
Abstract
Metagenomic projects generate short, overlapping fragments of DNA sequence, each deriving from a different individual. We report a new method for inferring the scaled mutation rate, θ = 2Neu, and the scaled exponential growth rate, R= Ner, from the site-frequency spectrum of these data while accounting for sequencing error via Phred quality scores. After obtaining maximum likelihood parameter estimates for θ and R, we calculate empirical Bayes quality scores reflecting the posterior probability that each apparently polymorphic site is truly polymorphic; these scores can then be used for other applications such as SNP discovery. For realistic parameter ranges, analytic and simulation results show our estimates to be essentially unbiased with tight confidence intervals. In contrast, choosing an arbitrary quality score cutoff (e.g., trimming reads) and ignoring further quality information during inference yields biased estimates with greater variance. We illustrate the use of our technique on a new project analyzing activated sludge from a lab-scale bioreactor seeded by a wastewater treatment plant.
Metagenomics applies shotgun-sequencing techniques to DNA extracted from a microbial community with the goal of learning about the ecological dynamics of the constituent microorganisms. Population genetics provides a theoretical basis to make inferences about population structure and evolution given samples of DNA sequences from individuals.
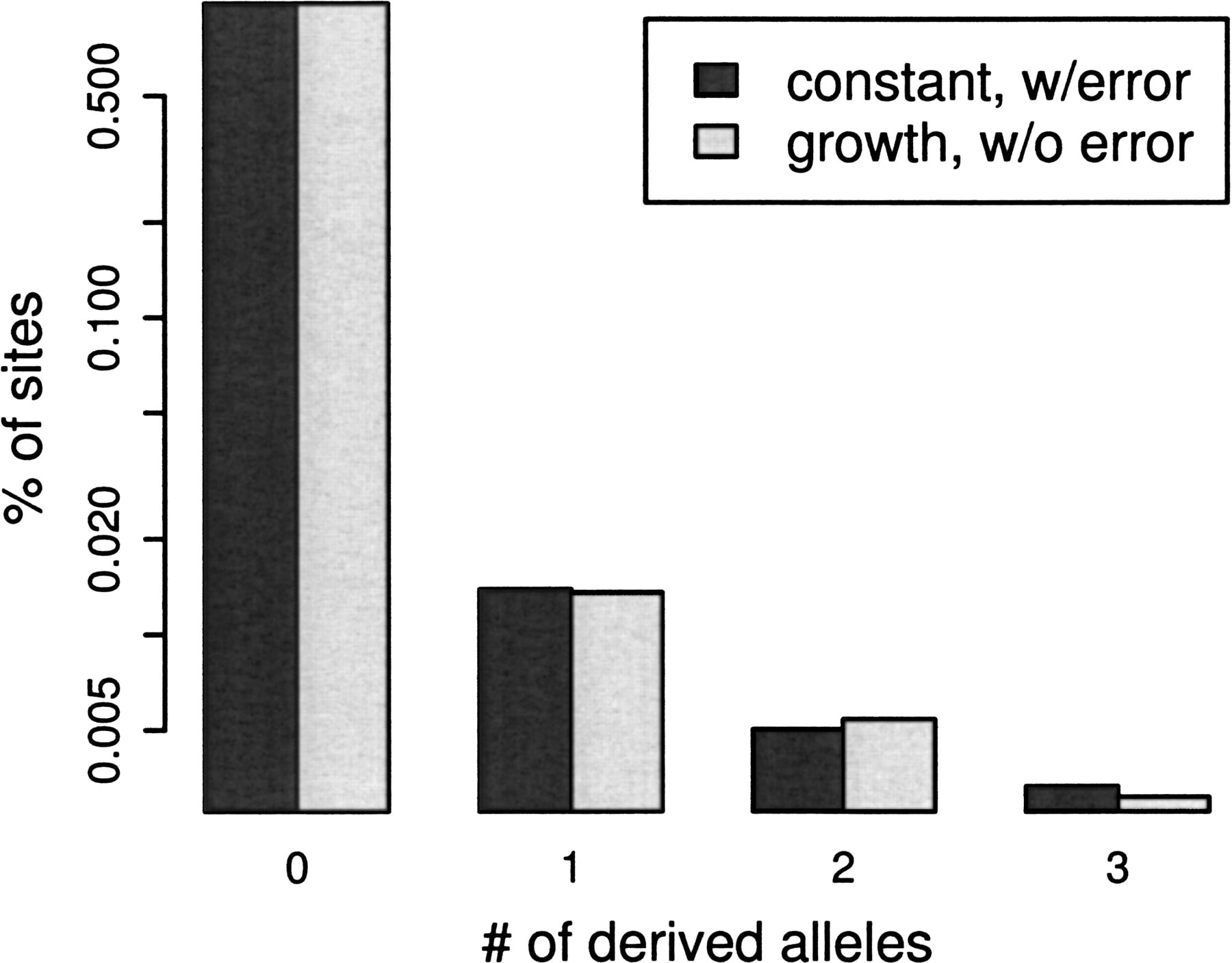
Although recent large-scale metagenomics sequencing projects (Tyson et al. 2004; Venter et al. 2004; Tringe et al. 2005; DeLong et al. 2006) provide genome-wide population samples seemingly ideal for population genetic analysis (Whitaker and Banfield 2006), challenges arise from their variable sample depth (i.e., read coverage) and variable sequence quality. Both of these factors must be taken into account during inference to yield unbiased estimates of population parameters. For instance, sequencing error produces an excess of singletons (apparent polymorphic sites in which only one individual in the sample has the derived allele), but such an excess is also a signature of population growth (Fig. 1)—a difference that can only be distinguished at higher sample depths.
Similarity of sequencing error in constant size population (dark bars: θ =0.01, R= 0, q = 0.001) to population growth (light bars: θ = 0.02, 1, q = 0) with low-read depth (n = 4).
In addition, researchers cannot afford to throw away experimental data. Even with modern high-throughput capillary sequencing technology, the cost and speed of DNA sequencing remain a limiting constraint on research (Shendure et al. 2004). Incorporation of sequence quality allows maximal use of the total sequencing output, leading to a greater yield for the same cost. Previous techniques for estimating population parameters from the site-frequency spectrum were designed to handle error- free samples of fixed size (Sawyer and Hartl 1992; Nielsen 2000; Polanski and Kimmel 2003) and cannot distinguish between population growth and sequencing error. While programs such as PolyBayes (Marth et al. 1999) integrate sequence quality information across assemblies with the goal of discovering polymorphic sites, this strategy has not been applied toward estimating population genetic parameters until now.
The key methodological step that makes population metagenomic analysis possible lies in the difference between metagenomic sequencing and traditional sequencing. Instead of sequencing reads from a single isolate organism, metagenomics projects sequence reads from a pool of DNA extracted from all individuals in the sampled community. Considering the large number of individuals in the sampled community relative to the number of reads sequenced, each read derives from a different individual microorganism with probability near one. Thus, we have a population sample equal to the “depth,” or number of reads, at every site in the assembly of overlapping reads. If only a few sites were sequenced or read depths were very low, this sample would not allow for meaningful statistical inference; however, with metagenomic assemblies spanning entire genomes and having average depths as high as 10 (Tyson et al. 2004), these population samples gain considerable power.
Estimated values for the apparent scaled mutation rate (θ) and the apparent scaled growth rate (R) will provide a glimpse into the evolutionary history of the sampled microbial community in a manner not previously possible. Beyond being a function of mutation and growth, these parameters will also reveal the action of natural selection with negative (background) selection lowering the apparent mutation rate and positive (Darwinian) selection increasing the apparent growth rate. Comparison of these parameter estimates across different groups of genes or genomic regions can then be used to guide experimental investigation into the biological role played by these genes.
Frequency spectrum
Given a population sample from n individuals (i.e., a constant read depth of n), the site-frequency spectrum represents the distribution of polymorphic sites that have a derived (as opposed to ancestral) nucleotide at a particular frequency in the sample. For example, in general, the most common type of polymorphic site will be one in which exactly one individual in the sample has the derived nucleotide; the next most common type of site will be one in which exactly two individuals in the sample have the derived nucleotide, and so on. Experimental data requires the sequence from an outgroup to distinguish which of the two nucleotides is ancestral and which is derived. If an outgroup is not available, the frequency spectrum must be “folded” to combine the indistinguishable categories (i.e., 1 and n − 1, 2 and n − 2, etc.).
With some assumptions about how mutations occur and how a population evolves, theory can predict the shape of this spectrum. Given an observed spectrum, maximum likelihood estimation can be used to work backward and infer the most likely parameters of the theoretical model. Relative comparison of these parameter estimates from different regions of the same genome provide useful information even when the underlying assumptions are questionable. This type of “genomic control” has been used in numerous studies when working with genome-scale data including some using the frequency spectrum for inference (Marth et al. 2004; Nielsen et al. 2005b).
Sawyer and Hartl have characterized the frequency spectrum as an explicit function of the selection coefficient for a constant-size population (Sawyer and Hartl 1992). However, their model makes no allowance for population growth and requires a number of strong assumptions regarding the mechanistic details of selection. Subsequent work has tested and relaxed some of these assumptions (Bustamante et al. 2001; Williamson et al. 2004; Zhu and Bustamante 2005), but strong population growth remains problematic for their model. Since microbial populations can easily undergo exponential growth, we will not consider this model further.
Instead, this study makes use of frequency spectrum formulas arising from two types of neutrally evolving populations, i.e., one that experiences exponential growth (Polanski and Kimmel 2003; Polanski et al. 2003) and one that maintains a constant size (Wright 1931, 1969). Both of these models assume a panmictic Wright-Fisher population with nonoverlapping generations (Wright 1931) conforming to an infinite-sites mutation model (Kimura 1969), where a mutation never hits the same location twice.
In reality, the frequency spectrum arising from evolutionary processes (e.g., mutation, selection, drift) is obscured by errors stemming from the data collection process. Raw data from automated sequencing machines provides a prior distribution for these error probabilities when using data from metagenomics sequencing projects.
Sequence quality
The Sanger method for DNA sequencing depends on chemical reactions that contain stochastic elements, which lead to varying output quality (Ewing et al. 1998).
Base-calling software converts the analog fluorescence output of automated sequencers into a string of digital nucleotides and attempts to quantify the probability of a given base being called in error. The widely used base-calling software Phred reports a quality score based on the shape of the peak and the shape of neighboring peaks calibrated to a particular sequencing chemistry (Ewing and Green 1998; Ewing et al. 1998). Ewing and Green have defined the Phred quality score, S, to be inversely related to the probability of error via the function Pr(err) =10− S /10. Since metagenomics projects deal primarily with haploid organisms and, in any event, clone their DNA fragments into bacterial vectors before sequencing, Phred's base-calling process will not be biased by heterozygous samples (Stephens et al. 2006).
A more refined estimate of sequence quality can be obtained by combining information across multiple aligned reads. If the reads all derive from a single haploid individual or are otherwise expected to be nonpolymorphic, then an algorithm need only consider the various error probabilities (Li et al. 2004). If the reads derive from a diploid individual or from multiple individuals, then the algorithm must distinguish between single nucleotide polymorphisms (SNPs) and sequencing errors. The first program to tackle this task, PolyBayes, used a Bayesian technique incorporating prior probabilities of polymorphism and adjusting the posterior based on the set of quality scores at a given position (Marth et al. 1999). Many others have followed, creating programs focused on SNP discovery, such as the program of Irizarry et al. (2000), or improving sequence assemblies, such as AutoEditor (Gajer et al. 2004).
Despite these tools for quantifying error rates, to the best of our knowledge, most studies performing population genetic analysis use human visual confirmation of quality in combination with a strict Phred quality score cutoff of 30 or 25, which corresponds to between one and three errors per thousand bases (e.g., Brown et al. 2004; Neafsey et al. 2004; Nielsen et al. 2005a).
Here, we avoid picking an arbitrary quality threshold and instead explicitly incorporate quality scores into the likelihood function used to estimate the parameters θ and R. Note that this strategy of integrating error probabilities into the likelihood calculation has general utility and has previously been applied to account for incorrect inference of the ancestral state from an outgroup (Williamson et al. 2005).
Below, we demonstrate the significant advantage of our approach over a cutoff-based method through the use of analytic and Monte Carlo techniques. After establishing that the method works in principle, we proceed to apply it to initial experimental data from a recent metagenomics sequencing project of activated sludge from a wastewater treatment plant (Martin et al. 2006).
Results
Analytic
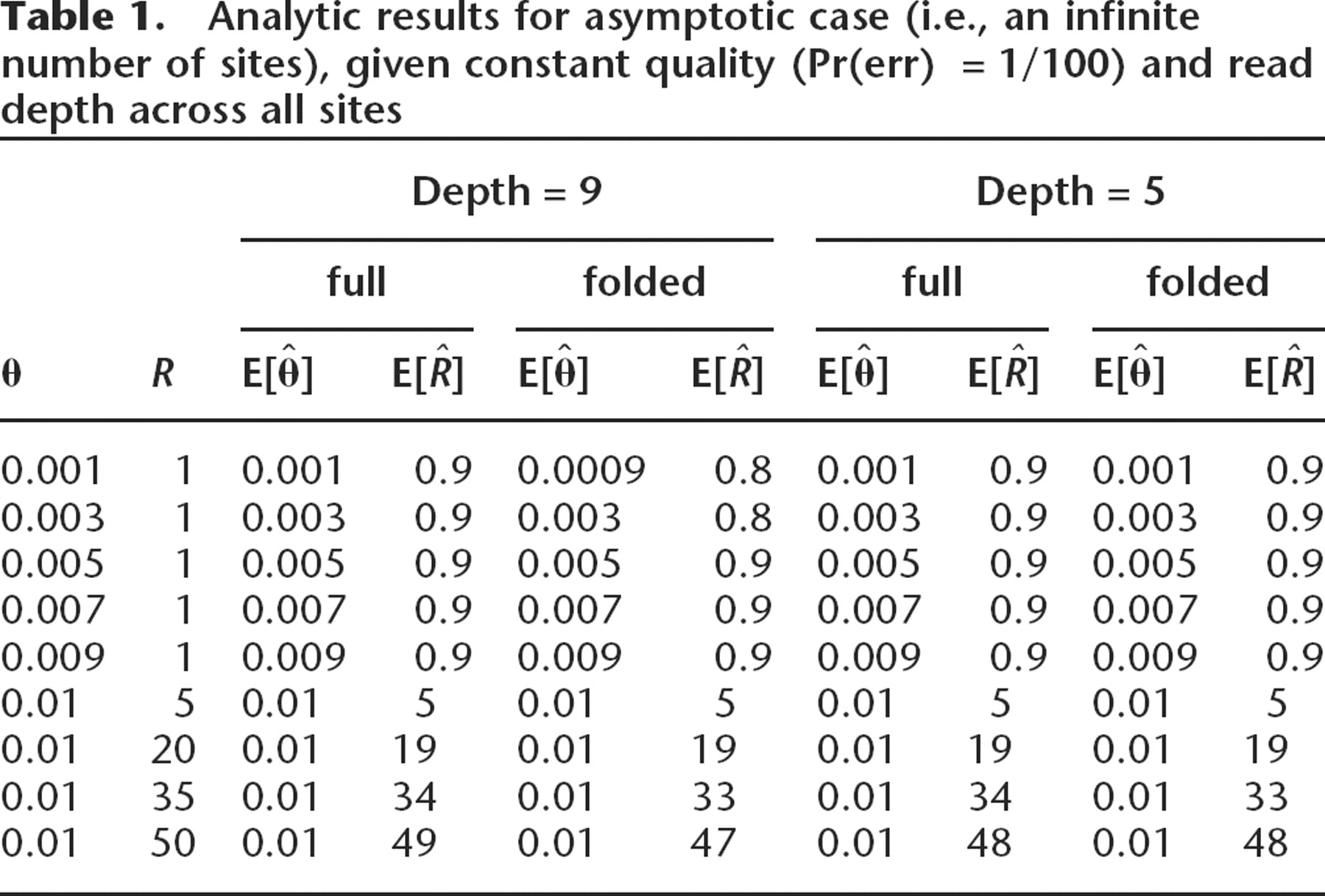
We first verified analytically that, for the restricted case of constant quality and depth across all sites, our method yields a nearly unbiased estimate for θ and R over reasonable parameter ranges. These results assume independence of sites and that sequencing errors cause a switch to any other nucleotide with equal probability. With the constant population size model, this result can be proven explicitly (e.g., if n = 5 and Pr(err) = 0.01, then
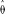 = 1.007θ; further results not shown). For R > 0, we numerically solved for asymptotic parameter estimates using a range of input parameter values (θ:0.001,…,0.01; R:1,…,50; n= 5,9; Pr(err) = 0.001, 0.01) covering two orders of magnitude in polymorphism rate (0.00016 to 0.017). As detailed below, the sludge parameter estimates fall within this range. Since metagenomics projects provide the first large population samples of microbial sequence, previous estimates for the range of microbial polymorphism rates are essentially nonexistent. Although mutation rates in Escherichia coli have been estimated via long-term laboratory evolution experiments to be on the order of 10−10 per site per generation (Lenski et al. 2003), calculation of the polymorphism rate would also require knowledge of the effective population size.
= 1.007θ; further results not shown). For R > 0, we numerically solved for asymptotic parameter estimates using a range of input parameter values (θ:0.001,…,0.01; R:1,…,50; n= 5,9; Pr(err) = 0.001, 0.01) covering two orders of magnitude in polymorphism rate (0.00016 to 0.017). As detailed below, the sludge parameter estimates fall within this range. Since metagenomics projects provide the first large population samples of microbial sequence, previous estimates for the range of microbial polymorphism rates are essentially nonexistent. Although mutation rates in Escherichia coli have been estimated via long-term laboratory evolution experiments to be on the order of 10−10 per site per generation (Lenski et al. 2003), calculation of the polymorphism rate would also require knowledge of the effective population size.
The numerical solutions consistently recovered the input parameter values with the exception of the case with low quality (Pr(err) ≥ 0.01), low read depth, and low polymorphism (i.e., low θ or high R; see Table 1). However, our estimate of R was always more sensitive to sampling perturbations than our estimate of θ; for example, using the full spectrum with θ =0.01, R=1, depth of 9, and an error probability of 0.001, we find the variance of
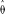 to be 0.00002% of its mean, while the variance of
to be 0.00002% of its mean, while the variance of
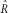 is 0.13% of its mean.
is 0.13% of its mean.
Analytic results for asymptotic case (i.e., an infinite number of sites), given constant quality (Pr(err)=1/100) and read depth across all sites
Simulation
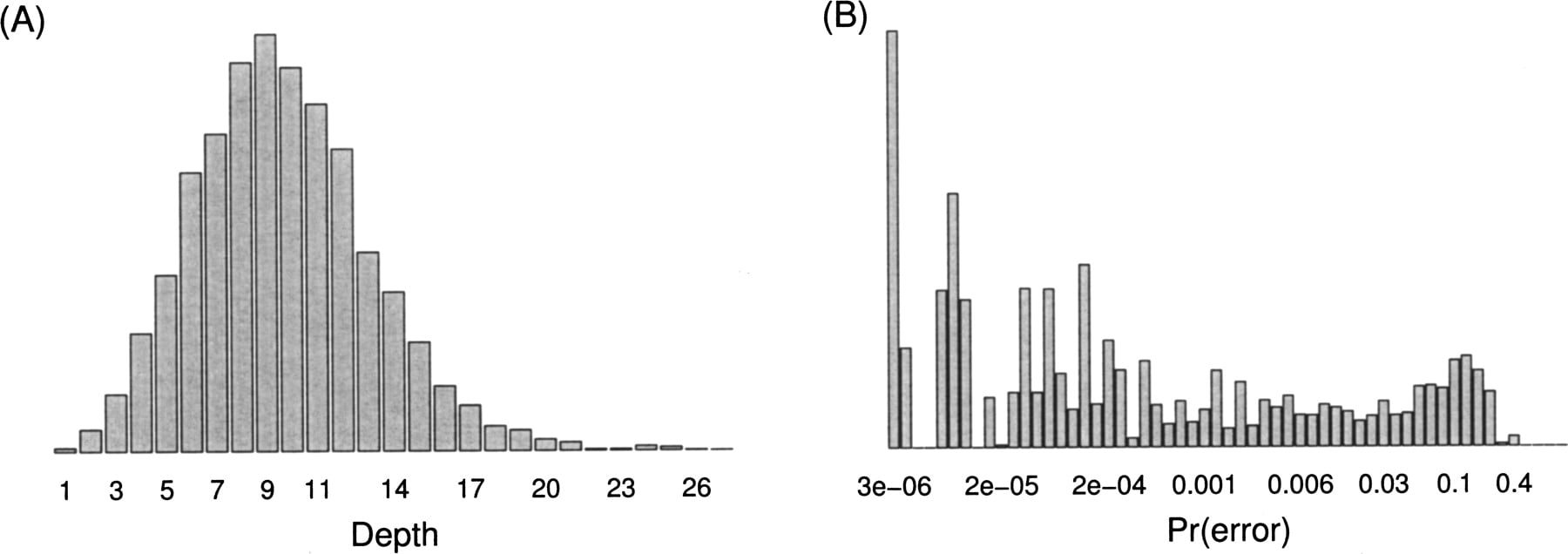
To test the utility of our method in a more realistic situation with variable depth and quality, we simulated data from populations experiencing a range of scaled mutation and growth rates. Each simulation sampled 100,000 independent sites from the same quality score distribution and a slightly modified version of the depth distribution as found in the largest contiguous sequence (170 kb) from the sludge metagenomics project (Fig. 2). We modified the depth distribution by trun cating the right-hand tail to a maximum of 20 to increase computation speed when performing replicate simulations. When a “sequencing error” occurred, the simulation switched the given nucleotide to one of the other three with probability 1/3. In addition to comparing our method against the true simulated value, we also used a simple cutoff scheme that ignores all bases below a threshold quality and completely trusts all bases above the threshold. This cutoff technique approximates the approach taken by previous studies in which low- quality bases are ignored and the rest are approved by a human (Brown et al. 2004; Neafsey et al. 2004; Nielsen et al. 2005a). Following these studies, we applied a Phred quality threshold of 30, which corresponds to a 0.001 chance of an error (and, as can be seen from Fig. 2, an elimination of approximately one-third of the data).
For largest contiguous sludge sequence (170 kb), distributions of read depth (A) and quality scores converted to error probabilities (B).
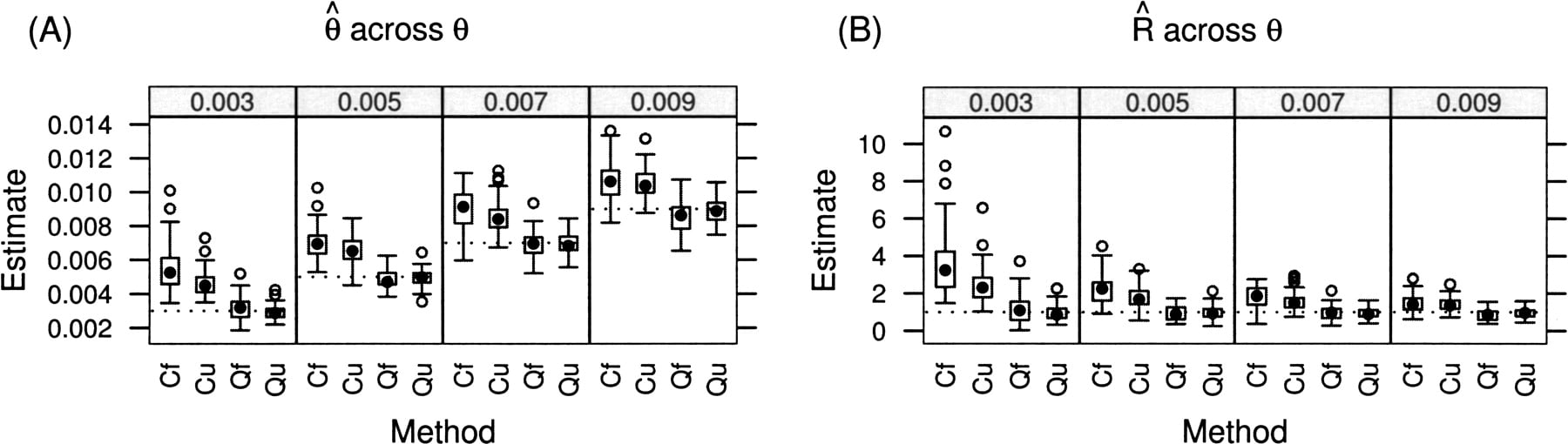
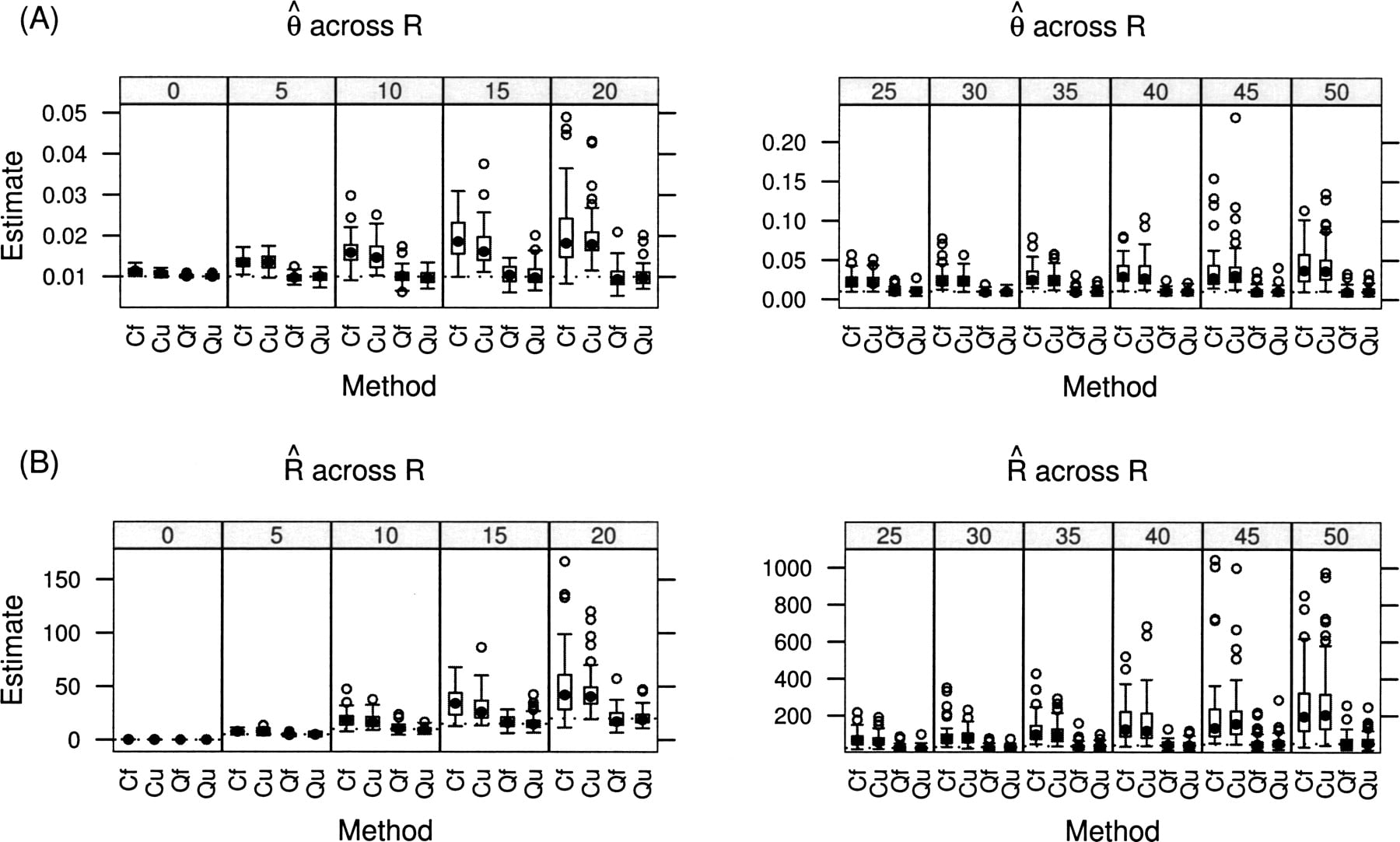
Using the same parameter ranges as above, we initially vary θ from 0.001 to 0.01 while keeping R fixed at 1 (Fig. 3). Taking the average sludge depth of nine, this range for θ corresponds to the rate of polymorphism ranging from 0.0017 to 0.017. Next, we vary R from 0 to 50 while keeping θ fixed at 0.01 (Fig. 4), which corresponds to a polymorphism rate ranging from 0.027 to 0.0024. In all cases, the cutoff estimates are distinctly biased in addition to having much greater variance than the estimates incorporating quality scores. Given our sample size (100,000 sites) and our depth distribution (Fig. 2), the folded estimates performed nearly as well as the full estimates.
Performance of the four techniques (Cf = Cutoff of 30 with folded spectrum, Cu=Cutoff with unfolded spectrum, Qf=Quality scores with folded spectrum, Qu =Quality with unfolded spectrum) across a range of true values of θ (labeled at top) with R = 1. (A) Estimated value for θ; (B) estimated value for R. True values for each parameter drawn as dotted lines. Box-and-whisker plots show quartiles and extreme values.
Performance of the four techniques (Cf= Cutoff of 30 with folded spectrum, Cu = Cutoff with unfolded spectrum, Qf =Quality scores with folded spectrum, Qu = Quality with unfolded spectrum) across a range of true values of R (labeled at top) with θ = 0.01. (A) Estimated value for θ; (B) estimated value for R. True values for each parameter drawn as dotted lines. Box-and-whisker plots show quartiles and extreme values.
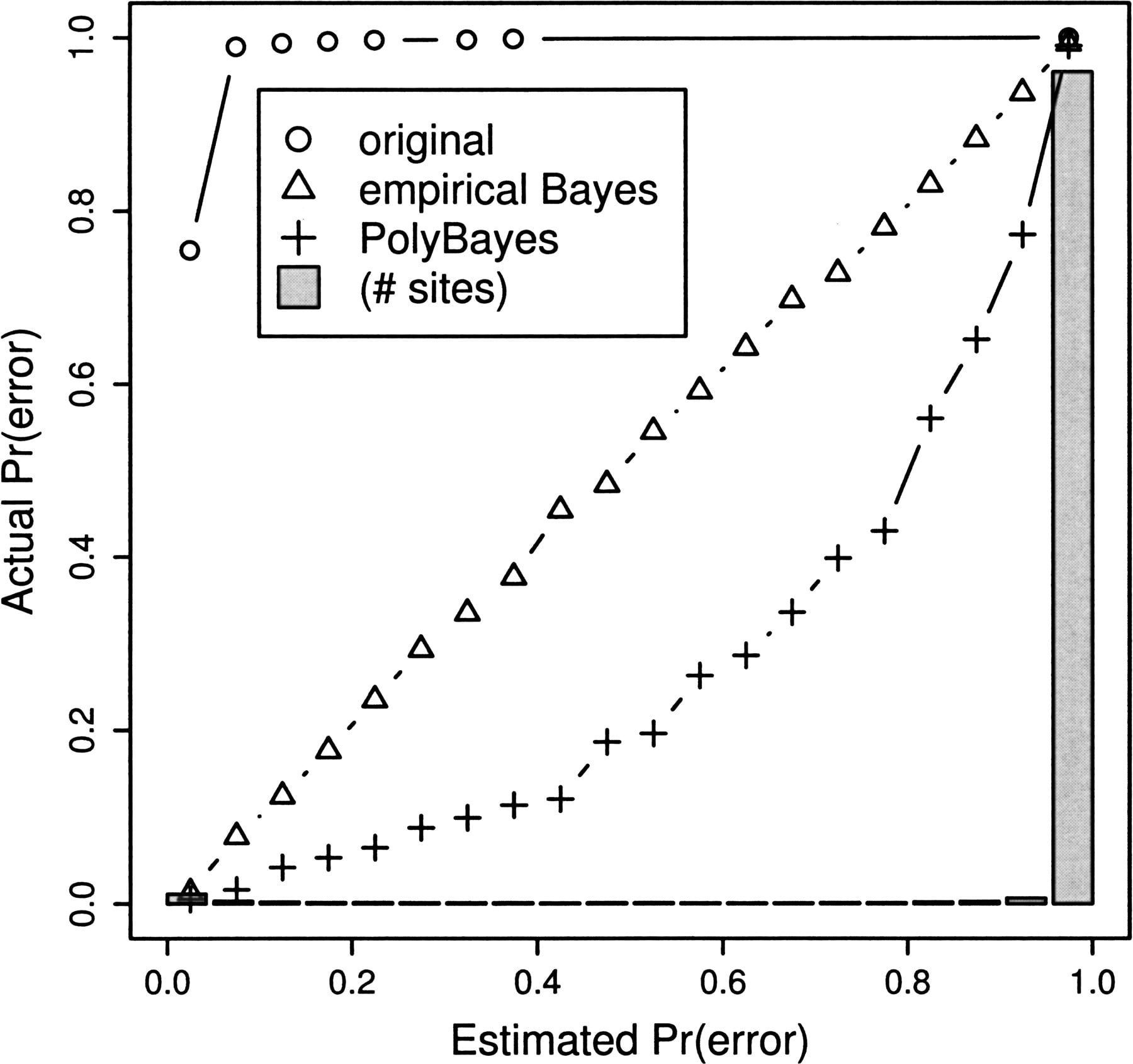
After obtaining quality-based parameter estimates, we can also calculate an empirical Bayes quality score for apparent polymorphic positions (see Methods, equation 8) that much more closely predicts the true quality of those bases than the original quality score. In addition, our model provides a slight advantage over the posterior SNP probability reported by PolyBayes (Marth et al. 1999). In Figure 5, we graph the three estimated error probabilities (original, PolyBayes, and our empirical Bayes) against the actual probability that a site with the given estimated quality is in error. Ideally, the quality scores should form a straight line through y = x, such that an error probability of, say, 0.7 corresponds to an error exactly 70% of the time. The original quality score accurately reflects the error probability across all sites; however, by restricting ourselves to apparent polymorphic positions, we are also selecting for sites with sequencing error. The Poly- Bayes curve illustrates the significant improvement gained by simply combining quality scores across aligned reads. Only in the middle probability ranges, which account for a small proportion of the total sites (note gray bars), does the site-frequency spectrum model contribute information and avoid the overestimate of error made by PolyBayes.
Comparison of original quality score, PolyBayes, and our empirical Bayes quality score (folded spectrum) for apparently singleton polymorphic positions. Sites are binned according to quality score along x-axes and true rates of error (known from simulation) are plotted on the y-axis. Gray bars show distribution of PolyBayes estimated probabilities of error.
Sludge
Application of the folded version of our technique to the largest contiguous sequence in the sludge data set yielded
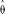 = 0.0012 for the constant population size model and
= 0.0012 for the constant population size model and
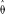 =0.0015 and
=0.0015 and
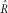 = 49 for the population growth model. Using the likelihood ratio test statistic and calculating its empirical distribution (see Methods), we find the growth model fits significantly better (P< 0.01) even with no recombination. With infinite recombination (i.e., independent sites), the P-value decreases to less than 10−16. Since bacterial recombination rates vary widely (Feil and Spratt 2001) and we have no prior estimate of this rate for the sludge population, we are fortunate that the growth parameter is significant regardless of recombination rate. Thus the model may be detecting very recent growth stemming from manipulation of the laboratory bioreactor prior to DNA sampling; following standard enrichment protocols (Seviour et al. 2003), some effort was made to increase the proportion of the phosphorus-accumulating bacteria to the final level of 80% of the biomass (Martin et al. 2006).
= 49 for the population growth model. Using the likelihood ratio test statistic and calculating its empirical distribution (see Methods), we find the growth model fits significantly better (P< 0.01) even with no recombination. With infinite recombination (i.e., independent sites), the P-value decreases to less than 10−16. Since bacterial recombination rates vary widely (Feil and Spratt 2001) and we have no prior estimate of this rate for the sludge population, we are fortunate that the growth parameter is significant regardless of recombination rate. Thus the model may be detecting very recent growth stemming from manipulation of the laboratory bioreactor prior to DNA sampling; following standard enrichment protocols (Seviour et al. 2003), some effort was made to increase the proportion of the phosphorus-accumulating bacteria to the final level of 80% of the biomass (Martin et al. 2006).
Our estimate for the polymorphism rate in the sludge (i.e., 1 − Pr(d= 0| θ, R,n)), using the average depth of nine, comes to the surprisingly high rate of 0.36%—noticeably higher than the approximate human rate of 0.1% (Cargill et al. 1999). Clearly, microbes have the potential for elevated polymorphism via a much larger effective population size than humans; however, clonal microbial populations with an effective population size near one also exist (Strous et al. 2006).
Discussion
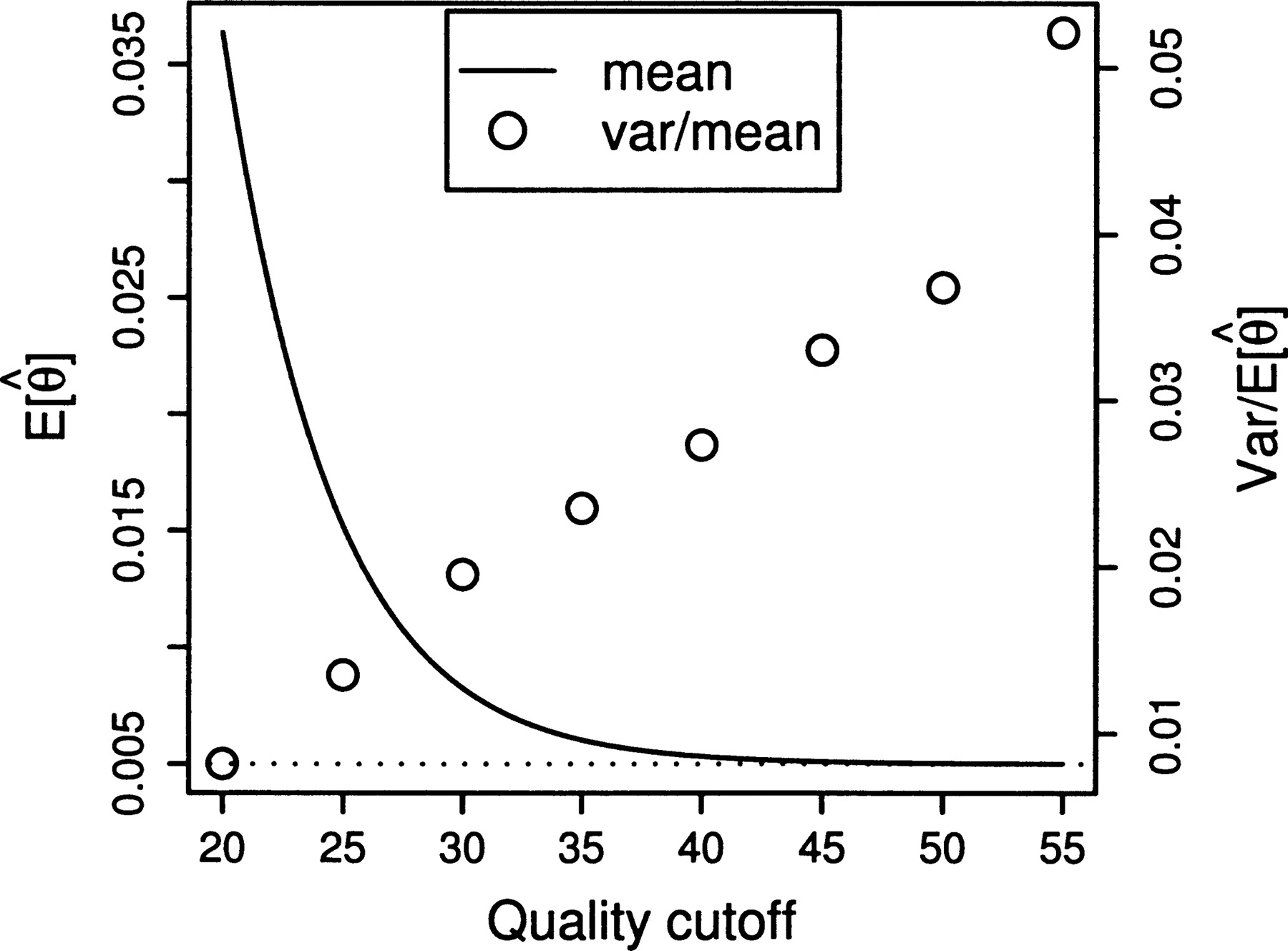
Figures 3 and 4 reveal the cutoff-based estimates to be consistently biased upward relative to the truth. This phenomenon arises from the fact that the cutoff error probability of 0.001 leaves a significant amount of error relative to the amount of signal, leading to an overall excess of apparent polymorphic sites (overestimating θ) and, in particular, an excess of sites with a single apparent derived nucleotide (overestimating R for reasons detailed below). If the cutoff were raised to a level sufficient to avoid the error, the increased variance due to lower sample size would eliminate the signal (Fig. 6).
Estimating θ by using an arbitrary quality cutoff (throwing out data below, trusting data above). Solid line shows (analytic) best-case estimate, assuming no sampling error for a depth of nine and constant quality at the cutoff level. Dotted line shows true value (θ = 0.01). Open circles plot standard deviation/mean when run on a finite sample of 170,000 sites with depth of nine and constant quality, but where the cutoff further reduces the number of sampled sites according to the sludge quality distribution.
While the results are quite promising, we have some caveats.
First, the analytic work and simulations treat sequencing error as causing a switch to any other nucleotide with uniform probability 1/3, which, given the sludge score and depth distributions, leads to a prediction that: ∼ 1.5% of sites would have three or four different nucleotides. However, only 0.3% of sites in the actual sludge sequence are tri- or quadrallelic. This discrepancy implies that at least one of our assumptions is incorrect; possibilities include either that sequencing error causes nonuniform transitions or that sequencing errors at the same site in different reads are nonindependent. However, determining the root cause is beyond the scope of this study. Instead, given the small number of these sites, we decided to first transform them into two-class “biallelic” sites and then use a likelihood function that treats sites as exclusively biallelic (see Methods). Alternatively, we could have either thrown out nonbiallelic sites or made the likelihood function more general. The former was rejected as being significantly biased—a minimum of two errors are required to change a fixed site into a triallelic site, while only one error is required to turn a polymorphic site into a triallelic site. An implementation of the latter approach added significant complexity for only a small gain in estimation power. The chosen route strikes a compromise between these two options, although it leads to the slight bias seen in the analytic results. If quality were lower or depth higher, the likelihood function might need to be adjusted to allow for a more general model of sequencing error.
Second, decreasing the number of sites or depth leads to an increase in the variance of the estimators (particularly
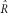 ) for all techniques. A problem arises with few sites, low depth, and a folded spectrum. Namely, a minimally polymorphic population with a lot of sequencing error creates a frequency spectrum similar to a quickly growing population with the exception of those few sites with derived nucleotides present in almost all the reads (right tail of Fig. 1). Folding the spectrum cuts the effective depth in half, which makes accurate estimation difficult and can lead to the maximum likelihood being found with θ and R going toward infinity.
) for all techniques. A problem arises with few sites, low depth, and a folded spectrum. Namely, a minimally polymorphic population with a lot of sequencing error creates a frequency spectrum similar to a quickly growing population with the exception of those few sites with derived nucleotides present in almost all the reads (right tail of Fig. 1). Folding the spectrum cuts the effective depth in half, which makes accurate estimation difficult and can lead to the maximum likelihood being found with θ and R going toward infinity.
Third, when we multiply the per-site likelihoods to form the total likelihood (equation 7), we assume sites segregate independently. Estimation of recombination rate from metagenomic data remains a challenge. Dependence due to low levels of recombination will bias these estimates on an absolute scale, but, as with the other model assumptions, relative comparison of estimates from different regions of the same genome will still provide insight into the evolutionary processes at work.
Finally, the validity of these parameter estimates depends entirely on the accuracy of the given metagenomic assembly on which they are based. The very polymorphism that makes population genetic analysis possible also poses a challenge to the assembly process, making metagenomic assembly algorithms an area of active research (Chen and Pachter 2005). To allow assessment of assembly accuracy, we strongly encourage future metagenomic projects to deposit assemblies and the accompanying traces in publicly available archives such as the National Center for Biotechnology Information's Assembly Archive and Trace Archive.
Given a constant-size or exponentially growing panmictic recombining population, our technique provides reliable estimates for θ =2Neu and R= Ner from metagenomic sequence in the face of sequencing error and limited data. The resulting empirical Bayes quality score can then be used to investigate other questions that require accurate assessment of whether a putative polymorphism is real or not. Many aspects of natural selection and population structure influence the apparent mutation and growth rates experienced by groups of genes. Thus, estimation of these parameters provides a key new tool for analyzing the evolutionary history of microbial communities.
In implementation of our Population genetic Inference In Metagenomics (PIIM) technique is freely available for download from http://ib.berkeley.edu/labs/slatkin/software.html
Methods
Statistical inference
Likelihood calculation
We begin with an assembly of metagenomic reads, FASTA sequences for the reads, and quality scores for every base call in each read. More generally, the data consist of a set of aligned reads containing K sites with a vector of quality scores for each site. Since the read depth varies across the assembly, we denote the read depth at a particular site k ∈ {1,…,K} to be nk . We assume independence of sites and that, at most, two different nucleotides are observed at any given site. If a given site actually has more than two nucleotides, we group the nucleotides into two classes—ancestral and nonancestral (if the ancestral is known) or the single most frequent nucleotide and everything else (if the ancestral is not known). For the duration of the Methods section, we will refer to these two classes of nucleotides as “ancestral” and “derived”; if an outgroup is not available, these references should be interpreted as “major” and “minor,” along with the adjustments noted below.
First, we establish some notation. Starting with a single site where n reads align (i.e., a depth of n), we need two pieces of information—the set of error probabilities, Q= {q 1,…,qn } and the subset of these probabilities corresponding to the observed derived nucleotides, Do ⊂ Q. Let do represent the number of observed derived nucleotides, do = |Do | . To convert from Phred quality scores, Si , to error probabilities, let qi = 10− Si /10 where i Î {1,…,n} (Ewing and Green 1998). Assuming only two types of sequencing errors (true ancestral to apparent derived, and vice versa), we define δ to be the difference between the observed number of derived nucleotides do and the true number d:

Where d − is the number of apparent derived nucleotides that are actually ancestral and d + is the number of apparent ancestral nucleotides that are actually derived. For simplicity of notation, since the order of the reads is irrelevant, take the first do reads to be derived and the rest as ancestral (i.e., Do = {q 1,…,qdo }).
For example, a site with depth n= 4 might have nucleotides (A, A, A, T) and quality scores (q 1,q 2,q 3,q 4). If ‘A’ is derived, then Do = (q 1,q 2,q 3), and thus, do = 3.
Now, given these error probabilities and the set of frequency spectrum parameters (denoted by [H9024] and discussed in detail in the next section), we want to know the likelihood of the observed configuration of nucleotides at a single site: Pr(Do | Q,n,Ω). By conditioning on the true number of derived nucleotides, d, we can split this likelihood into a sequencing error term (i.e., the probability of the observation given the truth and the error probabilities) and the frequency spectrum (the probability of the truth given the parameters):
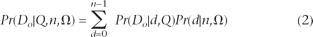
Note that this sum stops at d = n − 1 since frequency spectrum theory makes no statement regarding the fixation probability of the derived nucleotide (d = n). When we lack an outgroup to orient the polymorphism, we must fold the spectrum by changing the first term in the above sum to be (Pr(Do | d,Q) + Pr(Do | n − d,Q))/2.
Looking in more detail at the sequencing error term of equation 2, we condition on D, which we define in a parallel fashion to Do such that it is the assignment of the d true-derived nucleotides to a subset of the Q error probabilities:
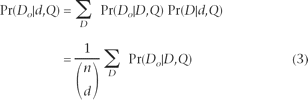
Where D ⊂ Q and contains exactly d elements. There are n choose d possible ways of assigning these d derived nucleotides to the n error probabilities. We assume that quality is independent of being ancestral or derived, so the probability of each of these assignments, Pr(D| d,Q), is the same and thus equal to
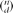 .
.
For reasons of computational efficiency outlined later, we calculate this quantity in an indirect fashion. Specifically, the sum in equation 3 is equivalent to Pr(δ| Do ,Q), the probability of observing a given deviation from the truth. The likelihood (equation 2) then can be rewritten as:
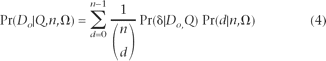
The probability of a given deviation from the truth is a function of the probability of its d + and d [H11502] components (equation 1). For instance, δ = 1 could arise from {d − = 1,d +=0} or {d − = 2,d +=1} any other combination with d [H11502] being one greater than d +. In general,
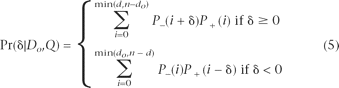
Where P − and P + give the conditional probability distributions for d − and d + , respectively. Note the probability distribution for d − (ancestral-to-derived) depends only on the error probabilities for the observed derived nucleotides (Do ) while the distribution for d + (derived-to-ancestral) depends only on the error probabilities for the observed ancestral nucleotides (Q\Do ). Intuitively, to calculate the probability that d − = x, we must sum the probabilities of all possible ways of having exactly x errors in the set of observed derived nucleotides. Formally, assuming errors are independent among reads, the random variable d − has conditional distribution:
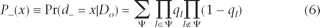
Where Ψ ⊂ {1,…,do
} and contains exactly x elements. The same formula holds for the conditional distribution of d
+ (≡P
+ (x)), except now Ψ ⊂ {do
+ 1,…,n}, and the probability is conditional on
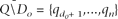 .
.
In the n= (A, A, A, T) example above, the probability that d − = 2 is the probability that the first two nucleotides were in error and the third was not (q 1 q 2(1 − q 3)), plus the probability that the first and third were in error and the second was not (q 1 q 3(1 − q 2)), plus the probability that the second and third were in error and the first was not (q 2 q 3(1 − q 1)). Here, Ψ, in turn, takes values {1,2}, {1,3}, and {2,3}.
Now, to find the total log likelihood across all K sites, we take a composite likelihood approach by assuming independence and summing the logarithm of the individual likelihoods (equation 4, substituting equation 5, equation 6, and the equation for P + analogous to equation 6). Below, we use superscripts to denote observations at a particular site, k ∈ {1,…,K}:
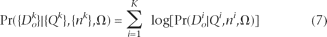
Frequency spectra
The last term in the per-site likelihood function (equation 4) is the frequency spectrum. Technically, the frequency spectrum refers to the distribution of polymorphic sites (i.e., those where the number of derived nucleotides, d, ranges from 1 to n − 1). However, through the use of an extra parameter, θ, we can also calculate the total proportion of sites that are fixed for the ancestral nucleotide such that d = 0 (Ewens 2004). We apply the following two frequency spectra, both of which assume a Wright-Fisher infinite-sites model.
The neutral, constant population size frequency spectrum has the form 1/d, where d = 1…n − 1. The expected proportion of all sites that are polymorphic, and thus participating in the 1/d formula, is
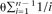 , where θ =2Neu is the product of the per-site mutation rate, u, and the effective population size, Ne
(Ewens 2004). As a result, this model contains only one parameter, θ:
, where θ =2Neu is the product of the per-site mutation rate, u, and the effective population size, Ne
(Ewens 2004). As a result, this model contains only one parameter, θ:
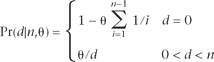
Since θ represents the scaled mutation rate per nucleotide, it must be very small in order to conform to the infinite-sites assumption that a mutation never happens twice at the same location. In fact, this probability distribution requires θ to be
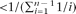 , which means that, for a reasonable maximum depth of n = 30, θ must be <0.25.
, which means that, for a reasonable maximum depth of n = 30, θ must be <0.25.
In analytic expression for the neutral, exponentially growing population frequency spectrum has recently been derived (Polanski and Kimmel 2003; Polanski et al. 2003), but, due to the complexity of its form, details are not presented here. This model becomes a function of θ and R, where R = Ner is the growth rate, r, multiplied by the effective population size, Ne . As a limiting case of this model, we recover the constant population size frequency spectrum when R = 0.
After finding the maximum likelihood parameter estimates for the two models, we compute the likelihood ratio test statistic (−2 log[lik const/lik growth]) to determine whether the more general model (population growth) fits significantly better than the more limited model (constant size). If the model assumption of independence is applicable, then this statistic follows a χ2 distribution with one degree of freedom. If not, we simulate a constant population with the observed amount of recombination using the program “ms” (Hudson 2002). After calculating this statistic for many simulated replicates, the critical value for our test statistic can be determined from the resulting distribution.
Revised quality score
In addition to the inherent utility of these parameter estimates, they can also be used to calculate the probability that an apparently polymorphic site (do
> 0) is in truth fixed (do
=0). Given our frequency spectrum parameter estimates,
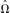 , this empirical Bayes quality score takes the form:
, this empirical Bayes quality score takes the form:
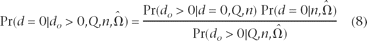
Note that, since this equation uses point estimates for the parameters, the resulting probabilities do not incorporate the variance in
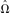 .
.
Computational complexity
Given the complexity of the likelihood function (equation 7), we resort to numerical methods for finding the maximum likelihood parameter estimates. Grid-based exploration of the likelihood surface with simulated data has revealed it to be consistent in shape with a single maximum (results not shown), so we have used the GNU Scientific Library (Galassi et al. 2003) implementation of the Nelder-Mead simplex algorithm (Nelder and Mead 1965).
Calculation of the probability distribution for d − (equation 6), along with the corresponding distribution for d + , forms the most computationally intensive step of this process by consuming O(2 n ) time, where n is the read depth at a given position. Although metagenomic projects tend to have low-average depths, some sites will range considerably higher, pushing 2 n beyond the range of reasonable calculation. To work around this problem, we took advantage of the fact that this function has only one maximum, which, for realistic quality scores, is found near the low end of the distribution (d small). Our algorithm calculates this function in order of increasing d until reaching the first value past the peak that falls below 10−10. From then on, all further values in the distribution are considered to be zero. The dynamic nature of this strategy prevents a rigorous analysis of the time savings; however, in practice, this simple change allows the program to run in a reasonable amount of time on a modern desktop computer (∼2 h for a 170-kb sequence with realistic depth/quality distributions having an average depth of nine).
Sludge metagenome
P. Hugenholtz at the DOE Joint Genome Institute generously shared pre-publication data from a recent metagenomics sequencing project of activated sludge from a wastewater treatment plant (available under Philip Johnson's Programs at http://ib.berkeley.edu/labs/slatkin/software.html, see also Martin et al. 2006). The enhanced biological phosphorus removal performed by the microbial community in this sludge is a little-understood process despite its significant ecological importance (Crocetti et al. 2000). The sludge we analyzed came from a laboratory biore- actor in Madison, Wisconsin that had been seeded from a local wastewater treatment plant. Fluorescent in-situ hybridization suggested that a single species dominated the sludge biomass, which meant it should dominate the sequenced metagenomic reads as well. We received the data in the form of a Phrap (http://www.phrap.org) assembly primarily consisting of that single species and restricted our analysis to the single largest contiguous sequence found in this assembly (170 kb). While this sequence has an average depth of nine and an average quality score of 36 (corresponding to a 0.0002 error probability), the respective distributions ranged from 1 to 27 and from 0 to 56 (Fig. 2). Since this species lacks a clear outgroup to distinguish derived from ancestral nucleotides, we applied the folded version of the analysis discussed above.
Acknowledgments
We thank Phil Hugenholtz and Victor Kunin at JGI for bringing this problem to our attention and sharing the sludge data that inspired this project. The excellent suggestions of three anonymous reviewers, Anna-Sapfo Malaspinas, John Novembre, Clair Null, Rachel Whitaker, and Weiwei Zhai improved earlier versions of this manuscript. This research was supported in part by National Institutes of Health grant R01-GM40282 to M.S.
References
- ↵G. Brown,G. Gill,R. Kuntz,C. Langley,D. Neale,(2004) Nucleotide diversity and linkage disequilibrium in loblolly pine. Proc. Natl. Acad. Sci. 101:15255–15260.
- ↵C. Bustamante,J. Wakeley,S. Sawyer,D. Hartl,(2001) Directional selection and the site-frequency spectrum. Genetics 159:1779–1788.
- ↵M. Cargill,D. Altshuler,J. Ireland,P. Sklar,K. Ardlie,N. Patil,N. Shaw,C.R. Lane,E.P. Lim,N. Kalyanaraman,(1999) Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat. Genet. 22:231–238.
- ↵K. Chen,L. Pachter,(2005) Bioinformatics for whole-genome shotgun sequencing of microbial communities. PLoS Comput. Biol. 1:106–112.
- ↵G. Crocetti,P. Hugenholtz,P. Bond,A. Schuler,J. Keller,D. Jenkins,L. Blackall,(2000) Identification of polyphosphate-accumulating organisms and design of 16S rRNA-directed probes for their detection and quantitation. Appl. Environ. Microbiol. 66:1175–1182.
- ↵E. DeLong,C. Preston,T. Mincer,V. Rich,S. Hallam,N. Frigaard,A. Martinez,M. Sullivan,R. Edwards,B. Brito,(2006) Community genomics among stratified microbial assemblages in the ocean's interior. Science 311:496–503.
- ↵W. Ewens,(2004) Mathematical population genetics: I. Theoretical introduction (Springer-Verlag, New York), 2nd ed..
- ↵B. Ewing,P. Green,(1998) Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8:186–194.
- ↵B. Ewing,L. Hillier,M.C. Wendl,P. Green,(1998) Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8:175–185.
- ↵E.J. Feil,B.G. Spratt,(2001) Recombination and the population structures of bacterial pathogens. Annu. Rev. Microbiol. 55:561–590.
- ↵P. Gajer,M. Schatz,S.L. Salzberg,(2004) Automated correction of genome sequence errors. Nucleic Acids Res. 32:562–569.
- ↵M. Galassi,J. Davies,J. Theiler,B. Gough,G. Jungman,M. Booth,F. Rossi,(2003) GNU Scientific Library Reference Manual (Network Theory Ltd, Bristol UK), 2nd ed..
- ↵R.R. Hudson,(2002) Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18:337–338.
- ↵K. Irizarry,V. Kustanovich,C. Li,N. Brown,S. Nelson,W. Wong,C.J. Lee,(2000) Genome-wide analysis of single-nucleotide polymorphisms in human expressed sequences. Nat. Genet. 26:233–236.
- ↵M. Kimura,(1969) The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 61:893–903.
- ↵R.E. Lenski,C.L. Winkworth,M.A. Riley,(2003) Rates of DNA sequence evolution in experimental populations of Escherichia coli during 20,000 generations. J. Mol. Evol. 56:498–508.
- ↵M. Li,M. Nordborg,L.M. Li,(2004) Adjust quality scores from alignment and improve sequencing accuracy. Nucleic Acids Res. 32:5183–5191.
- ↵H.G. Martín,N. Ivanova,V. Kunin,F. Warnecke,K. Barry,A.C. McHardy,C. Yeates,S. He,A. Salamov,E. Szeto,(2006) Metagenomic analysis of phosphorus removing sludge communities. Nat. Biotechnol. in press.
- ↵G. Marth,E. Czabarka,J. Murvai,S. Sherry,(2004) The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics 166:351–372.
- ↵G.T. Marth,I. Korf,M.D. Yandell,R.T. Yeh,Z. Gu,H. Zakeri,N.O. Stitziel,L. Hillier,P.Y. Kwok,W.R. Gish,(1999) A general approach to single-nucleotide polymorphism discovery. Nat. Genet. 23:452–456.
- ↵D. Neafsey,J. Blumenstiel,D. Hartl,(2004) Different regulatory mechanisms underlie similar transposable element profiles in pufferfish and fruitflies. Mol. Biol. Evol. 21:2310–2318.
- ↵J. Nelder,R. Mead,(1965) A simplex method for function minimization. Computer Journal 7:308–315.
- ↵R. Nielsen,(2000) Estimation of population parameters and recombination rates from single nucleotide polymorphisms. Genetics 154:931–942.
- ↵R. Nielsen,C. Bustamante,A. Clark,S. Glanowski,T. Sackton,M. Hubisz,A. Fledel-Alon,D. Tanenbaum,D. Civello,T. White,(2005a) A scan for positively selected genes in the genomes of humans and chimpanzees. PLoS Biol. 3:e170.
- ↵R. Nielsen,S. Williamson,Y. Kim,M.J. Hubisz,A.G. Clark,C. Bustamante,(2005b) Genomic scans for selective sweeps using SNP data. Genome Res. 15:1566–1575.
- ↵A. Polanski,M. Kimmel,(2003) New explicit expressions for relative frequencies of single-nucleotide polymorphisms with application to statistical inference on population growth. Genetics 165:427–436.
- ↵A. Polanski,A. Bobrowski,M. Kimmel,(2003) A note on distributions of times to coalescence, under time-dependent population size. Theor. Popul. Biol. 63:33–40.
- ↵S. Sawyer,D. Hartl,(1992) Population genetics of polymorphism and divergence. Genetics 132:1161–1176.
- ↵R.J. Seviour,T. Mino,M. Onuki,(2003) The microbiology of biological phosphorus removal in activated sludge systems. FEMS Microbiol. Rev. 27:99–127.
- ↵J. Shendure,R.D. Mitra,C. Varma,G.M. Church,(2004) Advanced sequencing technologies: Methods and goals. Nat. Rev. Genet. 5:335–344.
- ↵M. Stephens,J. Sloan,P. Robertson,P. Scheet,D. Nickerson,(2006) Automating sequence-based detection and genotyping of SNPs from diploid samples. Nat. Genet. 38:375–381.
- ↵M. Strous,E. Pelletier,S. Mangenot,T. Rattei,A. Lehner,M. Taylor,M. Horn,H. Daims,D. Bartol-Mavel,P. Wincker,(2006) Deciphering the evolution and metabolism of an anammox bacterium from a community genome. Nature 440:790–794.
- ↵S. Tringe,C. von Mering,A. Kobayashi,A. Salamov,K. Chen,H. Chang,M. Podar,J. Short,E. Mathur,J. Detter,(2005) Comparative metagenomics of microbial communities. Science 308:554–557.
- ↵G. Tyson,J. Chapman,P. Hugenholtz,E. Allen,R. Ram,P. Richardson,V. Solovyev,E. Rubin,D. Rokhsar,J. Banfield,(2004) Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428:37–43.
- ↵J. Venter,K. Remington,J. Heidelberg,A. Halpern,D. Rusch,J. Eisen,D. Wu,I. Paulsen,K. Nelson,W. Nelson,(2004) Environmental genome shotgun sequencing of the Sargasso Sea. Science 304:66–74.
- ↵R.J. Whitaker,J.F. Banfield,(2006) Population genomics in natural microbial communities. Trends Ecol. Evol. (in press).
- ↵S. Williamson,A. Fledel-Alon,C.D. Bustamante,(2004) Population genetics of polymorphism and divergence for diploid selection models with arbitrary dominance. Genetics 168:463–475.
- ↵S.H. Williamson,R. Hernandez,A. Fledel-Alon,L. Zhu,R. Nielsen,C.D. Bustamante,(2005) Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc. Natl. Acad. Sci. 102:7882–7887.
- ↵S. Wright,(1931) Evolution in Mendelian populations. Genetics 16:97–159.
- ↵S. Wright,(1969) Evolution and the genetics of populations, Vol. 2: The theory of gene frequencies. (University of Chicago Press, Chicago IL).
- ↵L. Zhu,C.D. Bustamante,(2005) A composite-likelihood approach for detecting directional selection from DNA sequence data. Genetics 170:1411–1421.
Notes
[1] Article published online before print. Article and publication date are online at http://www.genome.org/cgi/doi/10.1101/gr.5431206.