
Uneven distribution of expressed sequence tag loci on maize pachytene chromosomes
Abstract
Examining the relationships among DNA sequence, meiotic recombination, and chromosome structure at a genome-wide scale has been difficult because only a few markers connect genetic linkage maps with physical maps. Here, we have positioned 1195 genetically mapped expressed sequence tag (EST) markers onto the 10 pachytene chromosomes of maize by using a newly developed resource, the RN-cM map. The RN-cM map charts the distribution of crossing over in the form of recombination nodules (RNs) along synaptonemal complexes (SCs, pachytene chromosomes) and allows genetic cM distances to be converted into physical micrometer distances on chromosomes. When this conversion is made, most of the EST markers used in the study are located distally on the chromosomes in euchromatin. ESTs are significantly clustered on chromosomes, even when only euchromatic chromosomal segments are considered. Gene density and recombination rate (as measured by EST and RN frequencies, respectively) are strongly correlated. However, crossover frequencies for telomeric intervals are much higher than was expected from their EST frequencies. For pachytene chromosomes, EST density is about fourfold higher in euchromatin compared with heterochromatin, while DNA density is 1.4 times higher in heterochromatin than in euchromatin. Based on DNA density values and the fraction of pachytene chromosome length that is euchromatic, we estimate that ∼1500 Mbp of the maize genome is in euchromatin. This overview of the organization of the maize genome will be useful in examining genome and chromosome evolution in plants.
Maize (Zea mays ssp. mays) is both a genetic model and an economically important crop. Further progress exploiting maize as a model and a crop will be greatly aided by obtaining its complete genome sequence. However, the maize genome is large (2365 Mb vs. the 450-Mb rice genome) (Rayburn et al. 1993; Messing et al. 2004) and consists of ∼60%-80% repetitive sequence (Flavell et al. 1974; Hake and Walbot 1980; Messing et al. 2004). Both of these features make sequencing and assembling the entire maize genome difficult. Methods that concentrate on sequencing the gene space promise to provide important information on genetically active regions of the maize genome (Palmer et al. 2003; Whitelaw et al. 2003; Yuan et al. 2003) but, by their very nature, will not elucidate large-scale genome organization.
To date, several strategies have been used to provide insights into maize genome organization. These include the use of high-density genetic linkage maps (Davis et al. 1999; Cone et al. 2002; Sharopova et al. 2002), methylation filtration and high Cot selection to sequence the gene space (Palmer et al. 2003; Whitelaw et al. 2003; Yuan et al. 2003), and DNA contigs constructed from overlapping BACs (Coe et al. 2002; Cone et al. 2002). Many of these BAC contigs have been anchored by genetic markers to a commonly used linkage map (IBM2 Neighbors; http://www.maizegdb.org) and are useful both for estimating physical distances between genetic markers and as a resource for sequencing efforts. For example, Messing et al. (2004) used BAC contigs to organize ∼475,000 BAC end sequences that covered 13% (307 Mb) of the maize genome. From this snapshot of the genome, they estimated that there are ∼59,000 genes in maize, many of which are present in tandem arrays.
Sequence analysis of several genomic BAC-inserts has revealed that maize genes tend to be organized into clusters that are separated by large blocks of retrotransposons (SanMiguel et al. 1996; Keller and Feuillet 2000; Fu et al. 2001; Song et al. 2001; Walbot and Petrov 2001; Wicker et al. 2001; Rostoks et al. 2002). Because retrotransposons often insert into older retrotransposons, the “clumpiness” of genes within the genome can be further accentuated by successive rounds of retrotransposon amplification. Over time, retrotransposition, along with other mechanisms, can lead to extreme changes in gene spacing (Walbot and Petrov 2001). This process appears to be relatively rapid, because limited genomic sequencing has revealed differences in gene spacing, relative location, and even gene presence among maize inbred lines (Fu and Dooner 2002; Song and Messing 2003; Brunner et al. 2005; Lai et al. 2005). It is not yet clear, however, if the gene clustering evident in BAC-inserts is also evident on a chromosomal scale in maize.
Several methods have been used to bridge the gaps among the physical (DNA sequence), genetic (linkage maps), and cytological (chromosome) aspects of maize genome structure. The most straightforward technique is to locate genetically mapped markers onto maize chromosomes by using fluorescence in situ hybridization (FISH) (Harper and Cande 2000; Sadder et al. 2000; Sadder and Weber 2002; Koumbaris and Bass 2003). However, this method is technically difficult, which limits the type and number of probes that can be hybridized. Another method utilizes oat-maize addition lines and γ irradiation to introduce discrete maize chromosomal segments into an oat background. Once such radiation hybrids are produced, one can easily assay the presence or absence of markers on each maize chromosomal segment (Riera-Lizarazu et al. 2000; Kynast et al. 2004). This method is limited by the resources required to produce the large number of radiation hybrid lines necessary to segment the maize genome at a high level of resolution.
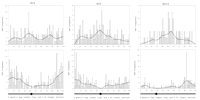
The distribution of ESTs from maize chromosomes 1, 5, and 10 in 1-cM bins on genetic linkage maps (top panel) and in 0.2-μm length bins on the physical structure of pachytene chromosomes (bottom panel). Underneath each graph is a representation of the pachytene chromosome (SC) with the short arm to the left and a circle marking the centromere. The genetic and chromosome maps of all three chromosomes are drawn to the same percentage scale. The genetic lengths have been normalized to fit the shorter RN map; otherwise the marker positions are the same as the IBM2 neighbors frame maps. The curve superimposed on each graph is a lowess smoothing function that shows the general trends of the distributions. For similar distributions for the other seven chromosomes, see Supplemental Figure 1.
A third approach is to map genes on maize pachytene chromosomes by relating linkage maps to the distribution of recombination nodules (RN-cM map) (Anderson et al. 2003, 2004). Recombination nodules (RNs) are multiprotein structures that mark crossover sites on synaptonemal complexes (SCs) at pachytene (for review, see Anderson and Stack 2005). The location and frequency of RNs on SCs provides a direct connection between genetic linkage maps and chromosome structure, and previous work has shown that one can accurately predict the chromosomal position of genetically mapped markers by using the maize RN-cM map (Anderson et al. 2004). This method is limited by the resolution of the RN-cM map, the number of genetically mapped markers, and the extent to which recombination patterns and gene locations among different maize lines are conserved. However, the advantage of this approach is that the RN-cM map is independent of, but related to, the genetic map so that every genetically mapped marker (and any markers mapped in the future) can be placed onto the physical structure of pachytene chromosomes in maize.
Here, we use the RN-cM map to estimate the positions of 1195 genetically mapped EST markers on the 10 pachytene chromosomes of maize. With estimates of the physical location of ESTs, we address these questions: (1) What is the relationship between the genetic linkage map and the physical (chromosomal) map of EST markers? (2) How are heterochromatin and euchromatin related to EST distribution? (3) What is the relationship between EST density and recombination rate along maize chromosomes? And (4) are ESTs clustered at the chromosomal level in maize?
Results
Distribution of ESTs on maize chromosomes
We used the RN-cM map developed in the inbred line KYS (Supplemental Table 1) to place 1195 ESTs from the IBM2 neighbors linkage map onto the physical structure of pachytene chromosomes (Fig. 1; Supplemental Table 2). We chose the IBM2 neighbors map because it has the largest number of genetically mapped EST markers. This linkage map incorporates several different genetic linkage maps from maize crosses of different backgrounds, including the UMC98 map (Lawrence et al. 2004; http://www.maizegdb.org). Previously, we showed that the RN-cM map accurately predicted the physical location on chromosome 9 of seven markers from the UMC98 linkage map (Anderson et al. 2004). To verify that the IBM2 neighbors map gave similar results, we compared the SC9 marker positions predicted from both linkage maps and found a very close relationship between the two (y = 0.99x–0.19, r2 = 0.997).
The distribution of markers along the IBM2 neighbors genetic linkage map differs substantially from their physical distribution on the chromosomes (Fig. 1; Supplemental Fig. 1). On the genetic maps, ESTs tend to cluster in regions of low recombination, particularly around centromeres. However, when the ESTs are mapped onto the physical structure of chromosomes, most ESTs shift to the distal regions of the chromosome arms, with relatively few markers around the centromeres. For example, a total of 428 (36%) ESTs were observed in the distal 20% of the chromosome arm lengths. This number is almost double what is expected if ESTs are distributed evenly along the lengths of the chromosomes. Conversely, only 84 (7%) ESTs were observed in the proximal 20% of the chromosome arm lengths.
Euchromatin and heterochromatin of maize pachytene chromosomes
We estimated the DNA density in euchromatin and heterochromatin and the positions of euchromatin/heterochromatin boundaries for each pachytene chromosome from the inbred line KYS by using chromosome squashes that had been stained with the fluorescent dye propidium iodide (PI) (Fig. 2; Supplemental Table 3). PI is a quantitative stain for DNA after double-stranded RNA has been removed (Noirot et al. 2002). The average area (PI staining intensity × width) of the fluorescence signal in euchromatin was 419 units (SD, 94) compared with 586 units (SD, 183) in heterochromatin. Therefore, the DNA density in heterochromatin is ∼1.4 (586/419) times that of euchromatin.
We were not able to accurately determine the DNA density of knobs directly because the intense PI staining of knobs saturated the digital camera, but we estimated the contribution that knobs make to total genome size indirectly. It appears that much of the variation in genome size among maize lines is attributable to knobs (Rayburn et al. 1985; Kato et al. 2005). Comparing the genome size of KYS (2700 Mbp) to a knob-less line (TKF, 2460 Mbp) suggests that ∼240 Mbp of the KYS genome is attributable to knobs (Rayburn et al. 1985, 1993; Ananiev et al. 1998). The inexact nature of these estimates is apparent since B73, the inbred line being used for whole-genome sequencing, has two small knobs but a smaller genome size (2365 Mbp) than does the knobless line (2460 Mbp). With this caveat, these data suggest that ∼9% (240 Mbp) of the KYS genome is present in knobs, with 55% (1474 Mbp) in euchromatin and 36% (986 Mbp) in pericentric heterochromatin (Supplemental Table 3).
Of the 1195 ESTs we analyzed, 1037 (87%) of the ESTs were in euchromatin, resulting in a density of about one EST per 1.4 Mbp in euchromatin. In comparison, 158 ESTs were in pericentric heterochromatin or about one EST per 6.2 Mbp. Based on this limited data set, gene density is about fourfold higher in euchromatin than heterochromatin. If maize has 59,000 genes (Messing et al. 2004) and the genes are distributed in the same manner as the ESTs analyzed here, then euchromatin is expected to have ∼51,199 genes per 1474 Mbp or an average of one gene per 28.8 kbp.
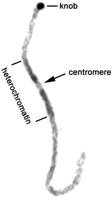
Reversed image of propidium iodide-stained chromosome 9 from a squash preparation. The positions of the centromere, knob, and heterochromatin/euchromatin boundaries are marked. The average amount of DNA per unit of pachytene chromosome length is ∼1.4 times higher for heterochromatin compared with euchromatin.
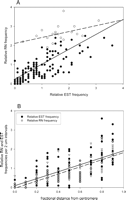
Relationship between the relative frequencies of ESTs and RNs on pachytene chromosomes. (A) For all 2-μm chromosome intervals. Open circles indicate telomeric intervals, and filled circles indicate all the other intervals. There is some overlap so not all points are visible in the graph. The regression line for all points is solid (y = 0.78x + 0.25, r2 = 0.47), and the regression line for the telomeric intervals only is dashed (y = 0.31x + 2.1, r2 = 0.20). (B) As a function of fractional arm distance of the interval from the centromere. Data for all 20 arms are pooled together, but the telomeric intervals have been omitted. The regression line for ESTs (filled circles) is solid (y = 1.81x + 0.10, r2 = 0.44), and the regression line for RNs (open triangles) is dashed (y = 1.86x–0.04, r2 = 0.64).
Recombination rate and EST frequency
We compared the relative frequency of ESTs and RNs in 2-μm arm length intervals for each maize chromosome (Fig. 3A). We used relative values because the number of observations of ESTs and RNs was not the same for each chromosome. Relative RN frequency is closely related to the relative frequency of ESTs per interval (r2 = 0.47). Eight of the 159 points used in this regression analysis were identified as outliers by residual analysis. Further examination showed that these “outlier” points were chromosomal intervals with many fewer ESTs than predicted from the regression equation based on the number of RNs. On the chromosomes, these outliers were the most distal (telomeric) intervals on the short arms of chromosomes 4, 6, 8, 9, and 10 and on the long arms of chromosomes 2, 5, and 8. Further examination revealed that the remaining 12 telomeric intervals were also located above the regression line. So, even though RN and EST frequencies are strongly correlated for the genome as a whole, there are conspicuous exceptions, especially near telomeres.
We then evaluated relative EST and RN frequencies per interval as a function of chromosomal position. Each 2-μm interval was converted to a fraction of chromosome arm length from the centromere, and the data for all 20 arms (except telomeric segments) were plotted together (Fig. 3B). Both EST and RN frequencies were low near the centromeres and progressively increased toward the distal ends of the chromosome arms. The slopes of the two regression lines were not significantly different from one another (P > 0.2), indicating a close relationship between relative EST and RN frequencies as a function of their location along chromosome arms.
EST clustering
We tested whether ESTs are evenly distributed along SCs, as expected for a uniform distribution, or whether they are clustered. We performed this test at two scales. The smaller scale, 0.2-μm SC length, is based on the highest resolution available from the RN-cM map. For this test, we applied a permutation procedure (see Methods). As shown in Table 1, each chromosome exhibited significant deviation from a uniform distribution. This result was expected since it is well known that heterochromatin has few genes compared with euchromatin. We then tested only the euchromatic regions of each chromosome. Again, all chromosomes were significantly different from random expectation at P < 0.05 (SC 10) or P < 0.01 (all other SCs). For the second scale, we pooled the EST data into 2.0-μm SC length segments. At this scale the number of observations per segment was sufficient to use the χ2 approximation. This test also showed that the distribution of ESTs was significantly different from uniform expectation for whole chromosomes (P < 0.005) as well as for euchromatic chromosome segments (P < 0.001). Altogether, our analyses at two different interval sizes indicate that genes are clustered both along entire chromosomes and within euchromatic regions.
Testing for EST clustering on maize chromosomes using 0.2-μm SC length segments
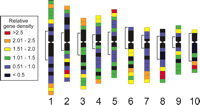
Gene clustering on maize chromosomes. Each chromosome has been divided into 2-μm intervals except where chromosome arm lengths are not evenly divisible by two. In those cases, the intervals around the centromeres are somewhat different from 2 μm. The color of each segment corresponds to its gene density ratio (observed gene density divided by expected average gene density for that chromosome), as provided in the key. The heterochromatin boundaries on either side of each centromere are indicated by brackets to the left of each chromosome.
To show more clearly the amount and location of gene clustering on maize chromosomes and to facilitate comparison with the wheat EST distribution data set (Qi et al. 2004), we determined the ratio of observed to expected EST frequency for each 2-μm segment, assigned a color code to the different values, and graphed the results on the 10 pachytene chromosomes (Fig. 4). As expected, segments near centromeres and within pericentric heterochromatin generally had lower EST densities than expected if genes were evenly distributed along chromosomes (indigo segments). However, there were a number of cases in which heterochromatic chromosomal segments had average (blue segments) to slightly above average (green segments) EST densities. In euchromatic regions of the chromosomes, the observed EST density of chromosomal segments varied over the entire range of values from ratios of less than one-half (indigo segments) to >2.5 times (red segments) the average gene density.
Discussion
Here we have addressed several questions relating to gene distribution and overall genome structure at the chromosomal level in maize. To do this, we estimated the positions of ∼1200 EST markers on pachytene chromosomes by using the RN-cM map and the IBM2 neighbors frame maps. These maps were generated by using different inbred lines. While differences among inbreds in intergenic lengths and gene locations have been reported at the DNA sequence level (Fu and Dooner 2002; Song and Messing 2003; Brunner et al. 2005), it is unclear how much this affects our results. Given the good correspondence between the predicted and observed locations of the chromosome 9 markers using different maps and inbred lines (Anderson et al. 2004), as well as the difference in resolution between DNA sequence and chromosome structure, it is likely that most of the sequence differences among inbreds reported to date would have little effect on the overall pattern of gene distribution that we have observed.
Chromosomal structure
In examining genome structure in relation to pachytene chromosomes, there are four obvious chromosomal landmarks: euchromatin, heterochromatin, centromeres, and telomeres. The karyotype of maize pachytene chromosomes has been well documented with regard to arm ratios and relative lengths (Freeling and Walbot 1994; references in Anderson et al. 2003). However, to our knowledge the relative positions of euchromatin-heterochromatin borders and the amount of DNA in euchromatin compared with heterochromatin have not been evaluated before. By using 2700 Mbp as the size of the KYS genome along with the chromosome length and DNA density of heterochromatin compared with euchromatin, we estimate that 1474 Mbp (55%) of the maize KYS genome is in euchromatin, 986 Mbp (36%) is in pericentric heterochromatin, and 240 Mbp (9%) is in knobs. The estimates for the amount of DNA in euchromatin and heterochromatin may represent basic genomic characteristics for maize because most differences between lines in genome size and karyotype appear to be due to differences in the size and number of knobs (Longley 1939; McClintock et al. 1981; Rayburn et al. 1985; Ananiev et al. 1998; Kato et al. 2005).
How do euchromatin and heterochromatin characteristics of maize compare with other plants? We can most directly compare our data with those from tomato, in which DNA density has also been assessed for pachytene chromosomes stained by using the quantitative Feulgen technique. Proportionally, the maize genome contains more repetitive DNA than does the tomato genome (60-80% vs. 30%) (Peterson et al. 1996, 1998), and the manner in which repetitive and single-copy sequences are arranged in the two plants are different. First, Peterson et al. (1996) found that pericentric heterochromatic segments were 1.23 times wider than were euchromatic segments and that the DNA density in heterochromatin was nearly five times higher than in euchromatin. In comparison, maize pachytene chromosomes differ little in width between heterochromatic and euchromatic regions, and the DNA in pericentric heterochromatin is only 1.4 times as dense as that in euchromatin. Second, the pericentric heterochromatin of tomato contains a large proportion of retrotransposons and repeated sequences but also contains most of the single-copy sequences, including some genes (Peterson et al. 1998; Budiman et al. 2004; Chang 2004; Guyot et al. 2005). Indeed, the majority (77%) of the tomato genome is located in heterochromatin. In contrast, pericentromeric heterochromatin in maize comprises only ∼36% of the genome, with a large proportion of retrotransposons and other repeated sequences (Mroczek and Dawe 2003) as well as substantial amounts of EST coding sequences (∼13% of our total EST sample). Third, the euchromatin of tomato is composed mostly of single-copy sequences with only a few retrotransposons (Chang 2004), while maize euchromatin harbors considerable amounts of retrotransposons (Meyers et al. 2001; Mroczek and Dawe 2003). Finally, crossing over is severely repressed in the pericentric heterochromatin of tomato (Sherman and Stack 1995), whereas crossing over occurs in maize heterochromatin, albeit at reduced levels (Fig. 1). Overall, this comparison between maize and tomato highlights remarkable differences among plant genomes in organization, chromosome structure, and recombination.
EST distribution on chromosomes
By sequencing genomic fragments of DNA, a number of investigators have shown that genes in maize and other cereals are clustered (Ware and Stein 2003). Work in barley and wheat indicated that genes are also clustered at the chromosomal level (for review, see Sandhu and Gill 2002; Erayman et al. 2004; Qi et al. 2004). We were interested in determining whether EST clustering could be detected at the chromosomal level in maize and, if so, how the clustering related to recombination patterns and chromosome structure. After placing the ESTs onto the structure of pachytene chromosomes, we determined that (1) ESTs are 4.4 times more frequent in euchromatin compared with heterochromatin, (2) ESTs are clustered within euchromatic chromosomal segments, and (3) the degree of EST clustering varies along the length of the chromosomes.
We recognize that the distribution of ESTs along maize chromosomes may not accurately represent the distribution of genes. ESTs can be genetically mapped only when there is a polymorphism between the two parental strains, and studies in several organisms (including tomato, Aegilops, and sea beet) have shown that the level of polymorphism is positively correlated with rate of recombination (see Tenaillon et al. 2002). If this correlation holds in maize, mapped ESTs are likely to be biased toward high recombination regions, contributing to observed correlations between EST and RN frequencies. However, no such correlation between recombination and polymorphism has yet been observed in maize (Tenaillon et al. 2002).
How does EST clustering on maize chromosomes compare to that for other cereals? The most direct comparison is with wheat chromosomes, in which EST density has been determined for bins delineated by deletion mapping (Qi et al. 2004). We have also used published data to develop a comparable bin map for rice chromosome 10 based on predicted genes in the genomic sequence (The Rice Chromosome 10 Sequencing Consortium 2003) and FISH localization of genetically mapped BACs to define bins (Cheng et al. 2001). Rice chromosome 10 is relatively uniform in gene density, with most bins having the expected or just slightly above the expected gene density ratios (Table 2). In contrast, maize and wheat chromosomes vary greatly in gene density along their lengths, with bins ranging from less than half to more than double the expected gene density ratios. Compared with wheat and rice, maize seems to have an overabundance of bins with very low gene frequencies. It is possible that some of these effects are a function of bin sizes, which are unequal among taxa. Smaller bin sizes should give a more nuanced view of gene distribution on chromosomes, and this may account for differences in gene density distribution for wheat chromosomes reported in different studies (cf. Qi et al. 2004 and Erayman et al. 2004). Similarly, smaller bin sizes for maize could also reveal additional variation; nonetheless, our conclusion that maize ESTs are clustered is evident for at least two bin sizes (0.2 μm and 2 μm).
Comparison of the frequency of chromosomal bins with different gene densities for wheat, maize, and rice
EST density and recombination rate
We found strong correlations between relative EST and RN frequencies for 2-μm intervals on maize pachytene chromosomes and also between chromosomal position and both EST and RN frequencies (Fig. 3). Together, these data indicate that recombination is closely associated with ESTs (genes) in maize.
Although EST and RN frequencies are closely related, our analyses also revealed distinctive differences. As a group, telomeric segments tend to have much higher levels of crossing over than predicted from their gene content. This disparity could be caused by procedural errors in merging the EST and RN maps. Similar to most linkage maps, the ends of the IBM2 neighbors maps are the least reliable portions. Because this is the region that has the most crossing over, differences in merging the linkage and RN maps are most likely to be accentuated in the distal, telomeric segments. Another possible source of error is underrepresentation of mapped ESTs in telomeric regions. A priori, it seems unlikely that there would have been systematic selection against ESTs from telomeric intervals since our only criterion for including ESTs in this study was that they were mapped genetically. However, evidence from wheat indicates that distal chromosomal segments have increased numbers of duplicated and derived (specialized) genes compared with the rest of the genome (Akhunov et al. 2003a,b; Dvorak et al. 2004). If this is also true for maize, then duplicate ESTs in telomeric regions may not have been mapped and/or EST libraries prepared from maize plants under “normal” conditions may not have been expressing these more specialized genes. Another possible explanation is that recombination in telomeric segments is regulated differently so that these regions normally have higher crossover rates than expected from their gene densities. Currently, the available data are insufficient to determine whether the altered relationship between ESTs and RNs in telomeric regions is real or an artifact.
In contrast to our results in maize, Akhunov et al. (2003b) found only a weak correlation between crossing over and gene density in their analysis of deletion stocks of hexaploid wheat. One possible reason for the lack of correlation observed by Akhunov et al. (2003b) is that the positions of deletion breakpoints in wheat were estimated from mitotic, not meiotic, chromosomes. Chromosome structure differs significantly at these two stages because the chromosomal length of euchromatin is proportionally much greater in meiotic compared with mitotic chromosomes (Stack 1984). This difference in chromosome structure could have obscured the close relationship that we and others have observed between genes and recombination.
Although we do not yet know all the factors involved in regulating recombination, the frequency and distribution of genes are clearly important features associated with the frequency and distribution of crossing over, accounting for ∼50% of the variation in recombination rates along the maize chromosomes. Previous work has shown that most recombination in plants occurs near or within genes and other low-copy sequences (Schnable et al. 1998; Fu et al. 2002; Wu et al. 2002), and the work presented here represents the first genome-wide support for this model in maize. However, genes are not necessarily recombination hotspots, and nongenic sequences can be hotspots for recombination (Yao et al. 2002). These observations, in combination with our data, strongly suggest that factors in addition to overall gene density are involved in determining recombination sites.
General considerations
Theoretically, ESTs mapped onto pachytene chromosomes should provide a “backbone” for the assembly of BAC contigs into a coherent chromosome sequence. For example, it may be possible to use the physical position of ESTs on chromosomes to estimate the size of gaps separating non-overlapping BAC contigs. Unfortunately, most maize BAC contigs are not yet long enough to characterize carefully the relationship between the physical length of BAC contigs and distances on pachytene chromosomes (data not shown). However, this situation should change rapidly with continued progress in the maize genome sequencing project.
In conclusion, our maize data suggest that (1) our localization of ESTs on pachytene chromosomes provides a more accurate picture of gene distribution than that previously offered by maize genetic maps; (2) despite this improvement, a full genomic sequence of maize will answer important questions about genome organization, particularly in telomeric regions; and (3) even complete genome sequences are not sufficient to understand pachytene chromosome structure and its attendant influences on crossing over and genome evolution.
Methods
Chromosomal mapping of EST markers
The RN-cM map (Supplemental Table 1; Anderson et al. 2004) was used to position a total of 1195 ESTs and genes from the IBM2 neighbors maps (http://www.maizegdb.org; Supplemental Table 2) onto the 10 pachytene chromosomes. Centromere positions on the IMB2 neighbors frame map were estimated by using cent markers for each chromosome from the UMC98 maps.
Euchromatin and heterochromatin: DNA density and position in chromosomes
We fixed anthers in 1:3 acetic ethanol and prepared squashes of pachytene chromosomes. The chromosome squashes were treated overnight with 200 μg/mL RNase A and 333 μg/mL PI in water. Nucleoli no longer stained with PI after this RNAse treatment, indicating that the RNA was completely removed. Experiments in which the squashes were treated first with RNase A then followed by PI staining gave similar chromosome staining results. A total of 76 chromosomes that did not appear to be stretched by the squashing procedure and that could be analyzed along their entire length (i.e., did not overlap much, if at all, with other chromosomes) were identified (Supplemental Table 3; Anderson et al. 2003). The average relative (%) positions of the heterochromatin-euchromatin borders were determined and then applied to our standardized SC karyotype to estimate the average heterochromatic and euchromatic length of each arm in micrometers.
An average DNA density was calculated for heterochromatic and euchromatic regions of the chromosomes by using digital photographs (8-bit) of 13 chromosomes selected based on their separation from all other chromosomes. After correcting for background, area measurements based on the fluorescence signal intensity and the width of the signal across the chromosome were determined at 60-80 sites along the length of each chromosome by using the image analysis program Image-Pro Plus (version 4.5.1). Average DNA density was calculated for heterochromatin and euchromatin based on the area measurements. Centromeres were a consistent size and shape and were stained uniformly, so they were used as an internal control to adjust for any variation in photographic and/or staining parameters. The total euchromatic or heterochromatic length was multiplied by its appropriate relative DNA density to estimate the fraction of each type of chromatin in the genome.
Relationship between EST and RN frequencies
Each chromosome arm was divided into segments 2 μm in length starting from the telomeres. The fraction of any arm length that was not evenly divisible by two was included in the segment closest to the centromere since few ESTs and RNs occur in this region. Because the number of observations for RNs and ESTs was not the same for each chromosome, relative RN and EST frequencies were determined by dividing the total number of observations for each segment by the average number of observations expected for the segment length. Relative RN and EST frequencies were compared by using regression analysis (Minitab version 14).
EST clustering
The average number of ESTs per segment was calculated by dividing the total number of ESTs mapped by the total number of 0.2-μm SC length segments for each chromosome. For each chromosome, we generated 1000 random, uniform distributions of ESTs along the entire chromosome length by using the SAS statistical program. We repeated these same operations for only euchromatic regions of the chromosomes after adjusting for the number of ESTs and the number of 0.2-μm SC length segments in euchromatic regions. Absolute differences between the average number of ESTs per segment and the number of ESTs observed from the data and between the average number of ESTs and the number of ESTs from the randomly generated uniform distributions were calculated for each segment. If the cumulative sum of these observed differences was greater than the 99th percentile for the randomly generated data, then the observed cumulative difference of ESTs was considered to be significantly different from a uniform distribution. Clustering was also assessed by using χ2 tests of the number of ESTs in 2-μm SC length segments.
Rice chromosome 10
The genetic and physical positions of rice chromosome 10 markers are given in Cheng et al. (2001) and listed at http://rgp.dna.affrc.go.jp/publicdata/geneticmap98/chr10pre.html. The nucleotide sequences of these markers are available at GenBank (accession numbers C23566, C23583, D13526, D15077, D15084, D15184, D15261, D15343, D15481, D15705, D15941, D22541, D22579, D24098, D24428, D24445, D24492, D24564, D24576, D24612, D24816, D25368, D25474, D25480, D28190, D28204, D28322, D38785, D39197, D40056, D40246, D40397, D46309, D46380). The rice chromosome 10 version 3.1 sequence was downloaded from http://www.tigr.org on April 19, 2005. A BLASTn was performed between the marker sequences, as well as the complete nucleotide sequence of chromosome 10, using default parameters without a filter. BLASTn hits of >80% similarity were retained, and their positions on the rice10 sequence were identified. Multiple markers were available for four of the 18 mapped positions. In these cases, we calculated the Mb position on the rice10 sequence as the midpoint among multiple markers. The marker positions defined 17 bins in which we counted the number of annotated genes, ignoring genes that had annotations related to transposable elements.
Acknowledgments
We thank Dr. Mike McMullen and anonymous reviewers for their helpful comments on the manuscript. We also thank Mr. Jim zumBrunnen for his assistance with the statistical analyses. This work was supported by grants from the National Science Foundation (MCB-314644 to L.K.A., MCB-9728673 to S.M.S., and DBI 0321467 and DBI 0320683 to B.S.G.).
Footnotes
-
[Supplemental material is available online at www.genome.org.]
-
Article published online ahead of print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.4249906.
-
↵3 Corresponding author. E-mail lorinda.anderson{at}colostate.edu; fax (970) 491-0649.
-
- Accepted August 24, 2005.
- Received June 9, 2005.
- Cold Spring Harbor Laboratory Press








