Calling cards for DNA-binding proteins
Abstract
Identifying genomic targets of transcription factors is fundamental for understanding transcriptional regulatory networks. Current technology enables identification of all targets of a single transcription factor, but there is no realistic way to achieve the converse: identification of all proteins that bind to a promoter of interest. We have developed a method that promises to fill this void. It employs the yeast retrotransposon Ty5, whose integrase interacts with the Sir4 protein. A DNA-binding protein fused to Sir4 directs insertion of Ty5 into the genome near where it binds; the Ty5 becomes a “calling card” the DNA-binding protein leaves behind in the genome. We constructed customized calling cards for seven transcription factors of yeast by including in each Ty5 a unique DNA sequence that serves as a “molecular bar code.” Ty5 transposition was induced in a population of yeast cells, each expressing a different transcription factor–Sir4 fusion and its matched, bar-coded Ty5, and the calling cards deposited into selected regions of the genome were identified, revealing the transcription factors that visited that region of the genome. In each region we analyzed, we found calling cards for only the proteins known to bind there: In the GAL1–10 promoter we found only calling cards for Gal4; in the HIS4 promoter we found only Gcn4 calling cards; in the PHO5 promoter we found only Pho4 and Pho2 calling cards. We discuss how Ty5 calling cards might be implemented for mapping all targets of all transcription factors in a single experiment.
Transcription factors program gene expression by binding to specific sites in the genome and regulating chromatin-modifying enzymes and the transcriptional apparatus. Knowledge of the sites in the genome bound by each transcription factor is necessary for a full understanding of transcriptional regulation. Chromatin immunoprecipitation can be used to identify the sites in the genome to which any DNA-binding protein binds by using the DNA that coprecipitates with it to probe a microarray of DNA fragments that tile the genome (called the “ChIP-chip” method; Ren et al. 2000; Horak and Snyder 2002). However, there is currently no realistic way to do the converse––identify all the proteins that bind to a particular region of the genome. To fill this gap in technology, we developed a new method for identifying protein–DNA interactions.
Our method exploits the retrovirus-like transposon Ty5 of bakers’ yeast. After Ty5 mRNA is reverse transcribed and converted into a double-stranded cDNA, the Ty5 integrase carries it to the nucleus and catalyzes its insertion into the genome (Voytas and Boeke 2002). Copies of Ty5 are found in the Saccharomyces cerevisiae and Saccharomyces paradoxus genomes near telomeres and the silent copies of the mating-type genes (Zou et al. 1995, 1996) because the Ty5 integrase interacts with Sir4, an integral component of the chromatin in these regions of the genome (Xie et al. 2001; Zhu et al. 2003). Fusion of Sir4 to a DNA-binding protein causes Ty5 to integrate into DNA near the binding sites for that protein (Zhu et al. 2003) (Fig. 1A). We have exploited this property of Ty5 to develop a method for identifying the proteins that bind to any selected region of the yeast genome. This method also provides a convenient alternative to the ChIP-chip technique for identifying the targets of any selected DNA-binding protein.
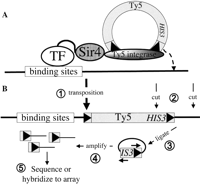
Identification of genomic targets of DNA-binding proteins using Ty5. (A) Sir4 fused to a DNA-binding protein causes Ty5 to integrate into the genome near the binding sites for that transcription factor (TF). (B) After Ty5 transposition (➀), genomic DNA is cleaved with a restriction enzyme that cuts near the end of Ty5 (➁) and is ligated in dilute solution to favor recircularization of the fragments (➂). This is followed by amplification of the circular DNA that contains the end of the transposon and flanking genomic DNA by an “inverse PCR” (➃), and the identity of the flanking genomic DNA is determined by DNA sequencing or hybridization to a DNA microarray (➄).
Results
Principle of the method
When a DNA-binding protein fused to Sir4 binds to a site in the genome, it recruits the Ty5 integrase and thereby directs insertion of Ty5 into the genome. If the Ty5 carries a unique sequence “bar code,” it becomes a “calling card” that uniquely identifies the transcription factor (TF) that directed its insertion. If we provide each DNA-binding protein with a bar-coded Ty5 calling card and induce transposition in a mixture of such strains, each carrying a different TF-Sir4 fusion and its matched Ty5 calling card, we should be able to identify all the proteins that bind to a particular region of the genome by recovering the Ty5 elements that were deposited there and reading the bar code sequences they carry (see Fig. 3, below).
Identification of targets of individual transcription factors
Before attempting to implement this method, we had to confirm that DNA-binding proteins reliably direct the insertion of Ty5 near their binding sites in the genome. We did this for Gal4, a DNA-binding protein with well-characterized targets in the genome. We fused the Gal4 DNA-binding domain (Gal4DBD) to a fragment of Sir4 (amino acids 951–1200) that contains its Ty5 integrase-interacting domain (Xie et al. 2001; Zhu et al. 2003). This Gal4DBD-Sir4 fusion protein was expressed in a yeast strain lacking SIR4 and carrying a Ty5 element under the control of the GAL1 promoter. Growth of this strain on galactose results in transcription of the Ty5 element, which is reverse transcribed into DNA that is competent to integrate into the genome (Zou et al. 1996). The Ty5 also carries a HIS3 gene with an artificial intron that interrupts its coding sequence and which therefore becomes functional only after this artificial intron is spliced out of the mRNA, thereby providing a selection for cells in which the Ty5 has integrated into the genome (Curcio and Garfinkel 1991; Zou et al. 1996).
To identify the regions of the genome into which Gal4DBD-Sir4 directed Ty5 insertion, we recovered the DNA immediately flanking the Ty5 and determined its nucleotide sequence. Genomic DNA from each His+ FOAr colony was cleaved with the restriction enzyme HinP1I, which cuts near the end of Ty5, and the resulting fragments were ligated in dilute solution to favor their recircularization (see Methods). The sequence of the junction of the Ty5 and genomic DNA was determined after its amplification by inverse PCR (Ochman et al. 1988) (Fig. 1B). Among 96 independent transposition events in cells expressing Gal4DBD-Sir4, 76 occurred in promoters of known targets of Gal4: 39 upstream of GAL1–10, 35 upstream of GAL7, one upstream of GCY1, and one upstream of FUR4. Almost all of these insertions are within 35 bp of a Gal4 binding site (CGGN11CCG). The 15 genes not known to be bound by Gal4 into whose promoters we found Ty5 to transpose are not likely to be bona fide Gal4 targets because only one contains a Gal4 binding site, and their known or predicted functions do not make them good candidates for targets of Gal4. Five Ty5 transposition events occurred in the telomeres and into other Ty elements in the genome, as previously observed (Zhu et al. 1999). The strong enrichment of Ty5 integration events near known Gal4 binding sites validated the use of Ty5 to mark TF binding sites.
The relatively small number of transposition events analyzed in this initial experiment makes it difficult to determine if the transpositions within promoters not known to be regulated by Gal4 represent previously unrecognized Gal4 targets or are the background false positives of this method. To enable analysis of many more Ty5 insertions, we employed a more efficient method to identify their sites of insertion. Yeast cells, representing ∼5000 Ty5 transposition events directed by Gal4-Sir4, were pooled, and their genomic DNA was extracted and digested with three different restriction endonucleases with 4-bp recognition sequences that are present 300–1000 bp from the end of Ty5. The resulting fragments (containing Ty5 sequence on one end and the adjacent genomic sequence on the other end) were ligated in dilute solution to favor their circularization and amplified by inverse PCR using primers complementary to the end of Ty5. The PCR products (of variable size) were labeled with Cy5 and used to probe a microarray of oligonucleotides that tile the yeast genome to identify regions of the genome flanking the Ty5 insertions (see Methods for details).
Seven regions known to be bound by Gal4 (GAL1-GAL10, GAL7, GAL2, GAL3, FUR4, GCY1, PCL10) (SCPD, http://rulai.cshl.edu/SCPD; TRANSFAC, http://www.gene-regulation.com/pub/databases.html#transfac; Ren et al. 2000) are among the top 20 hybridization signals (see Methods for details of the analysis of the hybridization signals); two other known Gal4-regulated genes, MTH1 and GAL80, rank in the top 60 hybridization signals. (The data for all the genes that pass our significance criteria are provided in Supplemental Table 1).
Eight of the 13 promoters among the top 20 hybridization signals on the array that are not known to be Gal4 targets contain at least one Gal4-binding site (CGGN11CCG) (Table 1). In an attempt to validate binding of Gal4 to these 13 promoters that are not known to be Gal4 targets, we immunoprecipitated Gal4 (via the Myc epitope it carries) and tested for coprecipitation of those regions of the genome. Three of the 13 promoters (SFL1-RUP1, YPL067C-YPL066W, YOR084W) were clearly enriched in the sample immunoprecipitated from cells with the Myc-tagged Gal4 compared to cells with an untagged Gal4 (Fig. 2A). Indeed, Gal4 regulates expression of these genes (Fig. 2B). Expression of the divergently transcribed genes flanking two of these promoters (SFL1–RUP1 and YPL067C–YPL066W) is induced by galactose via Gal4 (Fig. 2B, cf. lanes 3 and 1, and lanes 4 and 2); interestingly, Gal4 regulates expression of YOR084W in an unexpected way: It seems to repress its expression (cf. lanes 4 and 3, and lanes 6 and 5). Although 10 of the 13 potential Gal4 targets were not confirmed by the chromatin immunoprecipitation experiments, five of them have Gal4-binding sites and therefore could be Gal4 targets.
Top 20 targets of Gal4 and Gcn4
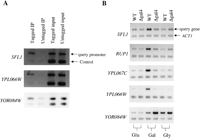
Verification of novel Gal4 targets. (A) ChIP assay for Gal4 binding. Chromatin was cross-linked to protein by treatment with formaldehyde, and Gal4 was tagged with the 18-myc epitope, which was precipitated with anti-myc antibody. The precipitated DNA was released from the protein and detected by PCR as described in the Methods, using primers specific for sequences upstream of the indicated putative Gal4 targets (query promoter) and primers specific for the GAL4 promoter (control promoter) that amplify a 150-bp fragment. (B) RT-PCR analysis compared the expression of novel Gal4 target genes in wild-type FM393 cells vs. gal4Δ cells grown on different carbon sources. Cells were grown to saturation in YPD and then diluted 100 times in fresh 2% glucose, 2% galactose plus 5% glycerol, or 5% glycerol. Cells were harvested once they reached mid-log phase (OD600 = 1.5–2.0), total RNA was prepared, and RT-PCR was performed on the indicated targets. Control reactions lacking reverse transcriptase produce no PCR products (data not shown).
To estimate the sensitivity and specificity of the method, we turned to Gcn4, because it has a well-characterized DNA-binding specificity (Oliphant et al. 1989), many known targets in the genome (Natarajan et al. 2001; Pokholok et al. 2005), and many genes are known that are unlikely to be its target (Pokholok et al. 2005). In addition, Gcn4 was used to determine the sensitivity and specificity of the ChIP-chip method (Pokholok et al. 2005), enabling a direct comparison of the two methods. Using the same approach as for Gal4, we determined where in the genome Gcn4-Sir4 deposits Ty5. About 300 regions of the genome displayed significant hybridization to the array (see Methods for the criteria for significance). Twelve known Gcn4 targets are among the top 20 signals; the remaining eight all have perfect or recognizable Gcn4-binding sites (several of these genes are especially propitious Gcn4 targets because they encode enzymes involved in amino acid biosynthesis) (Table 1).
To estimate the specificity and sensitivity of this assay, we determined how many known Gcn4 target genes (defined by Pokholok et al. 2005) were not identified by our method (“false negatives”) and how many regions of the genome that are unlikely to be Gcn4 targets (also as defined by Pokholok et al. 2005) turned up in our assay (“false positives”). Fifty-one percent of the known or likely Gcn4 target genes hybridized strongly enough to probes on the DNA microarray to pass our criteria for a positive signal. This false-negative frequency of 49% comes at a false-positive frequency of 2.5%. This is somewhat higher than the 25% false-negative frequency of the ChIP-chip method (at a false-positive frequency of 1%), which is perhaps not surprising since the reference sets of Gcn4 target genes chosen by Pokholok et al. (2005) are partially based on results from ChiP-chip experiments. It should be noted, however, that this false-positive rate means that a substantial proportion of our 300 potential Gcn4 targets are likely to be false positives (2.5% of 6000 = ∼150 false positives). Some of these are derived from recombination of the Ty5 calling card with Ty5 elements and LTRs, and can be easily recognized by their location (usually near the telomeres) in the genome. (The data for all the genes that pass our significance criteria are provided in Supplemental Table 2).
Identification of the proteins that bind to any selected region of the genome
With the confidence these results provided that DNA-binding proteins carrying Sir4 direct insertion of Ty5 into the genome near where they bind, we proceeded to test if the calling cards can be used to reveal which proteins bind to a particular region of the genome. We manufactured Ty5 calling cards containing 20-bp oligonucleotides that serve as “molecular bar codes” for seven transcriptional regulators fused to Sir4: Gal4, Gal80, Ste12, Bas1, Pho2, Gcn4, and Pho4. Yeast cells were cotransformed with a plasmid encoding a TF-Sir4 fusion and a plasmid carrying its matched Ty5 calling card (Fig. 3). These seven strains were pooled, and Ty5 transposition was induced by growing them on galactose-containing medium. We recovered the calling cards deposited in three different promoters by performing PCR with oligonucleotide primers complementary to Ty5 and to the regions flanking the promoters of interest (Fig. 3, see Methods for details). The identity of the “bar codes” in these PCR products was determined by using them to probe a mini-array of the bar code sequences (Fig. 3, see Methods for details). In each of the three promoters we analyzed, we found calling cards for only those proteins known to bind to them (Fig. 4): in the GAL1–10 promoter, we only found Ty5 elements carrying the Gal4 bar code (Fig. 4A); in the HIS4 promoter, we found only Gcn4 bar codes (Fig. 4B) (Tice-Baldwin et al. 1989). In the PHO5 promoter, we found only bar codes corresponding to Pho4 and Pho2, and only when transposition was induced in cells starved for phosphate (Fig. 4C,D), as expected because Pho4 and Pho2 bind to DNA only when phosphate is scarce (Barbaric et al. 1996; Oshima et al. 1996). This pilot experiment suggests that Ty5 can be used to identify proteins that bind to any region of the genome.
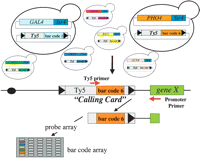
“Calling cards” for DNA-binding proteins. For each of seven transcription factors fused to Sir4 (Gal4, Gal80, Ste12, Bas1, Pho2, Gcn4, and Pho4), a unique 20-bp oligonucleotide was inserted into Ty5 to serve as a “molecular bar code,” thereby transforming Ty5 into a “calling card” that the TF leaves behind when it visits a site in the genome. Each strain was cotransformed with a plasmid encoding a TF-Sir4 fusion and a plasmid carrying its matched Ty5 calling card. After transposition, the calling cards deposited in the promoters of interest were recovered by a PCR with Ty5- and promoter-specific primers.
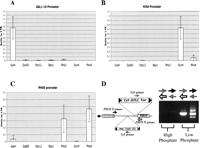
“Calling cards” deposited in three promoters. The PCR products from three promoters were hybridized to the bar code array. Shown is the ratio of the intensity of hybridization of each bar code to the intensity of hybridization to an LTR probe on the array. (A) In the GAL1–10 promoter, only the Gal4 bar code is enriched. (B) In the HIS4 promoter, only the Gcn4 bar code is enriched. (C) In the PHO5 promoter, only Pho2 and Pho4 bar codes are enriched. (D) When transposition was induced in media rich in phosphate (YPD), the PHO5-specific primers produced no PCR product, but when transposition was induced in cells grown in low-phosphate media the PHO5-specific primers produced abundant PCR products, which contain only Pho4 and Pho2 bar codes, as revealed by hybridization to the bar code array.
Discussion
We have exploited the properties of the Ty5 transposon to provide DNA-binding proteins with “calling cards” that reveal the places in the genome they visit. We validated this method with seven different DNA-binding proteins, and found that we could successfully identify the proteins that bind to different promoters. The method proved to be robust: It identified the proteins known to bind to the GAL1–10, HIS4, and PHO5 promoters. Based on these results, we are confident that we can implement calling cards for all ∼200 DNA-binding proteins of yeast, which would enable identification of all the proteins that bind to any particular region of the genome under a variety of growth conditions by a simple PCR followed by hybridization to a microarray of oligonucleotide bar codes. We are confident that calling cards can also be implemented for non-DNA-binding, chromatin-associated proteins, because we used calling cards to identify a known target of Mth1, which is recruited to promoters of HXT genes by the Rgt1 transcriptional repressor (data not shown).
This method fills a gap in technology for characterizing DNA-binding proteins. Currently, we can identify the targets of any particular DNA-binding protein with the ChIP-chip technique, but to do the converse—identify the proteins that bind to a particular region of the genome—one would have to perform a ChIP-chip experiment on all DNA-binding proteins of an organism. Our calling card method promises to make this feasible.
Our method also provides an alternative to the ChIP-chip method for the genome-wide identification of targets of transcription factors, and can serve as an independent verification of the results obtained with the ChIP-chip method. Indeed, we were able to discover previously unidentified targets of Gal4, probably the best characterized transcription factor of yeast, perhaps because our method is very different from those that employ chromatin immunoprecipitation.
The calling card technology could be improved in several ways. Probably most important is to increase the number of transposition events sampled. For practical reasons we have been harvesting 3000–5000 independent transposition events in each experiment, but it should not be difficult to scale up the experiment and obtain more. This may be necessary because we did not find in the HIS4 promoter bar codes for Pho2 and Bas1, which are known to bind there (Tice-Baldwin et al. 1989). We identified two Pho2 bar codes among 18 that we analyzed by direct DNA sequencing in a preliminary experiment, suggesting that binding of these proteins would have been detected by hybridization to the microarray with a larger number of Ty5 transposition events. The number of transposition events could also be increased by improving the Ty5 transposition efficiency, which is relatively low compared with other Ty elements. This could also allow a shorter time of induction of transposition. Second, expression of the Ty5 calling card from the GAL1 promoter limits the conditions that can be tested. It would be better to use a different promoter, such as one that is activated by a gratuitous inducer such as tetracycline (Belli et al. 1998; Berens and Hillen 2003). Third, it has been speculated that the region of Sir4 that interacts with the Ty5 integrase also interacts with other proteins, which might interfere with the method in some cases. A clever solution to this potential problem—use of a heterologous pair of protein interaction domains on the DNA-binding protein and the integrase—was implemented by Zhu et al. (2003). That would also allow the method to be applied with a SIR4 strain, which would avoid the possibility of disruption of chromatin structure in certain regions of the genome. Fourth, fusing Sir4 to a DNA-binding protein could interfere with its ability to bind to DNA. This problem can be minimized by fusing Sir4 to each end of the protein (in different constructs). Finally, insertion of a calling card into a promoter could, in some cases, disrupt expression of the gene, which might prevent recovery of those cells. This problem can easily be solved by using a diploid strain.
We would like to reduce the false-positive and false-negative rates of our method. We empirically determined the significance cutoff using lists of genes that are likely or unlikely to be Gcn4 targets, as was done for the ChIP-chip method (Pokholok et al. 2005). This cutoff was applied to all experiments. We arbitrarily chose a significance cutoff that yielded 2.5% false positives, which results in a 49% false-negative rate. Similar performance (4% false positives and ∼24% false negatives) was sufficient for application of the ChIP-chip method to genome-wide analysis of transcription factor targets in yeast (Harbison et al. 2004). Of course, the false-positive rate can be reduced by increasing the cutoff, but that comes at the expense of a higher false-negative rate. Advances in the experimental approach are likely to be necessary for significant improvement in the specificity and sensitivity of our method (Gabriel et al. 2006; Wheelan et al. 2006). One reason for this high false-positive rate might be the large number of cycles of the inverse PCR required to provide enough probe for hybridization to the DNA microarrays, which may result in over-amplification of some of the nonspecific insertions. Stochastic amplification of nonspecific insertions in the inverse PCR (“jackpotting”) could also contribute to the problem. Both problems should be ameliorated by performing the inverse PCR on individual molecules in a water/oil emulsion (Griffiths and Tawfik 2006). In addition, the low transposition efficiency of Ty5 in our experiments may exacerbate the “jackpotting” problem, so the false-positive rate will likely be improved if we can sample more transposition events.
By coupling the calling card method to next-generation (massively parallel) sequencing technologies, it may be possible to identify genome-wide the binding locations of all yeast transcription factors in a single experiment. Induction of transposition of the calling cards in a library of strains representing all ∼200 DNA-binding protein–Sir4 fusions with their corresponding calling cards, followed by recovery of each calling card along with the adjacent genomic DNA would enable determination of the sequences of both the bar code identifiers of the DNA-binding proteins and the adjacent genomic sequence, thereby revealing both where in the genome proteins bind and which proteins bind there. This would be equivalent to performing a ChIP-chip experiment for each of the 200 DNA-binding proteins. Several novel DNA sequencing methods have recently been developed that offer the throughput needed for this implementation of the calling card method (Margulies et al. 2005; Shendure et al. 2005). This would enable us to examine the regulatory network of yeast under a large number of different conditions. Finally, we note that transposons are present throughout the tree of life, so it may be possible to implement calling cards for DNA-binding proteins in species other than yeast.
Methods
Strains and growth media
The sir4 deletion mutant yDV561 (MATa, ura3-52, trp1-63, his3-200, leu2-1, lys2-801, ade2-101, sir4∷KanMX) obtained from Dan Voytas (Zhu et al. 2003) was the host strain for Ty5 transpostion. Chromatin immunoprecipitation was done from extracts of strain Z1319 (MATa, ade2-1, trp1-1, can1-100, leu2-3,112, his3-11,15, ura3, GAL+, psi+, GAL4∷18-Myc) (Ren et al. 2000). Yeast strain BY4743 (MATa/MATα his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 ura3Δ0/ura3Δ0 met15Δ0/MET15 LYS2/lys2Δ0) and homozygous gal4 deletion strain (Saccharomyces Genome Deletion Project, no. 31044) (MATa/MATα his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 ura3Δ0/ura3Δ0 met15Δ0/MET15 LYS2/lys2Δ0 gal4Δ0/gal4Δ0) (Brachmann et al. 1998; Giaever et al. 2002) were used for reverse transcription PCR to measure gene expression. Yeast cells were grown in complete synthetic media with the addition of 2% glucose or galactose, unless described otherwise.
Construction of plasmids
To construct pBM5037 (Gal4DBD-Sir4-Myc), the region of SIR4 encoding amino acids 951–1200 was amplified in a PCR and fused to the Gal4 DNA-binding domain (amino acids 1–147 plus amino acids 877–881) in pOBD2 by “gap repair” (Ma et al. 1987; Wach et al. 1994). Three copies of the Myc epitope were amplified using PCR and fused to the C terminus of Gal4DBD by gap repair. The entire ORF of each transcription factor was amplified in a PCR and used to replace Gal4DBD by homologous recombination. Gal4DBD-Sir4-Myc was linearized by cutting with XhoI (cuts once C-terminal to the Gal4DBD coding sequence) to serve as the recipient plasmid for gap repair to construct all the other TF-Sir4 fusions.
The plasmid pSZ293 with Ty5 expressed from the GAL1 promoter was obtained from Dan Voytas (Zhu et al. 2003). The XhoI–NotI fragment that includes GAL1∷Ty5 was inserted between the XhoI and NotI sites of pRS316 (Sikorski and Hieter 1989) to generate pBM4735. AcaI and FseI sites were engineered adjacent to the 3′ long terminal repeat (LTR) to allow insertion of the 20-bp “bar codes.” The bar codes that identify each transcription factor were those developed for each gene in the Yeast Gene Knockout (YKO) collection (Yuan et al. 2005). Double-stranded oligonucleotides with the bar code sequences were inserted between the engineered AcaI and FseI sites of the Ty5.
Induction of Ty5 transposition and inverse PCR
Since Ty5 is driven by the GAL1 promoter, transposition was induced by culturing cells in galactose medium for 2–3 d at room temperature. After induction, cells were plated on Glu −His media to select for cells with transposition events. His+ cells were replica plated on −His, FOA-containing media to eliminate His+ colonies due to recombination of reverse-transcribed Ty5 with the transposon donor plasmid.
To map sites of Ty5 integration directed by Gal4-Sir4, 96 His+ FOAr colonies were grown in YPD, and their genomic DNA was extracted and digested by HinP1I (1 μg in a 20-μL reaction). Five microliters of digested DNA was then ligated overnight at 15°C in 100 μL to encourage self-circularization. Five microliters of the ligated DNA was used as template for inverse PCR with primers that anneal to Ty5 sequences (OM6313 and OM6188 were used to amplify the genomic region on the right side of Ty5 integration; OM6458 and OM4960 were used to amplify the genomic region on the left side).
For hybridization of the inverse PCR products to the yeast genome tiling array, we pooled 3000–5000 His+ FOAr colonies for each sample, extracted the total DNA, digested it with three different enzymes (HinP1I, HpaII, and Taq1), and ligated them in dilute solution. Using two pairs of primers, the genomic region on the left side (primers OM6609 and OM6458) and right side (primers OM6610 and OM6456) of Ty5 was amplified from each enzyme-digested sample. The PCR products were purified (using the Qiagen PCR purification kit), and the same amount of product from digestion with each restriction endonuclease was pooled. A total of 1.6 μg of PCR product was labeled with Cy5 (using Invitrogen’s BioPrime Array CGH Genomic Labeling Module); and the genomic DNA, sonicated into 0.5- to 1-kb fragments, was labeled with Cy3. The Cy3- and Cy5-labeled samples were combined and hybridized to an Agilent yeast whole-genome tilling array.
For the experiments employing “bar-coded” Ty5 elements, we cultured in glucose medium lacking uracil and tryptophan seven strains, each carrying a different TF-Sir4 fusion and its matched bar-coded Ty5 element. Once the OD600 of each culture reached ∼1, 100 μL of cells of each strain were pooled and Ty5 transposition was induced and selected for as described above. Genomic DNA was extracted from ∼3000 His+ FOAr colonies and used as the template in a PCR with promoter-specific primers. To amplify all the calling cards deposited within a particular promoter, we used a primer that anneals to sequences flanking the promoter and a primer (OM6606) that anneals to sequences within Ty5. Six hundred nanograms of PCR products for each promoter was purified, labeled with Cy5, and hybridized to a mini-array of bar code oligonucleotides, using Genisphere’s Array 900DNA Cy3 and Cy5 labeling kits. Probes on the mini-array are 60-bp oligonucleotides consisting of three copies of the 20-bp bar code sequence. Each probe was printed in quadruplicate on the array. In addition, oligonucleotides of the LTR sequence were printed to serve as a positive control; three unrelated bar code oligonucleotides served as negative control.
Primers for PCR
OM6313: TAAGCTCGGAATTCGAGC TC; OM6188: ACAAGGAAAACATAGAG CAGC; OM6458: AGGTTATGAGCCCT GAGAG; OM4960: CGTAGTGAATTAC GATCTAGC; OM6609: CTTTTGGGTT ATCACATTCAAC; OM6610: ATCGTA ATTCACTACGTCAAC; OM6456: CCC ATAACTGAATACGCATG; OM6606: AAGATCGAGTGCTCTATCGC.
DNA sequencing
The ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction kit was used for DNA sequencing. One hundred nanograms of PCR product or 1 μg of plasmid DNA was used as the template. The products of the reaction were separated and detected on an ABI 310 genetic analyzer.
Microarray analysis
We used two methods to identify the regions of the genome where calling cards were deposited due to the binding of the TF-Sir4 fusion protein. Each method requires a different type of hybridization control.
The Rosetta error model
We used the Rosetta error model to analyze the transcription factors Gal4 and Gcn4. In these experiments, our control was a sir4D strain containing a plasmid expressing Ty5 (pBM4735), but with no plasmid expressing a TF-Sir4 fusion. We induced transposition and performed inverse PCR as described above. We labeled the control reaction with the Cy3 (green) dye, the experimental reaction with the Cy5 (red) dye, pooled the reactions, hybridized them to the microarray, and imaged the slide. For each probe, we subtracted the intensity value observed in the control channel from the intensity value observed in the experimental channel. We then assigned each probe a P-value that gives the probability of the observed intensity difference, assuming no calling card was deposited at that location. As did Pokholok et al. (2005), we used the Rosetta error model to calculate this P-value. In this model, the difference in intensities between two technical replicates is assumed to be normally distributed, and the variance of this distribution increases with average probe intensity (see Supplemental Information for more details).
We chose our significance cutoff empirically by using the published test sets of positive and negative targets for Gcn4 (Pokholok et al. 2005). We selected a P-value threshold that minimized the rate of false negatives at a false-positive rate of 2.5%. This cutoff resulted in a false-negative rate of 55%. If a gene is within 250 bp of a significant probe, then it is considered a target of the transcription factor that is being analyzed.
The Maximum Likelihood Estimate of DNA Concentration (MLEDC) method
The Rosetta error model works well when the distribution of intensities in the control channel is similar to the distribution of background intensities in the experimental channel. However, we observed a significant increase in integration “hot spots” when no TF-Sir4 fusion protein is present, rendering the Rosetta error model inadequate. Therefore, we developed a second way to analyze the calling card data. Using labeled genomic DNA as a control, we estimated the concentration of DNA present at each locus after recovery of calling cards and flanking genomic DNA (see Supplemental Information). The maximum likelihood value of DNA concentration is proportional to the average ratio of experimental to control intensities. We ranked the probes based on their average ratio and empirically selected a cutoff as described above. We selected a threshold that minimized the rate of false negatives at a false-positive rate of 2.5%. This cutoff resulted in a false-negative rate of 49%. Since this is slightly better than the Rosetta error model, the data were analyzed using the MLEDC method.
To understand better the nature of our false negatives, we manually examined the intensities of these genes in the MLEDC analysis—the majority of these features displayed little to no fluorescence in the red channel, suggesting that these features were categorized as negatives because no transposition event had occurred in these samples, and not due to inaccurate assumptions in our error model. Data from probes covering telomere regions were ignored (because Ty5 can insert into these regions of the genome due to homologous recombination with Ty5 elements that reside there). HIS3 probes were also excluded because HIS3 sequences from the Ty5 calling cards are present in the inverse PCR product.
For the bar code array experiments, the raw intensity of each probe on the array was normalized by dividing it by the raw intensity of a probe containing LTR sequence. To eliminate the random hopping background, we applied a stringent criteria: If the probe gets a ratio >0.1 only in one experiment out of three biological replicates, we count it as a random event and exclude it from the data.
Chromatin IP
Chromatin immunoprecipitation was performed as previously described (Aparicio 1999; Orlando 2000). Cultures were grown in minimal medium with galactose. Bound proteins were cross-linked to DNA in vivo by addition of formaldehyde, followed by cell lysis and sonication to shear DNA. Individual transcription factors were immunoprecipitated with antibody to their Myc epitope tag, followed by reversal of the cross-links. DNA immunoprecipitated from a Myc-tagged strain and from a control strain with no Myc tag were used as template to amplify the promoter of interest.
Reverse transcription PCR
Wild-type and Gal4 deletion strains were cultured in 50 mL of YP medium with 2% glucose, 2% galactose plus 5% glycerol, or 5% glycerol as carbon source. When the cultures reached an OD600 of 1.5, the cells were harvested and their RNA extracted. The same amount of RNA from each sample was treated with DNase and then reverse transcribed into cDNA using SuperScript II Reverse Transcriptase from Invitrogen. The cDNA served as the template in a PCR employing primers that amplify 200–300 bp of coding sequence of the genes of interest. Twenty-five cycles were used for each PCR. As a loading control, the ACT1 locus was amplified by RT-PCR for each sample.
Acknowledgments
We thank Dan Voytas (Iowa State University) for generously providing reagents and advice, and Seth Crosby and Michael Heinz (Washington University) for their expert assistance with the use of DNA microarrays. We also thank Doug Chalker (Washington University) for helpful suggestions, and Rick Young and Nancy Hannett (Whitehead Institute, MIT) for providing strains expressing Myc-tagged Gal4 and Gcn4. This work was supported by funds provided by the James S. McDonnell Foundation, and by NIH grants R21RR023960 and 5P50HG003170-03.
Footnotes
-
↵1 Corresponding author.
↵1 E-mail rmitra{at}genetics.wustl.edu; fax (314) 362-2157.
-
[Supplemental material is available online at www.genome.org.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.6510207
-
- Received March 17, 2007.
- Accepted June 11, 2007.
- Copyright © 2007, Cold Spring Harbor Laboratory Press







