse correlation between BAC size and average transcription signals (Pearson correlation = −0.237; P < 1.0 × 10−12), indicating a subtle effect where the larger the heterologous clone, the less the overall transcription from the genes it encodes. Comparing results from H. influenzae clones Q and S assayed alone versus in the context of the pool reveals the impact of pooling on signal strength. Loss of signal in the “pooled” experiment is expected given that the same total amount of RNA is used for hybridization, but the H. influenzae RNA content of the sample will be lower than the E. coli content by a factor of 18. We observed that up to 38% and 58% of genes that gave a signal above background (SNR > 1) with clone Q and S when assayed individually were undetectable in the pool. Differences in transcription signals between the pool and individual BAC samples is significant for both Q and S but more notable for the former (clone Q in pool, mean SNR = 8.4 vs. clone Q alone, mean SNR = 24.1; Student’s t-test, P = 1.0 × 10−3) compared with the latter (clone S in pool, mean SNR = 10.2 vs. clone S alone, mean SNR = 32.3, Student’s t-test, P = 4.2 × 10−2). Thus, due to dilution of H. influenzae RNA upon pooling, our observation of approximately half of all H. influenzae genes being transcribed in E. coli under control of their native promoters is an underestimation. An internal negative control for these experiments is provided by gaps in the clone tiling path (Fig. 2A, gray shades). Because DNA from these segments of the H. influnzae genome is not present in the pool, there should be no transcription sigal from genes annotated in these regions. Consistent with this, in gap regions we observe signal below background levels (SNR = 0.63 ± 0.09; mean ± SD).
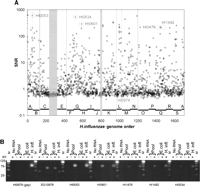
(A) H. influenzae global transcription profile. Eighteen H. influenzae tiling path clones were individually transformed in E. coli, the RNA prepared as described in Methods, pooled, and assayed. Clone D was found to be rearranged and was therefore omitted from the pool. Lack of signal in this region further illustrates high probe specificity. Highlighted genes were amplified from the sample by RT-PCR (B). HI0978 was chosen as a negative control since it is located in the tiling path gap between clones K and L. (B) RT-PCR from the H. influenzae RNA pool, targeting the five hyper-transcribed genes highlighted in A. This SYBR Green-stained agarose gel image demonstrates that amplification occurs only when reverse transcriptase is included. EG10978 represents a positive control for E. coli.
For validation purposes, six of the highly transcribed genes in the pool sample were interrogated by RT-PCR to confirm expression (circled data points in Fig. 2A, RT-PCR Fig. 2B). In addition, E. coli gene EG10978 (SNR = 5.7) was chosen as internal positive control of transcript abundance because it is transcribed at consistent levels across experiments, and H. influenzae gene HI00978, found in the tiling path gap between clones K and L, was chosen as a negative control. As shown in Figure 2B, we detect transcripts from all six highly transcribed H. influenzae genes in RNA from native H. influenzae culture, and in RNA from the pool of E. coli clones transformed with H. influenzae BACs. Further, by RT-PCR we detect transcripts from the E. coli positive control gene, but not the negative control H. influenzae gene located in the tiling path gap.
We observed that in E. coli, transcription levels of H. influenzae genes and their E. coli orthologs are weakly correlated. There are 1286 1:1 orthologous genes (best reciprocal BLASTN matches) between E. coli and H. influenzae, 1222 of which are found on our 18 tiled H. influenzae BACs. Transcription levels of this set of 1222 H. influenzae:E. coli orthologs show a weak but highly significant correlation in the same pooled RNA sample (Pearson correlation = 0.104, Student’s t-test, P < 1.0 × 10−22). Looking only at the most highly transcribed genes, the 37 genes with SNRs >100 (Supplemental Table S1), there are 24 E. coli orthologs and again a weak but significant correlation in transcription levels (Pearson correlation = 0.179, P < 1.0 × 10−5). To explore the possibility of any shared features of the H. influenzae genes most highly transcribed in E. coli (the 37 genes with SNR > 100), we looked at representation of predicted molecular function using GoMiner (Zeeberg et al. 2005). This analysis revealed that 48.6% (18 out of 37) of the most highly transcribed genes are involved in a cellular metabolic process (GO:0044237). Of significance, four are involved in cellular respiration (GO:0045333, P < 0.05), three of which are part of the TCA cycle (GO:0006099, P < 0.05). Also, there is enrichment for genes with oxidoreductase activity (GO:0016491, P < 0.05). These results suggests that promoter regions driving the expression of genes involved in metabolic processes might have more conserved regulatory elements across species; although as noted above, we only observe a weak correlation between transcription of these H. influenzae genes and their E. coli orthologs.
We hypothesized that highly transcribed H. influenzae genes possess strongly conserved E. coli-like σ70 (also known as RpoD) binding sites in their promoter regions that are readily recognized by the E. coli transcription machinery. σ70 is the most common RNA polymerase sigma factor in E. coli, and it is responsible for initiating the transcription of most genes (Gross et al. 1992). We used a well established E. coli σ70 binding site model (Shultzaberger et al. 2007) derived from experimentally verified promoters (Hershberg et al. 2001; Salgado et al. 2001) and the Multiscan program (Shultzaberger et al. 2007) to scan the 200 bases upstream of H. influenzae genes with SNRs of >10 and >100. We evaluated the number of putative E. coli σ70 binding sites and also, using a bit score, the relatedness to the E. coli σ70 binding site model. The bit score reflects the degree of conservation at a given position within a σ70 site (Schneider and Stephens 1990). For the 37 genes with SNR > 100, the number of sites and the sum of conservation information (total bits) were tested by bootstrapping one thousand sets of 37 randomly chosen promoter regions (Table 2). We find the enrichment for E. coli σ70 to be significant for the number of sites (P < 2.0 × 10−3) and overall conservation to E. coli (P < 3.0 × 10−4) at a cutoff of 5 bits/site. In contrast, for the 170 genes with SNR > 10, this approach did not reveal significant enrichment for σ70 promoters or σ70 conservation to E. coli (Table 2, tests A and B).
Analysis of bacterial promoters
It has been noted that highly transcribed genes have strong Shine-Dalgarno (SD) signal sequences (Karlin and Mrazek 2000). We searched the sets of H. influenzae genes that are highly transcribed in E. coli (the 37 genes with SNR > 100 and the 170 genes with SNR > 10) for SD sequences (purine runs ≥ 5 bp) and for strong SD sequences (GAGG or GGAG) within 20 bp of the translation start site. We have identified 34 and 18, as well as 158 and 71, such sites within the 37 and 170 H. influenzae promoters regions, respectively. We find these to be highly significant (P < 1.0 × 10−3, P = 0, P = 0, P = 0) when tested against 1000 sets of 37 or 170 randomly chosen weak promoter regions (Table 2, tests D and E). Motif finding using MEME (Bailey and Elkan 1994) confirms the presence of the word AGGAG or slight variation of it, predominantly −6 to −10 nt upstream of the translation start (data not shown).
To explore the effect of foreign DNA on the E. coli transcriptional profile, we compared the relative abundance of E. coli transcripts in untransformed E. coli to levels observed in E. coli transformed with the H. influenzae, P. aeruginosa, and human BACs. Growth conditions were identical in all cases. E. coli’s transcriptional response to the presence of heterologous DNA propagation is very similar across experiments and especially where the BAC DNA is from the same species (Supplemental Table S2). Notably, all P. aeruginosa clones elicit the differential transcription at least twofold above native levels of two gene clusters (flg and fli) that include 16 flagella biosynthesis genes (Supplemental Table S2; Fig. 3). E. coli also overproduces six of these transcripts in response to the invasion by the human clone. In contrast, neither H. influenzae clone Q nor S appears to trigger the transcription of these genes. Another striking overtranscribed gene cluster is that of the oligopeptide ABC transporter family (Fig. 3). The transformation of any of the three P. aeruginosa BACs induces transcription 30-fold over the native E. coli transcription levels. As observed with the flagella gene cluster, these transporter genes were also overtranscribed when the host was transformed with the human BAC but not with either H. influenzae BAC. The phage-shock-protein (psp) operon, suspected to play a role in maintaining cytoplasmic membrane integrity and/or the proton-motive force (Darwin 2005), is positively modulated by the presence of H. influenzae clone Q, P. aeruginosa clones E4–6, as well as human BAC P8 (Supplemental Table S2) at very similar values (threefold to ninefold over the native E. coli gene signal). H. influenzae clone S did not elicit a response in the host for genes of that operon. Overall, at cutoffs of plus or minus twofold expression, we find that far fewer E. coli genes are strongly modulated by the transformation of the H. influenzae S clone (138 modulated genes) compared with the H. influenzae Q or P. aeruginosa E4, E5, or E6 clones, which elicited the differential transcription of 408, 359, 280, and 436 genes, respectively. This difference is even more striking when transforming the human BAC clone, which induces 527 E. coli genes and represses 406 genes.
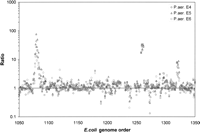
Host response to P. aeruginosa clone propagation. The transcription ratio between P. aeruginosa BACs–E. coli hybrid cells and native E. coli was calculated for a short contiguous section of the E. coli genome encompassing 300 genes. The point clusters at 1075, 1260, and 1325 correspond to the flagellar gene cluster, the oligopeptide ABC transporter operon, and the psp operon, respectively.
To explore transcription in E. coli of DNA from a species so phylogenetically diverged that there would be no expected regulatory conservation, we included in our array design probes for a segment of the human genome (chr 6: 108,550,285–108,670,449), and we probed the array with RNA from E. coli transformed with a human BAC that spanned these genome coordinates. Due to the higher repeat content in human versus bacterial DNA, probes designed for the array could not be evenly distributed across this region. Surprisingly, however, we detected widespread but punctuated transcription from the human DNA propagating in E. coli (Fig. 4A). Signal levels were substantially above the background levels observed when the array was probed with E. coli RNA alone, and in some regions, signal was comparable to the most highly transcribed H. influenzae loci (SNR > 100). To further test transcription of human DNA in E. coli, we selected five regions from the human segment that gave strong signals on the array, and tested these by RT-PCR, using as template an aliquot of the same RNA that was used for probing the array. As shown in Figure 4B, human transcript was detectable for all five regions tested. We hypothesized that transcription was being driven by cryptic sites in human DNA that by chance had homology with the E. coli σ70 binding site. To test the hypothesis, we ran Multiscan on 200-nt regions immediately flanking high-signal (SNR > 5) probes using the E. coli σ70 promoter model and found 76 potential binding sites (Table 2, test C). We bootstrapped these results against 1000 random sets of 46 random sequences drawn from a pool of human regions immediately flanking low-signal probes (SNR < 5) on the BAC. We could not find any significant enrichment of E. coli σ70 promoters nor were the 76 candidates more conserved in regions flanking high-signal probes.
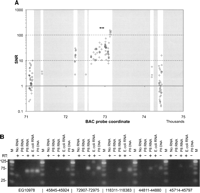
(A) Signal-to-noise ratio for human BAC P8 probes located on a 4-kbp stretch between coordinates 71 and 75 kbp. Transcription signals are detected throughout the BAC and are particularly strong in the 73-kb region. The double-sided arrow indicates the position of a -p RT-PCR product, amplified from an RNA preparation of DH10B transformed with human BAC P8. The gray areas are regions excluded from oligonucleotide design due to repeat content, self-folding, or elevated Tm. (B) Human BAC P8 regions exhibiting strong transcription signals were identified and used for RT-PCR primer design. This SYBR Green-stained agarose gel image shows corresponding RT-PCR amplicons from a DNase-treated RNA preparation from DH10B transformed with the whole human BAC P8.
Discussion
We have used programmable oligonucleotide microarrays and RT-PCR to explore the transcription of foreign DNA inserted into E. coli. We observe that in E. coli at least half of all genes from H. influenzae, a closely related commensal gamma-proteobacteria, are transcribed at some level. We call this transcription of heterologous DNA cross-expression. An immediate question is whether the cross-expression we observe is random or specific. There is likely some combination of both. Specific activation of H. influenzae promoters by the E. coli transcriptional machinery is supported by the observations that in H. influenzae, recognition sequences for the E. coli polymerase σ70 subunit, which drives transcription of most E. coli genes (Gross et al. 1992), are promoter region specific. Further, the H. influenzae genes that are most highly expressed in E. coli have, in their promoter regions, significant matches to the E. coli σ70 binding site. We also observe a weak but significant positive correlation between transcription levels in E. coli of native E. coli genes and their introduced H. influenzae orthologs. The notion of specific transcriptional activation is also supported in some respects by our limited observations on cross-expression using DNA from a more distantly related gamma-proteobacteria, P. aeruginosa. As with H. influenzae, E. coli σ70 recognition sequences detectable in P. aeruginosa are promoter region specific. It is interesting that there is substantial cross-expression of P. aeruginosa genes, but less so than observed for H. influenzae. P. aeruginosa may show restricted cross-expression due to weaker conservation of regulatory elements. However, one must also consider that the E. coli core promoter sequence is AT rich. Given that the H. influenzae genome is AT rich (61.9% AT) and the P. aeruginosa genome is AT poor (33.4% AT), the possibility remains that elevated cross-expression in H. influenzae reflects elevated AT content in addition to evolutionary conservation. Also, there must be at least some degree of nonspecific transcriptional activation in E. coli due to our observation of transcripts arising from heterologous human DNA. There are no obvious grounds for proposing shared functional elements between human and E. coli, so it is likely that human transcripts are arising from cryptic sites in the human genome that are recognizable by the E. coli transcriptional machinery. Since we did not find any significant E. coli σ70 site matches in our human BAC clone sequence, the identity of these cryptic sites remains unknown. It has recently been observed that in mammals, as much as 93% of genomic DNA is transcribed into primary transcripts (The ENCODE Project Consortium 2007). The biological relevance of ubiquitous transcription is unknown, but it appears from our data that pervasive transcription can also occur within and between bacterial genomes. We suggest that in the wild this may facilitate utilization by host species of horizontally transferred DNA elements.
A surprising observation from the present study was the elevated expression in E. coli of ancient bacteriophage Mu (FluMu) genes carried by the H. influenzae genome. There are only two E. coli orthologs for these FluMu genes, yet all the FluMu genes are more highly transcribed in E. coli than in their native H. influenzae (Supplemental Fig. S3A). To date, the relevance of the FluMu prophage for fitness of H. influenzae remains unclear (Morgan et al. 2002). Perhaps there are historical promoter elements that are dormant in H. influenzae but recognizable by E. coli. It is unclear, however, how specific sequences recognized by E. coli would be maintained in H. influenzae over evolutionary time without positive selection, but perhaps genetic drift has not been sufficient to completely erase the phage’s compatibility with other gamma-proteobacteria. Alternatively, CRISPR sequences (Sorek et al. 2008) may be actively suppressing expression of FluMu genes, but we could not find any compelling evidence for these sequences in H. influenzae.
It is clear from the present study that heterologous DNA introduced into an E. coli host can be extensively transcribed by the host’s transcriptional machinery, but it is also clear that the presence of foreign DNA can induce a response that involves modulated expression of host gene content. The elevated transcription of genes involved in flagellum biosynthesis is striking (Fig. 3). Although modulation of these genes in response to a variety of physiochemical stress, including pH, temperature, oxygen, and osmolarity has been studied in E. coli (Soutourina and Bertin 2003; McCarter 2006), there have not been previous accounts linking the modulation of these genes to entry and propagation of heterologous DNA in the cell. Another set of genes showing a dramatic host response are those of the psp operon. The psp operon helps ensure the survival of E. coli in late stationary phase under extracytoplasmic stress (Model et al. 1997), and there is now a consensus that induction of the psp operon occurs through any extracytoplasmic stress that lowers the cell energy levels (Darwin 2005). We observe a marked decrease in transcription of genes related to the menaquinone and nitrate biosynthetic pathways, both key to anaerobic respiration in E. coli (Haddock and Jones 1977; Bentley and Meganathan 1982), consistent with the notion that over-transcription of psp genes is related to energy depletion.
Of the DNA constructs we studied, it appears that P. aeruginosa DNAs most strongly modulated E. coli transcription, with E. coli’s response to transformation being largely consistent for all three P. aeruginosa BACs tested (Supplemental Table S2). While the human clone and H. influenzae clone Q elicited a somewhatconsistent response to P. aeruginosa clones, H. influenzae clone S had less of a modulatory effect. It is unclear why H. influenzae clone S should be more benign, but this may be due to the fact that this clone has the most extensive synteny with E. coli. More than three quarters of its genes are 1:1 E. coli orthologs. Perhaps then, host response is not related simply to the presence of heterologous nucleic acid in the cell, but rather to the dissimilarity of gene content. While it is tempting to speculate that in some cases a host transcriptional response may be defensive, for example, a flight reflex mediated by up-regulation of flagellar genes, in many cases altered host transcription in response to foreign DNA may simply reflect nonspecific disruption of host transcriptional homeostasis.
In closing, we consider some implications of the findings of the present study. Cross-expression between bacteria has not previously been studied on a whole genome scale. We have focused exclusively on transcriptional cross-activation, and while a gene must be transcribed before it can be translated, the proportion of heterologous genes transcribed in E. coli that are translated into functional protein remains unknown. Given the long-standing observation of unclonability of toxic genes in E. coli (Holt et al. 2007; Sorek et al. 2007) it is certain that at some unknown frequency translation is successful. Further, we are now aware that lateral transfer of genetic material among bacteria has produced complex phylogenies that are not interpretable in the context of linear descent (Ochman et al. 2000; Chen et al. 2005). This tangled web of relationships could not have arisen without a mechanism of cross-expression of mobile genetic material. We must also take note that in any case where E. coli is transformed with heterologous DNA, even in routine molecular cloning experiments, cross-expression of the introduced DNA means hybrid organisms are created.
Finally, there are implications in the present study for the field of synthetic biology. One of our motivations for undertaking this work was to explore a model system whereby an H. influenzae genome is engineered by assembling it an E. coli host, followed by removal of the host genome (Holt et al. 2007). Recently, Lartigue and coworkers (2007) used whole genome transformation to introduce the Mycoplasma mycoides genome into host Mycoplasma capricolum cells, displacing the host genome in a single step and creating nascent M. mycoides cells. This is an important proof of principle, but the approach will likely be limited to Mollicutes where genomes are small and the lack of a cell wall allows whole genome exchange. Itaya et al. (2005) showed that it is possible to use a host genome as a vector to propagate that of another, if the donor genetic material is not extensively transcribed. To build synthetic microbes with utility, it will be necessary to establish methods for genome engineering using industrial host strains, and for most engineering projects, coresidence and coexpression of donor and host genetic material will almost certainly be necessary. Factors that influence coresidence, in particular the mechanisms of transcriptional regulation of required genes and mitigation of the effects of toxic or otherwise incompatible genes, will need further exploration. Here we demonstrate that extensive cross-expression of heterologous DNA in E. coli host cells can be achieved, that it is likely governed by both specific and nonspecific mechanisms of activation, and that it modifies the transcriptional profile of the host cell.
Methods
RNA isolation
Cells were grown to stationary phase at 37°C in 5 mL in brain–heart infusion broth (BHI) containing hemin (10 μg/mL), and NAD (2 μg/mL). H. influenzae 883 cells were grown in the presence of kanamycin (10 μg/mL), and E. coli DH10B T1 cells containing BACs were in the presence of chloramphenicol (10 μg/mL). Total RNA was purified from 1 mL of bacteria culture using RNAprotect reagent (Qiagen) and the RNeasy Mini Kit (Qiagen), including on-column DNaseI digestion, according to the manufacturer’s instructions. RNA yield and integrity were assessed by an RNA 6000 Nano Assay (Agilent Bioanalyzer). RNA concentrations were normalized to 1 μg/μL, and 40 μg of each RNA sample was submitted to NimbleGen for microarray analysis.
Oligonucleotide array design
We selected 60-bp sequences targeting open reading frames using filters designed to maximize sensitivity and specificity, as previously described (Maydan et al. 2007). Briefly, oligonucleotides containing 20-mers appearing more than once in the genomes of interest or having an overall identity above 75% with non-target sequences were eliminated from the pool of candidate oligos. Oligos presenting a significant self-folding energy or homopolymer longer than five bases in length were also eliminated, and only the oligos with GC content between 36% and 62% were kept for further consideration, which corresponds to a range of ∼10°C in melting temperature. In the case of the human BAC, the selection process also eliminated known DNA repeats. Final selection of oligonucleotides also considered the k-mer count, GC content, overlap between neighboring probes, and the number of probes per gene. The k-mer count of a probe is defined as the sum of the frequencies within the genomes of interest of all k-mers of length 14 or 15 present within that oligonucleotide. NimbleGen Systems manufactured the microarrays with each selected 60-mer oligonucleotide synthesized at random positions. The number of probes associated with each species is listed in Supplemental Table S3.
RT-PCR
E. coli, H. influenzae, and Homo sapiens sequences were screened against E. coli, H. influenzae, and H. sapiens genomes and masked for non-unique 20-mers as described in the oligonucleotide array design section. Primers for RT-PCR were designed using Primer Express Version 2.0 Software from Applied Biosystems and obtained from Invitrogen. H. sapiens primers were selected from the corresponding NimbleGen oligo sequence plus 10 flanking base pairs on each side. The list of primer sequences is presented in Supplemental Table S4.
RT-PCR for H. influenzae and H. sapiens sequences transcribed in E. coli was performed using one-step reactions with Applied Biosystems’ Power SYBR Green Master Mix on an ABI Prism 7900HT Sequence Detection System. RNA was from the same isolates used to generate NimbleGen data. Reactions were in a volume of 12.5 μL with 5 ng total RNA and a primer concentration of 100 nM each. Positive (DNA), negative (distilled water), and RT-negative controls were included for each primer pair. Reactions were performed in triplicate. Five microliters was run on 3% NuSieve 3:1 (Cambrex BioScience) agarose gels with Invitrogen’s 25-bp DNA Ladder and stained with a 1× solution of SYBR green nucleic acid gel stain (Lonza Group Ltd.) to visualize amplification products.
E. coli σ70 promoter discovery
Using the most recent H. influenzae Rd KW20 gene annotations from GenBank (gi NC_000907.1), a minimal promoter region representing 200 bp upstream of each ORF translation start was extracted from the completed genome sequence. Multiscan was used to identify conserved E. coli σ70 bipartite promoters in H. influenzae promoter regions, using a previously established E. coli σ70 promoter model that accounts for gap variations between the −10 and −35 promoter elements (Shultzaberger et al. 2007). Conserved σ70 promoters were identified from 37 and 170 H. influenzae promoter regions upstream of genes whose transcript abundance appeared elevated (SNR of >100 and >10, respectively). The number of promoters found, their degree of conservation from E. coli models (sum of all bits) and signal strength (most conserved promoter/site) were tested at bits/site cutoff of 0, 1, 3, 5, 7, 9, and 11 bits in n = 1000 bootstrap computations aimed at evaluating the significance of σ70 promoter enrichments in the regions upstream overtranscribed H. influenzae genes.
Shine-Dalgarno site search
Putative Shine-Dalgarno (SD) sequences are defined as purine runs ≥ 5 bp long within 20 bp upstream of the translation start (Karlin and Mrazek 2000). A strong SD sequence includes GAGG or GGAG. For genes showing SNR above or equal to 100 and 10, we scanned 37 and 170 promoter regions, respectively, for the regular expression [AG]{5} and G(AG|GA)G, 20 bp upstream of the translation start and counted the number of SD and strong SD sites, respectively. From a pool of promoter sequences upstream lowly transcribed genes (<1 SNR), we chose sets of 37 and 170 sequences at random and tested the null hypothesis. This process was bootstrapped 1000 times, using a different random set each time and a probability calculated.
Motif finding
MEME (Bailey and Elkan 1994) was run on sets of 37 promoter regions driving the expression of highly transcribed H. influenzae genes (parameters -dna -nmotifs 10 -minw 5 -maxw 30 -mod oops), as well as random sets from lowly transcribed H. influenzae genes.
Acknowledgments
We thank Rosie Redfield (University of British Columbia) for providing the H. influenzae Rd KW20 strain. R.A.H. is a Michael Smith Foundation for Health Research Scholar. Funding for this work was provided by Microsoft Research (award no. 15993).
Footnotes
-
↵3 Corresponding author.
↵3 E-mail rholt{at}bcgsc.ca; fax (604) 876-3561.
-
[Supplemental material is available online at www.genome.org. The array data have been submitted to Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE11051.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.080358.108.
-
- Received May 5, 2008.
- Accepted August 6, 2008.
- Copyright © 2008, Cold Spring Harbor Laboratory Press








