
Mapping of the juxtacentromeric heterochromatin-euchromatin frontier of human chromosome 21
-
1 These authors contributed equally to this work.
Abstract
Euchromatin and heterochromatin are functional compartments of the genome. However, little is known about the structure and the precise location of the heterochromatin–euchromatin boundaries in higher eukaryotes. Constitutive heterochromatin in centromeric regions is associated with (1) specific histone methylation patterns, (2) high levels of DNA methylation, (3) low recombination frequency, and (4) the repression of transcription. All of this contrasts with the permissive structure of euchromatin found along chromosome arms. On the sequence level, the transition between these two domains consists most often of patchworks of segmental duplications. We present here a comprehensive analysis of gene expression, DNA methylation in CpG islands, distribution of histone isoforms, and recombination activity for the juxtacentromeric (or pericentromeric) region of the long arm of human chromosome 21. We demonstrate that most HapMap data are reliable within this region. We show that high linkage disequilibrium between pairs of SNPs extends 719–737 kb from the centromeric α-satellite. In the same region we find a peak of histone isoforms H3K9Me3 and H3K27Me (715–822 kb distal to the α-satellite). In normal somatic cells, CpG islands proximal to this peak are highly methylated, whereas distal CpG islands are not or very little methylated. This methylation profile undergoes dramatic changes in cancer cells and during spermatogenesis. As a consequence, transcription from heterochromatic genes is activated in the testis, and aberrant gene activation can occur during neoplastic transformation. Our data indicate that the frontier between the juxtacentromeric heterochromatic domain and euchromatic domain of the long arm of chromosome 21 is marked by a heterochromatic peak located ~750 kb distal to the α-satellite.
The division of the genome into functional chromatin compartments is a universal property of eukaryotic genomes. In humans, regions surrounding centromeres and telomeres are heterochromatic whereas large parts of the chromosome arms consist of transcriptionally competent euchromatin. Except for neocentromeres, functional human centromeres are embedded within megabase-long tandemly repeated DNA. The transition between this centromeric satellite DNA and the sequence of the chromosome arms usually consists of a patchwork of segmental duplications with copies on several chromosomes (Bailey et al. 2002). Several large-scale analyses have elucidated the epigenetic structure, i.e., the distribution of histone isoforms and the nature of DNA methylation along the chromosome arms in model organisms and in humans (ENCODE Project Consortium 2004; Rakyan et al. 2004; Bernstein et al. 2005). However, the complex genomic structure of human juxtacentromeric regions has so far hampered detailed mapping analyses of their epigenetic features. ChIP-on-chip for instance, which would be the method of choice for a comprehensive study, cannot be applied because of the high sequence similarities between juxtacentromeric regions of different chromosomes. Instead, immunofluorescence studies of mouse and human metaphase chromosomes have shown, at a chromosome-wide level of resolution, that heterochromatin in centromeric regions is associated with specific histone modifications such as H3 trimethylation in Lys9 (H3K9Me3) and monomethylation in Lys27 (H3K27Me) (Peters et al. 2003). Acetylated histone H4 (H4Ac) is enriched on the chromosome arms but is virtually absent from the region around the primary constriction (Boggs et al. 2002). In addition, centromeric and juxtacentromeric satellite DNA has been shown to be highly methylated (Ehrlich 2002). Impaired methylation of this satellite DNA is often observed in cancer cells and also found in a hereditary disease, ICF syndrome (immunodeficiency, centromere instability, and facial anomalies; OMIM 242,860), in which DNA hypomethylation occurs predominantly in juxtacentromeric satellite 2 (Miniou et al. 1994). It is so far not known to what extent the methylation of satellites spreads to the adjacent nonsatellite sequences and to the nearby CpG islands. This is due in part to the fact that the complex structure of juxtacentromeric segmental duplications has hindered systematic analysis of DNA methylation profiles. Specifically, the high sequence identity between duplicated regions presents a major obstacle for all hybridization-based approaches, and these regions are systematically excluded in such studies. To better understand the processes that control the structural and functional integrity of chromosome architecture it is, however, necessary to identify what separates constitutive heterochromatin from euchromatin. A first step toward the determination of what separates these chromatin domains would be to accurately localize the corresponding border region.
We had proposed that the frontier between juxtacentromeric heterochromatin and adjacent euchromatin of human chromosome 21q lies between a 1.1-Mb duplication-rich proximal region that is transcriptionally silent in somatic tissue and a chromosome 21-specific distal region that contains active genes (Fig. 1; Brun et al. 2003). Recent segmental duplications represent at least 5% of the human genome. Their biased distribution to subtelomeric and juxtacentromeric regions might be a byproduct of the spreading of constitutive heterochromatin within these regions: New insertions could be more easily tolerated here because both ectopic recombination between duplicated blocks and transcription of genes contained in the new copy would be repressed. This hypothesis entails that constitutive heterochromatin would spread over most or the entire juxtacentromeric duplication-rich region. In mammals, analysis of transgenic genes suggests that heterochromatic genes are generally not expressed due to the repressive structure of the compact chromatin, whereas the open structure of euchromatin appears to be a prerequisite, but mechanistically independent, condition for other regulatory mechanisms to activate transcription of euchromatic genes (Lundgren et al. 2000). In support of the hypothesis that juxtacentromeric duplications are of a heterochromatic nature, we showed earlier (Grunau et al. 2000) that the CpG island of a pseudogene (ψSLC6A8) embedded in the juxtacentromeric segmental duplications of chromosome 16 (16p11.1) (Eichler et al. 1996) is highly methylated. In contrast, its paralogue on Xq28 (SLC6A8) shows typical hypomethylation in its CpG island and is strongly expressed in various somatic tissues. The juxtacentromeric copy is not expressed in normal somatic cells but is activated in the testis, where demethylation occurs during spermatogenesis. Another consistent example is provided by the BAGE genes that map to the juxtacentromeric regions of chromosomes 13 and 21 (Ruault et al. 2002, 2003). BAGE genes are highly methylated and transcriptionally silent in normal tissues but are hypomethylated in cancers and testis, where they can be expressed (Grunau et al. 2005). Detailed transcriptional maps of duplication-rich regions are still rare; however, studies have shown that duplication-rich regions harbor few genes that are generally silent in normal cells but that become expressed in some tumors and in testis (Guy et al. 2000, 2003; Brun et al. 2003; Mudge and Jackson 2005). We decided therefore to focus our research on the duplication-rich region and the adjacent unique sequences.

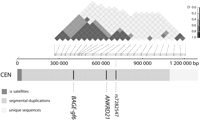
Sequence analysis of the juxtacentromeric region of chromosome 21. Grayscales of the horizontal bar correspond to α-satellites, to regions that are rich in segmental duplications, and to unique sequences (from proximal to distal). Genes and pseudogenes are indicated below the bar. In the upper panel representation of copy numbers of nearly identical sequences in the genome, in black is bioinformatics data complied from http://humanparalogy.gs.washington.edu/eichler/celera5/, with the approximate copy number on the left. Unique sequences are in gray. Hollow bars show the results of PCR on a panel of monochromosomal hybrid DNAs (PCR details on request). Copy number is above each bar. Dotted lines indicate positions of the last two PCR sites that give signals on multiple chromosomes and the first chromosome 21-specific PCR site. The x-axis is in base pairs in contig NT_011512.10/Hs21_11669.
Sequence variants between paralogous copies (PSVs) have been used as chromosome-specific anchors for typing nearby polymorphic microsatellite markers embedded within segmental duplications of 21q11 and 21p (Laurent et al. 2003). Linkage analysis showed that recombination is repressed across the most proximal 21q region but not in the more distal region in women, suggesting again that a transition between a proximal condensed chromatin domain and a distal open region occurs between 430 kb and 1.26 Mb from the centromeric α-satellite block. A recent haplotype map of the human genome may serve to refine this transition between repressed and active domains for recombination (Altshuler et al. 2005). Linkage disequilibrium (LD) and coalescent analyses were used to infer historical recombination rates between pairs of SNP markers spaced 1 kb on average from each other and along the sequenced human genome (Myers et al. 2005). However, SNPs in duplicated regions can be confounded with PSVs and multisite variations (Fredman et al. 2004). Consequently, and since no dedicated typing strategy had been applied in the HapMap project for the chromosome-specific detection of duplicated SNPs, the reliability of the haplotype map in juxtacentromeric regions is unclear. We sought to address this issue by developing a set of 21q11-specific SNPs, including SNPs typed in the HapMap project.
In the present work, we used the juxtacentromeric region of human chromosome 21 as a model system to map for the first time at ≤100-kb resolution a transition from constitutive heterochromatin to euchromatin. We based our mapping strategy on three characteristic chromatin features—histone modifications, DNA methylation, and competence for gene transcription—and on the analysis of recombination activity inferred from linkage disequilibrium along the region. We show that the chromatin frontier is located roughly 750 kb distal to the centromeric α-satellite DNA.
Results
Chromosome-specific physical and genetic markers within 21q11 segmental duplications
To identify specific markers of chromosome 21q11 that are suitable for ChIP experiments, we used the paralogy detector tool described by Bailey et al. (2002) for estimating the number of paralogous copies of any sequence along the 1.3-Mb juxtacentromeric 21q11 DNA. We extracted 11 subregion sequences that were duplicated less than four times per haploid genome, contained no dispersed repeats, and were at least 600 bp long. Their paralogous copies were retrieved from the human genome through BLAST searches, and multiple alignments were performed. We designed seven sequence-tagged sites (STS) that show a 21-specific paralogous restriction variant and two STSs that encompass a 21-specific junctional break point between duplicated blocks (Supplemental Table I). Two additional STSs were designed within the distal, chromosome 21-specific region of 21q11. These 11 STSs were validated experimentally as chromosome 21-specific elements by PCR amplification in a panel of commercially available monochromosomal hybrid DNAs (data not shown).
For the analysis of recombination frequency, twenty 21q11-specific SNPs were selected by combining PSV-specific detection of the 21q11 copy and SNP genotyping. The most reliable approach was PSV-specific amplification followed by RFLP genotyping (see Methods). These 20 SNPs cover a 673-kb region with the most proximal SNP lying 239 kb distal to the α-satellite (Supplemental Table II). After experimental validation of the single chromosome location of the amplified products, each SNP was typed in a panel of 87 European individuals also used in the HapMap project (CEPH-CEU HapMap panel). None of these 20 SNP markers showed any significant difference between observed and expected frequencies of genotypes under the Hardy-Weinberg equilibrium assumption, further reinforcing their single locus location.
High linkage disequilibrium extends 719 kb distal to the α-satellite
The linkage disequilibrium coefficient between pairs of our 20 SNP markers showed high values (D′ > 0.8) in the proximal 21q11 region; this strong LD extended up to 668 kb from the α-satellite (data not shown). Six SNPs among these 20 markers were also typed in the framework of the HapMap project. Comparison between the two sets of data showed an overall 98.9% agreement, indicating that most HapMap data available within segmental duplications are reliable. Strong LD (D′ > 0.8) extends up to 719 kb from the α-satellite, even between pairs of HapMap markers 200 kb apart (Fig. 2; Supplemental Table III). This contrasts with the distal domain, where very few pairs of adjacent SNPs show significant LD as found along the entire human haplotype map for such physical distance between markers (average spacing 47 kb). Selecting all available Hap-Map SNPs, polymorphic in Europeans, around the transition further narrowed the frontier between the proximal and the distal domain to within a 18-kb region, 719–737 kb from the α-satellite (between SNP IDs rs7282547 and rs2260407).
Haploview plot of HapMap linkage disequilibrium coefficient D′ for pairs of SNPs along 21q11. Twenty-five highly informative SNPs were selected along a 963-kb 21q11 region in the Hap-Map database, spanning the previously reported transition between repressed and active domains for meiotic recombination in women (Laurent et al. 2003) and with the most proximal marker 229 kb from α-satellite. High D′ values extend up to 719 kb (SNP ID rs7282547) from the centromeric α-satellite and indicate a repression of recombination in this region. The x-axis is as in Figure 1.
Histones H3K9Me3 and H3K27Me are enriched ~740 kb distal to the α-satellite
We analyzed the distribution of histone isoforms along the juxtacentromeric region by quantitative PCR amplification of immunoprecipitated 21q11 STSs. Native immunoprecipitation experiments using antibodies against the histone isoforms H3K9Me3, H3K27Me, H3K9Ac, and H4Ac were performed with human fibroblasts, two breast tumor cell lines and a multiple myeloma cell line. Real-time PCR amplification of 21q11-specific STSs, if necessary in conjunction with specific restriction digests, was used to quantify the DNA associated with different histone isoforms (Fig. 3; Supplemental Table IV). The euchromatic GAPDH locus served as reference. The two euchromatic histone isoforms H3K9Ac and H4Ac show ratios to GAPDH <1, indicating the entire region is noneuchromatic. The heterochromatic histones H3K9Me3 and H3K27Me are slightly enriched in the entire region, with a weak increase toward the centromere. However, these two histone isoforms were strongly enriched in two STSs close to each other, 739 and 741 kb from the centromeric α-satellite, respectively. Two STSs with relatively low enrichment in these two H3 isoforms delimit this heterochromatic peak, 714 kb and 821 kb from the α-satellite, respectively. While absolute values of enrichments versus GAPDH vary, the profiles of histone modifications are similar between cell types, and no significant difference could be detected between the shape of histone modification profiles of cancer cells and fibroblast.

Schematic representation of DNA methylation and histone modification status in the juxtacentromeric region of human chromosome 21. (A) The x-axis is as in Figure 1; the y-axis shows relative enrichment of histone isoforms compared to GAPDH. STSs and genes are represented along the x-axis. Error bars are omitted for sake of clarity; standard errors are in Supplemental Table IV. (B) CpG methylation is given for BAGE-gf6, ANKRD21, RBM11, ABCC13, STCH, and NRIP1, with 5mCpG as filled circles and unmethylated CpG as hollow circles for normal somatic tissue (skin 1), sperm (sperm 1), melanoma cell lines melanoma 1 (SK23-MEL), and melanoma 2 (LB1622-MEL). Average methylation of CpG pairs is given under each sequence stack. Arrowheads indicate positions of restriction sites used in COBRA. ANKRD21 exists in two alleles, and for heterozygous samples melanoma 2 and sperm, methylation in each allele is indicated. (C) Average methylation (%5mCpG) in three tissue groups determined by COBRA and corrected for PCR bias. For TPTE, BAGE, and ANKRD21 the average methylation in the corresponding gene families is shown since PCR primers used for COBRA do not distinguish between paralogous loci.
CpG island DNA methylation is high in the proximal domain and low in the distal domain
We sought to analyze DNA methylation and gene expression in the chromosome domains that are proximal and distal to the heterochromatic peak. We compared the degree of methylation in the CpG islands of four proximal loci (TPTE, BAGE2, BAGE-gf6, and ANKRD21) with four distal genes (RBM11, ABCC13, STCH, and NRIP1). TPTE (Tapparel et al. 2003), BAGE2 (Ruault et al. 2003), and ANKRD21 (Bera et al. 2002) are transcribed in the testis and in some cancers. All three genes possess several paralogous copies, including the nontranscribed gene fragment BAGE-gf6 located proximal to ANKRD21 on 21q11. LIPI, RBM11, ABCC13, STCH, and NRIP1 are chromosome 21-specific genes and are expressed in various cell types, including somatic tissues (Brun et al. 2003). LIPI and RBM11 share a common CpG island. Genomic DNA of eight arbitrarily chosen normal tissue samples, eight tumor cell lines and tumors, and four sperm and four testis samples (Supplemental Table V) was treated with sodium bisulfite followed by PCR amplification. The degree of methylation was determined by digestion with informative enzymes for all samples (Combined Bisulfite Restriction Assay [COBRA]). For a subset of samples (sperm, normal skin, and two melanoma cell lines), PCR products were subcloned and sequenced (Supplemental Table V; Fig. 3). In sperm, all CpG islands except those of the BAGE loci are completely demethylated. In normal somatic tissue, the CpG islands of RBM11, ABCC13, STCH, and NRIP1 are also free of methylation. In contrast, the CpG islands of TPTE, BAGE, and ANKRD21 are highly methylated. PCR conditions used for COBRA of these loci allow for amplification of all members of these gene families that are located in juxtacentromeric duplications of chromosome 21 and other chromosomes. Our data indicate that hypermethylation of these regions is not restricted to chromosome 21. COBRA data for these loci were corroborated by sequencing of subclones (data not shown). Interestingly, we identified two alleles in the ANKRD21 locus. The short allele was previously misassigned to chromosome 22 (Gen-Bank entry BX072566), but PCR on bisulfite-treated DNA of monochromosomal somatic hybrid cell clearly established that it is the short allele of ANKRD21 on chromosome 21. We noticed that methylated CpG tend to be more numerous in the long allele; further investigations are underway to corroborate this finding. The situation changes completely in DNA from tumor cells, where the CpG islands of the distal genes RBM11 and ABCC13 acquire significant methylation whereas methylation decreases in proximal genes TPTE and BAGE. The probability that methylation difference between normal and carcinoma tissue occurs by chance is 0.047 for RBM11, 0.005 for ABCC13, and 0.003 for BAGE (t-test). Interestingly, methylation of the two distal genes STCH and NRIP1, which lie farther, does not change in tumor cells. Sequencing of individual clones from bisulfite-treated and PCR-amplified genomic DNA confirmed the COBRA data (Fig. 3; Supplemental Table V). Our results show that in normal somatic cells, CpG islands in the proximal region are highly methylated, while CpG islands distal from the heterochromatic peak are free of methylation. This situation is clearly different in cancer cells.
Chromatin-decompressing drugs have an effect on proximal genes but not on distal genes
We hypothesized that drugs that repress chromatin-modifying enzymes whose activity is necessary for heterochromatinization would activate transcription of genes in the putative heterochromatic domain (TPTE, BAGE2, and ANKRD21), whereas euchromatic genes (RBM11 and ABCC13) would not be affected. Four cancer cell lines with different expression patterns were chosen: two breast cancer and two melanoma-derived cell lines. Cells were treated with trichostatin A (TSA), an inhibitor of histone deacetylase, and with 5-aza-cytidine (5aC), an inhibitor of DNA methyltransferase. One melanoma cell line that did not show transcription from any of the investigated genes before treatment showed transcription for all putative heterochromatic genes after TSA treatment. To exclude the possibility that gene activation is due to trans-acting factors, cell lines were incubated with TSA and cycloheximide (CX), an inhibitor of protein synthesis, and with TSA and hydroxyurea (HU), an inhibitor of replication. Neither co-incubation had an adverse effect on gene activation, indicating that inhibition of histone deacetylation alone is sufficient for gene activation and is independent of protein or DNA synthesis. DMSO, an unspecific gene activator, served as control. The treatment with TSA had the most pronounced effect on transcription patterns: TPTE transcription was induced in two of three cell lines. BAGE2 was activated in all BAGE-negative cell lines (three out of three). Transcription strength depended on TSA concentration (data not shown) and incubation time (Fig. 4). ANKRD21 was activated in the only nonexpressing cell line (SK23-MEL). RBM11 was expressed in only one of four cell lines (BT474), and ABCC13 could not be activated in any of the cell lines. Inhibition of DNA methylation with 5aC induced gene expression from BAGE and TPTE in SK23-MEL. COBRA analysis of the CpG islands confirmed DNA methylation decrease in this cell line (TPTE ± 0%; BAGE, −9%; RBM11, −4%; ABCC, −6%). Epigenetic states must, by definition, be heritable, and we therefore analyzed in one cell line transcription 30 d after removal of the drugs (Fig. 4). Two of three proximal genes, BAGE and TPTE, were still transcriptionally active, confirming the epigenetic nature of control of gene expression in this region. Taken together, our results show that the proximal genes can be activated by chromatin-decondensing drugs, whereas distal genes, in general, cannot.

(A) Bands of RT-PCR products in agarose gels after electrophoresis and staining with ethidium bromide. Four different cell lines were incubated with the drugs indicated above each panel. A testis cDNA library served as positive control. Genes are indicated on the left side. On the right side of the upper panel are enzymes that were used to identify loci on chromosome 21; on the right side of the lower panel is the size of PCR products. (B) Kinetics of gene induction by TSA: The x-axis is the duration of treatment with 20 μM TSA in hours; y-axis is relative transcription strength determined by quantitative RT-PCR (ng RNA target/ng RNA ABL1).
Discussion
Despite the growing knowledge of chromatin structure in mammalian cells, how this chromatin structure is precisely organized along the chromosomes remains largely unknown. Here, we describe the linkage disequilibrium pattern, chromatin structure, CpG methylation, and gene expression of a chromosome region linking a constitutive heterochromatic domain to a euchromatic region.
Linkage disequilibrium analysis showed that the recombinationally inert proximal domain, subjected to the centromere repressive effect, extends 719–737 kb from the α-satellite block in chromosome 21q.
Independently, histone modification mapping revealed a peak of heterochromatic histone isoforms 715–822 kb from the α-satellite. These concurrent frontiers strongly indicate that the distal boundary of juxtacentromeric heterochromatin lies within a short region (<100 kb), presenting probably a highly condensed chromatin structure. Interestingly, this peak is conserved in various human cell lines and in primary cells, but not in a monochromosomal hybrid cell line (WAV17) containing human chromosome 21 in a background of mouse chromosomes (data not shown). This finding suggests that the heterochromatin peak is maintained through a species-specific mechanism. Species specificity was also observed in other examples of chromatin organization: The human β-globin locus, for instance, lacks the heterochromatic block that is flanking transcriptionally active chromatin present in the chicken β-globin locus (Labrador and Corces 2002). Given that high levels of H3K9Me3 and H3K27Me characterize heterochromatin in human centromeric regions, their absence in the proximal 700-kb domain of chromosome 21 is unexpected, but not contradictory, as the previous analyses were done using a cytogenetic level of resolution. Moreover, we showed that proximal genes could be activated by treatment with TSA, a drug that inhibits histone deacetylation, while distal genes could not. Therefore, while little H3K9Me3 is present in this 700-kb region, the reaction to TSA implies that other histone modifications that require deacetylation are present in this most proximal zone. We suggest that the most proximal 700-kb region is indeed heterochromatic but that this juxtacentromeric heterochromatin presents histone modifications different from those in canonical heterochromatin of the more proximal α-satellite DNA. Further experiments are needed to establish the exact nature of these features. Obvious candidates to which this chromatin mapping analysis should be extended include mono-and dimethylation of Lys9 of H3, marks of facultative heterochromatin in mouse cells, trimethylated Lys20 of H4, and HP1α, the hallmark of heterochromatin. Since mammalian juxtacentromeric heterochromatin formation is also dependent on RNA species (Maison et al. 2002), it will be necessary to screen the model region for sites of active transcription in intergenic regions and their response to TSA treatment.
In further support of a juxtacentromeric heterochromatic domain spanning 700 kb, we showed that in normal somatic cells, CpG islands are highly methylated within this proximal region, whereas CpG islands are free of methylation in the distal region. The density of known CpG islands in this region necessarily limited the resolution of our analysis. We have analyzed all known promoter CpG islands, but the discovery of further CpG islands might allow for a more detailed study in the future. Hypomethylation of the proximal loci occurs only in cancer cells. Since the location of the peak of heterochromatic histone isoforms on chromosome 21 is identical in cancer and normal cells, the most likely explanation is that in cancer the DNA methylation machinery is uncoupled from the system regulating the chromatin structure.
Taken together, four independent lines of evidence (histone modifications, DNA methylation, transcription, and recombination) point toward the same location as a frontier between heterochromatin and euchromatin. It is, for the moment, not clear by which mechanism its location is defined. The frontier coincides neither with the border between the duplicated and the single copy genomic sequence nor with any of the repeat clusters that we previously described in the region (Brun et al. 2003): (1) a 56-kb DNA stretch containing 89% LTR elements located 900–1000 kb from the α-satellite, and (2) a 100-kb DNA stretch that contains HSREP522 elements and CAGGG array clusters and that is located very close to the transition between the duplication-rich and the single-copy sequence. This finding suggests that clustering of repetitive sequences is not responsible for the constitution of the heterochromatic peak. Alternatively, boundary elements may define the frontier. Boundary elements, responsible for the separation of chromatin forms, have been identified in a number of organisms. CTCF binding sites are the best-analyzed example of boundary elements in mammalian systems. For the β-globin locus, Burgess-Beusse et al. (2002) showed that CTCF colocalizes with a peak of histone H4 acetylation and it was proposed that the recruitment of histone acetylation activity prevents the spreading of heterochromatin through the boundary element. We used an online bioinformatics tool for the prediction of CTCF binding sites but found no indication of the presence of the corresponding boundary element (data not shown). However, the analogy of the observed H3 methylation peak with the CTCF-associated H4 acetylation peak is striking, and it might well be that increased histone methylation activity is actually shielding the centromeric heterochromatin from the euchromatin. Our findings fit equally well into the picture that is emerging from investigations using the mardel(10) neocentromere. Using ChIP, Chueh et al. (2005) observed peaks of CENP-A enrichment and concluded that CENP-A binding is not homogeneously distributed along the neocentromere but is rather organized in clusters of up to 52 kb, roughly corresponding to the size of the observed H3 methylation peak and maybe hinting at a functional unit size. The same neocentromere was recently analyzed for its DNA methylation: Wong et al. (2006) showed that after centromere formation, DNA methylation of CpG islands sharply decreases at the frontier of the neocentromere. This corresponds to what we observed in the case of the natural centromere of chromosome 21.
In summary, our findings show that the juxtacentromeric region of the long arm of chromosome 21 is composed of a proximal compartment that is separated from the euchromatic compartment of the chromosome arm by a cluster of histone isoforms H3K9Me3 and H3K27Me 715–822 kb from the centromeric α-satellite block. CpG islands in the proximal compartment are densely methylated, recombination frequency is very low, and in normal somatic cells transcription is repressed. In addition, our data establish the PSV-based mapping strategy as an accurate method to determine the epigenetic status of juxtacentromeric regions rich in segmental duplications. The method provides a better resolution than cytogenetic approaches. Since our strategy can be applied to any juxtacentromeric candidate region, further insight into the factors that determine the location of the heterochromatin boundary will come from the mapping of other human heterochromatin–euchromatin frontiers.
Methods
Cell culture and tissue material
Cell lines SK23-MEL, LB23-MEL, and LB1622-MEL DNA were a gift of Pierre van der Bruggen (Ludwig Institute for Cancer Research, Brussels, Belgium) and Elfriede Nößner (National Research Center for Environment and Health, Munich, Germany). Cell lines BT20 and BT474 were a gift of Charles Theillet (EMI 229 INSERM, Montpellier, France), and primary foreskin fibroblasts were a gift of Jean-Pierre Molès (IURC, Montpellier, France). Cell lines were maintained in DMEM-Glutamax (Gibco) with 10% fetal calf serum and penicillin/streptomycin. OPM-2 cells were a gift of Brigitte Sola (Université de Caen, France). These cells were grown in RPMI 1640 medium with 10% fetal calf serum, 2 mM L-glutamine, penicillin/streptomycin. Normal tissue material came from routine autopsies at the Institute of Pathology of the Friedrich Schiller University (Jena, Germany). DNA of the ovarian carcinoma F, and sperm 3 and 4 were a gift of Melanie Ehrlich (Tulane Medical School, New Orleans, LA). CEPH DNA was provided by Gilles Vergnaud (IGM, Orsay, France).
Design of chromosome-specific markers within genomic duplications
Copy number of 5-Kb windows along the most proximal sequence contigs of 21q (AL163202; AL163203; AL163204; AP001660) was estimated using the paralogy detector tool at http://humanparalogy.cwru.edu/eichler/celera5 as published (Bailey et al. 2002). Regions that had less than four copies per haploid genome were selected. Sequences without dispersed repeats and at least 600 bp long were extracted from these regions using RepeatMasker (http://repeatmasker.org/). Copies of these sequences were retrieved by BLAST analysis against finished (nr) and unfinished (htgs) human BAC sequences. Multialignment of copies showing >90% nucleotide identity was produced using ClustalW (http://www.infobiogen.fr/services/analyseq/cgi-bin/clustalw_in.pl). When no other reliable chromosome assignation annotation was available, sequences with >99.5% nucleotide identity (disregarding tandem repeats polymorphisms) over >10 kb were considered allelic copies, and, conversely, sequences showing <98.5% identity were considered ectopic copies. Paralogous specific variant bases creating restriction differences between ectopic copies were found by visual inspection of the multialignment aided with a restriction difference-seeking tool at http://insilico.ehu.es/restriction/two_seq/. Primers were designed using the BeaconDesigner 2.0 program (PREMIER Biosoft International). The predicted chromosome location of designed amplicons was experimentally verified with an extended chromosome mapping DNA panel. This includes a commercially available rodent/human fusion hybrid cells mapping panel (mapping panel 2.3, Corriel Cell Repository) plus a supplemental set of DNA preparations from six such hybrid cell lines, each containing another human chromosome for pairs 13 (GM11689), 14 (GM104798), 15 (GM11715), 21 (GM08854), and 22 (GM13258).
When both allelic and paralogous sequences were available, SNPs could be distinguished from PSVs by visual inspection of ClustalW multialignment between all copies. We used three methods for genotyping duplicated SNPs: (1) PSV-specific PCR followed by allelic RFLP genotyping (PSV − PCR + RFLP; 11 cases), (2) PCR amplification combining a PSV-specific primer on one strand with an allele-specific primer on the other strand (PSV/AS-PCR; nine cases), and (3) restriction digest of genomic DNA using a paralogous restriction variant followed by PCR amplification of the uncut 21q11 copy and RFLP genotyping (three cases). Primer sequences, PCR conditions, and restriction enzymes used are shown in Supplemental Table II. All PCR reactions were performed in 5 μL of 45 mM Tris-HCl (pH 8.8), 11 mM ammonium sulfate, 4.5 mM MgCl2, 6.7 mM β-mercaptoethanol, 4.4 μM EDTA (pH 8.0), 1 mM each dNTP, and 113 μg/mL BSA (Jeffreys et al. 1990). Restriction digests were performed in 10 μL with 4 U of enzyme for 2 h in buffers and at the temperature recommended by the manufacturer. PCR or restriction digest products were separated on 2% 0.5 × TBE agarose gels, and genotypes were called manually for 87 individuals among the 90 Europeans used in the HapMap project (CEPH-CEU-HapMap panel). Amplification and RFLP genotyping of PSV-based digested genomic DNA produced callable genotypes but with spurious background, shadow bands most likely arising from amplification of incompletely digested paralogous copies. Linkage disequilibrium coefficients were estimated and plotted from genotypes of 59 unrelated individuals (unphased haplotypes) by JLIN (McCaskie et al. 2005; Carter et al. 2006). CEPH-CEUHapMap genotypes available in dbSNP for six SNPs of 20 were retrieved and compared with genotypes established by a PSV-specific approach. Two discrepancies were found among 238 pairs of established genotypes for three SNPs typed with PSV − PCR + RFLP and nine discrepancies among 240 pairs were found for three SNPs typed with PSV/AS-PCR. Retyping these 11 events showed three typing errors in each of two SNPs typed with PSV/AS-PCR, reducing to five the number of confirmed discrepancies between the two sets of data. Twenty-five HapMap SNPs with Het > 0.4 and average spacing of 40.1 kb along the 21q11 region were selected, and their linkage disequilibrium coefficients, previously calculated from phased haplotypes, were retrieved from the HapMap database and plotted using Haploview.
Treatment of cell lines with Trichostatin A, 5-aza-cytidine, and other drugs
Pilot studies were conducted to determine optimal concentrations of 5-aza-cytidine (5aC) (Sigma, A2385) and Trichostatin A (TSA) (Sigma, T8552) (1–10 μM 5aC, 20 nM–20 μM TSA) and to establish optimal incubation times (6 h–5 d 5aC, 2 h–24 h TSA). Eventually, 5aC (5 μM) was used for 3 d; culture medium was changed every day and supplemented with fresh 5aC. Incubation times and TSA concentration, necessary for transcriptional activation, were found to be relatively high (24 h and 20 μM TSA). Since TSA is suspected to be unstable, we wondered whether the need for high initial TSA concentration stems from the degradation of the drug during incubation. We therefore added TSA to a final concentration of 20 nM every 2 h for 24 h. We observed the same effect as with 20 μM starting concentration and concluded that the requirement for high initial TSA concentrations is indeed due to a successive degradation of the drug (Fig. 4). For ease of use, the incubation with 20 μM TSA for 24 h was maintained for subsequent experiments. The medium was not changed. Hydroxyurea (Sigma, H8627) was used at 2 mM; cycloheximide (Sigma, C7698), at 30 mg/L; and DMSO (Sigma, D8418), at 2%.
RNA extraction and RT-PCR
Total RNA was extracted with TriZOL (Invitrogen) and, if necessary, melanin was removed with a GenElute Mammalian Total RNA Miniprep kit (Sigma, RTN-10) to avoid inhibition of DNA synthesis. RNA was quantified by spectrophotometry (Eppendorf BioPhotometer), and 2 μg were used for cDNA synthesis with Omniscript RT (Qiagen) in the presence of 10 U RNAsin (Promega) in 20-μL reaction volumes. The cDNA was purified with a Qiagen PCR cleanup kit and eluted in 50 μL of 10 mM Tris-HCl. One microliter was used as a template for PCR in 45 mM Tris-HCl (pH 8.8), 11 mM ammonium sulfate, 4.5 mM MgCl2, 6.7 mM β-mercaptoethanol, 4.4 μM EDTA (pH 8.0), 1 mM each dNTP, 113 μg/mL BSA (Jeffreys et al. 1990), 2 pmol of each primer, and 0.5 U Taq-polymerase (Promega) in a total volume of 10 μL. PCR conditions are given in Supplemental Table VI. Paralogous sequence variants generating diagnostic restriction sites were used to identify transcription from genes on chromosome 21. RT-PCR products were digested with restriction enzymes that leave the chromosome 21-specific cDNA intact (Supplemental Table VI), and fragments were separated by electrophoresis.
Quantitative RT-PCR
One microgram of RNA was reverse transcribed using the Quanti Tech Reverse Transcription kit (Qiagen, 205311) according to the manufacturer's instructions. The cDNA was purified with the Qiaquick kit (Qiagen, 28704) and dissolved in 30 μL of 10 mM Tris/HCl.
Expression was quantified using a MyIQ PCR Biorad thermal cycler and the QPCR Rox-&Go Green kit (MP Biomedicals, EPQON480); PCR reactions were done in 25 μL final volume containing 400 nM of each primer and 1 μL of each template DNA (equivalent to 33 ng of RNA). PCR conditions were the following: 95°C for 15 min, followed by 50 cycles of 95°C 30 sec, 60°C 30 sec, and 72°C 1 min. We used the housekeeping ABL1 gene to normalize cDNA amounts. Primers are listed in Supplemental Table VI. Each analysis was done in triplicate, and assay efficiencies were calculated using cDNA dilution series. To calculate gene expression we used the relative standard curve method.
Since primers that amplify BAGE and ANKRD21 were not specific for the genes mapping to chromosome 21, we digested the PCR products with appropriate enzymes to quantify the expression of chromosome 21 genes (see Supplemental Table VI), and we separated the generated fragments on acrylamide or agarose gels. The relative amount of the 21-specific transcripts was estimated by gel image analysis using the ImageJ software (http://rsb.info.nih.gov/ij/).
DNA methylation analysis
Homogeneous tissue or cell culture material (200–500 mg) was washed in sterile water and incubated in 1 mL of buffer (20 mM TRIS at pH 8, 1 mM EDTA, 100 mM NaCl, 0.5% SDS) with 0.3 mg protease K (Boehringer) for 16 h at 55°C. DNA was extracted by phenol-chloroform treatment and precipitated with sodium acetate/isopropanol, washed with 70% ethanol, dissolved in 10 mM Tris-HCl (pH 8), and stored at −20°C. Bisulfite treatment of 300 ng genomic DNA was performed as described by Boyd and Zon (2004), but a higher sodium bisulfite concentration (541 g/L) and a shorter incubation time (4 h) were used (Grunau et al. 2001). DNA was recovered in 50 μL of 10 mM Tris/HCl and stored at −80°C. CpG islands were predicted with CpG island searcher (Takai and Jones 2003) (parameters: %GC ≥ 55, Observed CpG/ Expected CpG ≥ 0.65, length ≥ 200) and primers were designed with MethPrimer (Li and Dahiya 2002). One microliter of bisulfite-treated DNA was used for PCR amplification with nested primer sets (Supplemental Table VII). The specificity of the selected primers for the predicted paralogous loci was confirmed using bisulfite-treated DNA of rodent/human fusion cell lines containing single human chromosomes (Corriel Cell Repository). PCR products were either cloned into pGEM-T-Easy (Promega), and individual clones were sequenced or analyzed using the absence or presence of informative restriction enzyme sites as an indicator of methylation in the original genomic DNA (Combined Bisulfite Restriction Assay [COBRA] [Eads and Laird 2002]) (Supplemental Table VII). Such restriction sites were identified with Snake Charmer (http://methdb.igh.cnrs.fr/cgrunau/methods/snake_charmer.html). Digestion fragments were separated on 2% SB agarose gels (Brody and Kern 2004), and band intensities were quantified with ImageJ. Amplification bias was measured according to Warnecke et al. (1997) by using mixtures of human sperm DNA and Hela DNA, exhaustively methylated with M.SssI. Bias values b were used for the correction of the COBRA data: % methylation = (100 * COBRA score)/ (b * 100 − b * COBRA score + COBRA score) with COBRA scores the percentage of methylation estimated from the digest alone without bias correction.
Chromatin immunoprecipitation
ChIP followed a published protocol (Umlauf et al. 2004). Antibodies were purchased from Upstate: anti-hyperacetylated histone H4(penta) nr.06-946, anti-acetylated histone H3K9 nr.06-942, anti-trimethylated H3K9 nr.07-523 (lot 24,486), and anti-monomethylated H3K27 nr.07-448 (lot 24,439). Two micrograms of antibody were used for immunoprecipitation of 2 μg of DNA. DNA was extracted with phenol/chloroform, precipitated, and dissolved in 20 μL of 10 mM Tris-HCl (pH 7.8). Quantitative real-time PCR amplification was performed in a MyIQ PCR Biorad thermal cycler in 25 μL final volume of IQ SYBR Green supermix (Biorad), 300 nM of each primer, and 1 μL of template DNA. For each locus, a standard curve was established using serial dilutions of purified and spectrophotometrically quantified genomic DNA. Using these curves, we converted the real-time PCR data into nanograms DNA of the corresponding locus associated with each analyzed histone isoform (ng STS(B)). The amplified product was then submitted to adequate restriction digest, followed by electrophoresis into a 2% agarose gel in SB buffer (Brody and Kern 2004). The relative amount of 21q11-specific fragment was estimated by gel image analysis using the ImageJ software. Ratios of precipitated DNA in STS along 21q to that in a reference sequence in the 5′ region of GAPDH were calculated as follows: Enrichment factor = [ng STS(B)/ng GAPDH(B)]/[ng STS(I)/ng GAPDH(I)] with (B) for antibody bound and (I) for input. This ensures that if, unnoticed, a STS were amplified x-fold in the genome, the enrichment factor would not change ([x * ng STS(B)/ng GAPDH(B)]/[x * ng STS(I)/ ng GAPDH(I)] = [ng STS(B)/ng GAPDH(B)]/[ng STS(I)/ng GAPDH(I)]). The unbound fraction of mock-treated chromatin (i.e., precipitation in parallel with the other ChIP but without antibody) was considered as input. All experiments were done in duplicate or triplicate. Cell lines OPM-2, BT20, and BT474 and primary human fibroblasts were used for these experiments.
In silico prediction of boundary elements
We used the online tool (available at http://www.essex.ac.uk/bs/molonc/spa.htm) that allows searching for binding sites of one of the vertebrate insulator proteins, the CCCTC binding factor (CTCF) (Burgess-Beusse et al. 2002). Due to the lack of consensus motifs, prediction strength is limited and does not replace experimental proof.
Statistical analysis
T-test was calculated at http://home.clara.net/sisa/t-test.htm using COBRA data in Supplemental Table V.
ACKNOWLEDGMENTS
This work was supported by grants of the Association pour la Recherche sur le Cancer (ARC) and Ligue contre le Cancer to A.D. and grants of the Fondation Lejeune to J.B. and A.D. C.G. was financed in part by a European Grant HPRN-CT-2000-00089. We are grateful to Anne-Marie Laurent for excellent technical assistance, to Russell Eberhardt and Gary Burkhart for carefully reading the manuscript, and to the anonymous reviewers for valuable comments.
Footnotes
-
↵2 Present address: Centre de Biologie et d'Ecologie Tropicale et Méditerranéenne, UMR 5555 CNRS-UPVD, 66860 Perpignan, France
-
↵3 Corresponding author.
↵3 E-mail albertina.de-sario{at}igh.cnrs.fr; fax +33-4-99-61-99-01.
-
[Supplemental material is available online at www.genome.org. DNA methylation data are available at MethDB (http://www.methdb.net) under sequence IDs 14048–14053.]
-
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.5440306.
-
- Received April 26, 2006.
- Accepted August 2, 2006.
- Copyright © 2006, Cold Spring Harbor Laboratory Press








