
Analysis of 5′ junctions of human LINE-1 and Alu retrotransposons suggests an alternative model for 5′-end attachment requiring microhomology-mediated end-joining
- Nora Zingler1,
- Ute Willhoeft2,
- Hans-Peter Brose3,
- Volker Schoder3,
- Thomas Jahns2,
- Kay-Martin O. Hanschmann1,
- Tammy A. Morrish4,
- Johannes Löwer1, and
- Gerald G. Schumann1,5
- 1 Fachgebiet Pr2/Retroelemente, Paul-Ehrlich-Institut, D-63225 Langen, Germany
- 2 Zentrum für Bioinformatik, Universität Hamburg, D-20146 Hamburg, Germany
- 3 Institut für Medizinische Biometrie und Epidemiologie, Universitätsklinikum Hamburg-Eppendorf, D-20246 Hamburg, Germany
- 4 Department of Human Genetics and Department of Internal Medicine, University of Michigan Medical School, Ann Arbor, Michigan, 48105-0618, USA
Abstract
Insertion of the human non-LTR retrotransposon LINE-1 (L1) into chromosomal DNA is thought to be initiated by a mechanism called target-primed reverse transcription (TPRT). This mechanism readily accounts for the attachment of the 3′-end of an L1 copy to the genomic target, but the subsequent integration steps leading to the attachment of the 5′-end to the chromosomal DNA are still cause for speculation. By applying bioinformatics to analyze the 5′ junctions of recent L1 insertions in the human genome, we provide evidence that L1 uses at least two distinct mechanisms to link the 5′-end of the nascent L1 copy to its genomic target. While 5′-truncated L1 elements show a statistically significant preference for short patches of overlapping nucleotides between their target site and the point of truncation, full-length insertions display no distinct bias for such microhomologies at their 5′-ends. In a second genome-wide approach, we analyzed Alu elements to examine whether these nonautonomous retrotransposons, which are thought to be mobilized through L1 proteins, show similar characteristics. We found that Alu elements appear to be predominantly integrated via a pathway not involving overlapping nucleotides. The results indicate that a cellular nonhomologous DNA end-joining pathway may resolve intermediates from incomplete L1 retrotransposition events and thus lead to 5′ truncations.
The human LINE-1 (Long interspersed nuclear element 1, L1) is one of the best characterized members of the extensive group of non-LTR retrotransposons (Malik et al. 1999). Roughly 520,000 L1 copies account for ∼17% of the human genome (Lander et al. 2001). Additionally, L1 retrotransposons are indirectly responsible for the generation of ≥13% of the human genome by mobilizing Alu elements (Dewannieux et al. 2003) and by creating processed pseudogenes in trans (Esnault et al. 2000).
A functional full-length L1 element is ∼6 kb long (Fig. 1) and includes a 5′-untranslated region (5′-UTR) bearing an internal promoter, two open reading frames (ORFs) separated by a 63-nt intergenic region, and a 3′-UTR terminating in a poly(A) tail (Dombroski et al. 1991). ORF1 encodes an RNA-binding protein that has nucleic acid chaperone activity in vitro (Kolosha and Martin 1997, 2003; Martin and Bushman 2001), but no known specific role in the L1 replication mechanism. The ORF2-encoded protein (ORF2p) contains three domains critical for L1 propagation: endonuclease (EN) (Feng et al. 1996), reverse transcriptase (RT) (Mathias et al. 1991; Dombroski et al. 1994), and a 3′-terminal Zn finger-like domain (Fanning and Singer 1987). There are several variations in the structure of genomic L1 elements. Only 15% of all intact L1 insertions flanked by target site duplications (TSDs) represent full-length insertions, 85% are 5′-truncated, and 19% are both 5′-truncated and 5′-inverted (Szak et al. 2002).
Retrotransposition of a new L1 copy into the degenerate genomic consensus sequence 5′-TTTT/A-3′ (Cost and Boeke 1998; Gilbert et al. 2002; Szak et al. 2002) is initiated by a process termed “target-primed reverse transcription” (TPRT), in which ORF2p nicks the target DNA to generate a free 3′-OH. This hydroxyl acts as primer for reverse transcription using L1 RNA as template (Luan et al. 1993; Cost et al. 2002). The result is simultaneous reverse transcription and joining of the 5′-end of the first-strand cDNA with the genome. The second strand of the genomic target is nicked at variable distances downstream of the complementary sequence of the degenerate genomic consensus site, preferably within 15-16 bp (Jurka 1997; Szak et al. 2002). The subsequent steps of the integration process of elements that are both 5′-truncated and 5′-inverted can be satisfactorily explained by a variation of TPRT called “twin priming” (Ostertag and Kazazian Jr. 2001b), which is corroborated by the existence of short patches of complementary nucleotides at the junction between the 5′-TSD and the inverted L1 sequence. However, for both full-length and 5′-truncated L1 insertions, neither the means by which the 3′-end of the cDNA is attached to chromosomal DNA nor the mechanisms initiating second-strand synthesis have been elucidated so far. It is also yet unknown which mechanism leads to the generation of 5′-truncated L1 copies. L1 truncation has long been explained by an inability of the L1 RT to copy the entire L1 RNA, either by prematurely dissociating from the RNA or by competing with a cellular RNAse that digests the RNA before completion of reverse transcription (Ostertag and Kazazian Jr. 2001a).
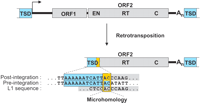
Retrotransposition of a functional human L1 element frequently results in 5′-truncated copies with overlapping nucleotides at the 5′ junctions. These microhomologies (yellow box) between the genomic target site duplication (TSD) and the 5′-end of the adjacent L1 sequence make a precise assignment of the 5′ boundary of the L1 insertion ambiguous. A 13-bp TSD sequence (blue) observed after de novo L1 retrotransposition (Symer et al. 2002) serves as a representative example. Nucleotide sequences of the genomic pre- and post-integration sites as well as the L1 consensus sequence at the junction region are shown. (ORF1/2) Open reading frame 1/2; (EN) endonuclease; (RT) reverse transcriptase; (C) cysteine-rich motif.
Several mechanisms have been suggested to explain the attachment of the 5′-end of non-LTR retrotransposons to the chromosome, which are based on regions of microcomplementarity found at the junctions between the 5′-end of the retrotransposon and the 3′-end of the adjacent TSD (Fig. 1). These overlapping nucleotides have been described initially for L1 insertions in the mouse genome and for Cin4 elements in maize, and led to replication models that require bridging of chromosomal double-strand breaks (DSBs) by L1 RNA (Voliva et al. 1984; Schwarz-Sommer et al. 1987). However, these gap repair models that require two hybridization events of the retrotransposon RNA to the target DNA are not compatible with what is currently known about L1 EN activity and the mechanism of TPRT. Therefore, a different mechanism has been proposed, called double-TPRT, in which second-strand synthesis is primed by annealing of the newly synthesized cDNA to complementary sequences in the genomic target. The sequential TPRT reactions were suggested to be carried out by the element-encoded protein machinery (Supplemental Fig. 1). This model was originally developed to explain R1Bm replication (Feng et al. 1998), and was subsequently adapted to account for the insertion mechanism of mouse and human L1. As L1 EN is much less sequence-specific than R1 EN, L1 RT was proposed to use fortuitous matches between the L1 cDNA and the target sequence to prime second-strand synthesis (Martin and Bushman 2001; Symer et al. 2002; Martin et al. 2005).
In the course of our efforts to investigate the means by which the L1 5′-end is attached to the chromosomal DNA, we evaluated whether there is a preference for overlapping nucleotides (nt) between the 5′-end of pre-existing L1 insertions and the 3′-end of the adjacent genomic TSDs, as previously reported for a small number of de novo L1 integrants obtained from tissue culture experiments (Symer et al. 2002). To this end, we performed a comprehensive genome-wide analysis of TSDs flanking extant genomic L1 and Alu insertions. Characterization of the junctions between genomic target sequences and the 5′-ends of L1 and Alu insertions revealed that, in contrast to full-length insertions and 5′-truncated Alu elements, 5′-truncated L1s preferentially exhibit features that are observed after DSB repair by alternative, error-prone nonhomologous end-joining (NHEJ). Based on our observations, we propose that there are at least two different mechanisms responsible for attachment of the 5′-end of L1 and the initiation of second-strand synthesis.
Results
Microhomology patches in the range of 1-12 nt are significantly overrepresented at the 5′ junctions of endogenous 5′-truncated L1s
In order to gain insight into the enigmatic mechanism of 5′-end attachment of human L1 to the genomic target DNA, we initiated a genome-wide analysis of TSDs flanking endogenous L1 insertions. For this purpose, we applied a recently developed computer program called TSDfinder (Szak et al. 2002) that was designed to identify flanking TSDs longer than 8 nt and poly(A) tails of 3′ intact L1 insertions. The program was run against nonredundant human DNA-sequence contigs (NT_* records) assembled at NCBI by April 10, 2003 (build 33) constituting ∼99% of the euchromatic genome. To identify predominantly young L1s, we used the DNA sequence of the highly active L1.3 element (Dombroski et al. 1993), containing a polymorphic 131-nt sequence at a defined position in the 5′-UTR (Hattori et al. 1985) as the query sequence.
The program identified 10,034 L1 insertions with an intact 3′-end, a poly(A) tail, and TSDs, with the majority (90%) belonging to the younger, primate-specific subfamilies L1Hs, L1P1, or L1P2 (Smit et al. 1995). This set of L1 insertions was further refined to remove insertions with noncanonical features including: inversions, nucleotides of unknown origin added to the 5′-end of the L1 sequence, and large internal insertions or deletions. The resulting set of standard insertions comprised 573 full-length and 2427 5′-truncated L1 insertions.
In our TSDfinder output file we reproduced the previously described distribution of TSD lengths (Szak et al. 2002) with peaks at 9 and 15 nt (Supplemental Fig. 2). It was suggested that a significant fraction of the 9-11-nt TSDs may consist of false positives (Szak et al. 2002). Indeed, when we analyzed the refined data set of 3000 standard insertions, the peak at 9 nt decreased drastically and the second peak shifted slightly from 15 to 16 nt. Thus, by removing L1 insertions with noncanonical features, we concomitantly eliminated the majority of false-positive TSDs deemed to be responsible for the high abundance of short TSDs in the TSDfinder output file.
Analysis of TSDs flanking 5′-truncated L1 insertions uncovered microhomologies between the 3′-end of the TSD and the 5′-end of their adjacent L1 insertion in 62% (1503/2427) of all cases (Fig. 2A). The regions of microcomplementarity covered 1-12 consecutive nucleotides. Using three different methods, we evaluated statistically whether the observed frequencies of microhomologies at the 5′ junctions were significantly different from what was expected by chance. For that purpose, we initially applied methods similar to those described previously to investigate viral/host junction sequences (Roth et al. 1985). First, the expected frequencies of identical nucleotides overlapping by chance were calculated assuming an unbiased base composition of L1 and target sequences (Fig. 2, open bars). Second, in order to account for the insertion preference of L1 elements in A+T-rich regions, the actual base composition of the flanking genomic sequences was determined in a 20-nt window surrounding each TSD. The resulting probabilities for the occurrence of each base in the genomic target sequence and the base composition of L1 (A+T content: 57%) were used to calculate a biased distribution pattern (Fig. 2, gray bars) that predicts a higher number of randomly occurring microhomologies as both the L1 sequence and the genomic target DNAs are A+T-rich. In an additional approach, we determined the probability of microhomologies between each individual TSD sequence and any portion of its adjacent L1 sequence to assess the influence of the actual base composition directly at the TSD/L1 junction. We slid the TSD sequences against their respective adjacent L1 sequences and counted the maximum overlap lengths at the TSD 3′-end by “exact pattern matching” at each L1 sequence position (Fig. 2, striped bars). All three methods yielded similar distributions, which deviated from each other by <10%. Statistical comparison of the expected distributions with the observed distribution (Fig. 2, black bars) showed that in the case of 5′-truncated L1 elements (Fig. 2A), the observed frequencies of contiguous overlapping nucleotides were significantly higher than those expected by chance (p1,2,3 < 0.0001). Thus, although the nucleotide bias of both the 5′-truncated L1 sequence and its preferred integration site does influence the frequencies of microhomologies expected by chance, this effect is minor compared to the actual distribution observed.
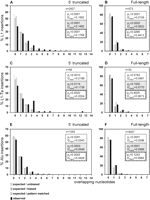
Statistical analysis of homologies at the junctions between the 5′-ends of pre-existing L1 and Alu elements and the 3′-ends of their flanking TSDs. The bar charts compare the relative numbers of expected versus observed endogenous L1- (A,B), L1 Ta (C,D), and AluY (E,F) elements with variable numbers of overlapping nucleotides shared between the 3′-end of the TSD and the 5′-truncated flank of the element. Within each of the three groups, 5′-truncated elements (A,C,E) are compared with full-length insertions (B,D,F). Open bars represent the expected distribution, assuming an unbiased base composition. Gray bars indicate the expected distribution after adjustment for the actual base composition of the elements and their target sequences (biased). Striped bars display the distribution obtained from sliding each TSD against its adjacent L1/Alu sequence and counting the hits found by “exact pattern matching.” The observed relative quantities of elements with the indicated numbers of overlapping nucleotides in their TSDs are represented as black bars. The x-axis is the number of overlapping nucleotides; the y-axis is the percentage of element insertions with the respective number of microhomologies; (n) number of endogenous elements analyzed; (p1, p2, p3) significance of the difference between expected/unbiased (expected/biased, pattern matched) and observed distribution of overlapping nucleotides (at a multiple significance level [Bonferroni-adjusted] of α = 0.025); (Dmax1, Dmax2, Dmax3) maximum absolute distance between the observed and the expected/unbiased (expected/biased, pattern matched) distribution function. The confidence intervals of the p-values are listed in Supplemental Table 1.
Microhomologies associated with full-length insertions had to be investigated in a separate study, as the mathematical basis for the calculation of the expected distribution of overlapping nucleotides differs between 5′-truncated and full-length L1s. Since in the case of full-length insertions the sequence of one of the joined ends is specified, the distribution function shifts toward fewer randomly occurring microhomologies. Also, considering the A+T-rich target sequences, patches of microcomplementarity are more rarely expected to form with the guanine-rich 5′-termini of full-length L1 insertions.
An unambiguous definition of the transcriptional initiation sites of full-length L1 elements is not possible as it has been found that initiation sites can vary in both downstream and upstream directions of nucleotide +1 (Athanikar et al. 2004; Lavie et al. 2004). YY1 was shown to direct transcription initiation to purine residues located within the first 5 nt of the L1 consensus sequence (Athanikar et al. 2004). Of the L1 copies listed in a database of 218 human-specific full-length L1 insertions provided in the same study, 90% (i.e., 196/218) were derived from parental elements characterized by the 5′-terminal sequences 5′-GGGGGAGG-3′, 5′-GAGGGAGG-3′, or 5′-GGAGGAGG-3′. For our study, we therefore defined full-length L1 insertions as elements starting within the first 5 nt of any of these three 5′-terminal sequences. We reasoned that by using this approach, the majority of all pre-existing full-length L1 insertions in the genome would be covered.
Statistical analysis yielded the surprising result that the distribution of microhomologies in endogenous full-length elements differs quite dramatically from the situation observed in 5′-truncated L1 insertions (Fig. 2A,B). While the observed distribution is clearly shifted toward longer patches of microhomology (1-12 nt) in the case of 5′-truncated L1s, this is not the case for full-length insertions, which display overlapping nucleotides roughly at the expected rates (Fig. 2B). Differences between the expected distributions derived from “exact pattern matching” and observed frequencies are not statistically significant as the p-value, p = 0.0268, is above the significance level of α = 0.025. Although p-values are much lower for unbiased and biased base compositions (p1 < 0.0001, p2 = 0.0002) (Fig. 2B), one has to be aware that this is at least partly caused by the high number of full-length elements analyzed (see paragraph on Ta elements and Fig. 2D). Also, the maximum deviation Dmax between expected and observed distributions, which serves as a measure for the biological relevance of the observed effect, is small for full-length L1 elements (Dmax = 0.0413-0.0726) when compared to the corresponding values for 5′-truncated L1s (Dmax = 0.1482-0.1952). This indicates that there is no preference for microhomologies at the 5′ junctions of full-length L1s.
To ensure that false-positive, short TSDs do not bias our results, we repeated these analyses with only those L1s from the initial data set that are flanked by TSDs at least 14 bp long. Although this led to minor reductions of microhomology frequencies observed for truncated L1s, the preference for 1-12-nt overlaps remained highly significant. The results for full-length elements did not change either (Supplemental Table 1). Similarly, we ascertained that the elimination of TSDs containing mismatches (see Szak et al. 2002) does not influence the outcome of our analysis (data not shown). Thus, the effect we observe is not an artifact created by the TSDfinder program.
The presented data lead to the conclusion that there are at least two mechanisms involved in the attachment of the 5′-end of an L1 copy to the chromosomal DNA: (1) 5′-truncated L1s preferentially use a mechanism involving complementary base-pairing; (2) full-length L1 insertions are attached to the chromosomal DNA predominantly by means of a second mechanism that does not require base-pairing. By analyzing our data set of 3000 endogenous standard L1 insertions for a correlation between insertion length and incidence of microhomologies (Supplemental Fig. 3), this hypothesis of two distinct mechanisms being responsible for the formation of full-length and 5′-truncated elements was supported: While microhomologies were only found in 25% of L1 insertions starting at positions +1 to +6, 50%-65% of all insertions with 5′-truncations exhibit overlapping nucleotides, regardless of the extent of the truncation.
L1 Ta elements display the same distribution of microhomologies at their 5′ junctions as evolutionary older L1s
Although the preference for microhomologies at the 5′-ends of pre-existing 5′-truncated standard insertions (62% observed vs. 44%-50% expected) is statistically significant, it is much more pronounced in previously described 5′-truncated de novo standard insertions of wild-type L1s (93%) isolated from HeLa and HCT116 cells (Symer et al. 2002). Of the reported set of 13 de novo standard insertions flanked by 3-23-bp TSDs, 12 contained 1-5-nt microhomologies. However, these de novo integration events came from retrotransposition of derivatives of L1.3 (Dombroski et al. 1993; Sassaman et al. 1997), a member of the youngest subfamily of L1 elements collectively termed Ta (Skowronski et al. 1988), which arose about 4 million years ago (Mya) (Boissinot et al. 2000) and spans the vast majority of functional L1 elements in the human genome. In contrast, more than 95% of elements included in our genome-wide analysis of pre-existing L1 insertions were members of older classes of L1s (L1P1, L1P2, L1P3, and L1P4) that were generated between 5 and 60 Mya (Smit et al. 1995; Lander et al. 2001). To examine whether Ta elements have a stronger bias for microcomplementarities than the older classes of L1s, we analyzed L1 Ta elements separately. For this purpose, we screened our 3000 standard L1 insertions selected from the TSDfinder output file with the L1-specific 19-bp oligonucleotide 5′-CCTAATGCTAGATGACACA-3′ (Myers et al. 2002), whose 3′-terminal ACA sequence is diagnostic for the L1Hs Ta subfamily (Dombroski et al. 1991). A subset of 56 5′-truncated and 52 full-length L1 Ta elements was identified.
Statistical evaluation showed that the observed frequency of overlapping nucleotides at the 5′-end of L1 Ta insertions (Fig. 2C,D) reflects our findings observed for the initial, larger data set including the older classes of L1 elements (Fig. 2A,B). While 59% (33/56) of the 5′-truncated L1 Ta insertions are characterized by 1-12 overlapping nucleotides at their 5′-end (extensive set of L1s: 62%, 1503/2427), only 25% (13/52) of the full-length insertions have short stretches of overlapping nucleotides (extensive set of L1s: 24%, 137/573). The similarities in observed frequencies of overlapping nucleotides between our initial set of L1 insertions and the subset of young L1 Ta elements are also consistent with the similarly high Dmax-values reflecting the biological relevance of the data (Fig. 2A-D). However, owing to the comparatively low number of elements included in our L1 Ta study, the p-values differ from those calculated for the extensive set of L1s. The results suggest that the difference between observed and expected distributions of microhomologies is only significant for 5′-truncated elements (p1,2,3 ≤ 0.0114), but not for full-length insertions (p1,2,3 ≥ 0.1502) (Fig. 2C,D) and thus concurs with the aforementioned interpretation of the data obtained from the extensive set of L1s. As the features of 5′ junctions of the Ta subfamily reflect the situation we described for older L1 families, we conclude that differences in the age of L1 insertions cannot be the cause for the observed higher frequency of microhomologies at the junctions of the comparatively small set of de novo integrants in tissue culture (Symer et al. 2002).
Alu elements are mainly inserted through a mechanism not requiring complementary base-pairing
Since Alu retrotransposons were shown to be mobilized by the L1 protein machinery (Dewannieux et al. 2003), we asked the question whether the microhomology-mediated mechanism suggested for 5′-truncated L1 elements might also be involved in Alu integration. For this reason, we used the TSDfinder program to identify flanking TSDs and poly(A) tails of 3′ intact pre-existing genomic Alu insertions. Owing to the relatively young age (5-35 million years) and the high copy number (>200,000 copies) of the recently integrated AluY elements, we used a consensus sequence (Batzer and Deininger 2002) as query sequence for the RepeatMasker program. We identified 34,913 Alu insertions with intact 3′-ends, poly(A) tails, and flanking TSDs. We refined this Alu data set and eliminated inverted or rearranged Alu elements as well as insertions harboring nucleotides of unknown origin, and ended up with a set of 10,062 standard Alu insertions.
As described before for the L1 TSD data sets (Supplemental Fig. 2A), the length distributions of Alu TSDs from the TSDfinder output file and from the refined Alu TSD data set were compared with each other (Supplemental Fig. 2B). As expected, the distributions of Alu TSDs and L1 TSDs are almost identical, supporting the finding that L1s and Alus are mobilized by the same L1-encoded protein machinery (Dewannieux et al. 2003). Similar to L1 TSDs (Supplemental Fig. 2A), the peak comprising the 9-11-nt Alu TSDs disappeared after the data set was refined to include only TSDs from standard insertions.
Although it was shown that Alu transcription predominantly starts at the first nucleotide of the Alu repeat (Elder et al. 1981; Fuhrman et al. 1981), transcriptional initiation of Alus is not always precise (Liu et al. 1994). Analogous to full-length L1s, we therefore defined full-length Alu elements as those starting within the first 5 nt of the consensus sequence. We obtained a set of 9007 full-length and 1055 5′-truncated Alu insertions. As described before for L1 insertions, we calculated expected unbiased, biased, and pattern-matched frequencies of microhomologies at the 5′ junctions of Alu insertions (Fig. 2E,F). For 5′-truncated Alus, a minor shift toward longer stretches of overlapping nucleotides was seen (Fig. 2E). Although p-values were at (p1 = 0.0281) or below (p2[3] = 0.0003 [0.0043]) the significance level, this shift was much less pronounced (Dmax = 0.0343-0.0546) than the one observed for truncated L1s (Fig. 2A,C). Similar to full-length L1s, full-length Alu elements with overlapping nucleotides were found roughly at the expected rate (Fig. 2F). Although the low values for p1 and p2 (<0.0001), which are probably a consequence of the high number of elements analyzed, suggest a significant difference between expected and observed distributions, the low Dmax-values (Dmax1[2] = 0.0296 [0.0566]) indicate that the biological relevance of those differences is low. This is consistent with the fact that the observed distribution of microhomologies conforms to the more realistic frequencies obtained by pattern-matching (p3 = 0.1243, Dmax3 = 0.0069). Therefore, we conclude that the 3′-ends of new Alu cDNAs are predominantly attached to the chromosomal DNA via a pathway that does not depend on overlapping nucleotides.
Clearly, differences in the sequence composition between L1 and Alu elements (A+T content: 57% vs. 37%) increase the probability of finding a sequence match in the case of L1 relative to Alu in the same A+T-rich genomic insertion site. However, since we considered the differences in the base composition of L1s and Alus in our calculation of biased/expected and pattern-matched frequencies, our data indicate that different preferences of L1 and Alu for microhomologies are a phenomenon that is not caused by the base composition of these elements. If a microhomology-dependent pathway is involved in Alu integration at all, it is apparently used much less frequently than in L1 integration.
Discussion
Expanding a recent report by Martin and coworkers who analyzed 73 human and 215 mouse L1 elements (Martin et al. 2005), we performed a comprehensive, genome-wide analysis of junctions between pre-existing L1 sequences and their flanking TSDs and identified a significant preference for 1-12-nt microcomplementarities at 5′ junctions of 5′-truncated L1 standard insertions. Strikingly, inverted L1 insertions also often display similar patches of 1-6-nt microhomology, both at their 5′-end and at their inversion junction. A variation of the TPRT reaction, called twin priming (Fig. 3B), was suggested to be responsible for the formation of these 5′-truncated as well as inverted L1 insertions (Ostertag and Kazazian Jr. 2001b). In this model, microhomologies at the inversion junction are attributed to base-pairing of the resulting single-stranded cDNAs at small regions of complementarity. This mechanism of resolution is typical of DSB repair by “alternative” or “error-prone” NHEJ (also designated “microhomology-driven single-strand annealing” or “microhomology-mediated error-prone end-joining”), which was reported to use microhomology patches of 1-10 nt to rejoin DSB ends (Thacker et al. 1992; Göttlich et al. 1998; Pfeiffer et al. 2000). It is distinct from “classical” or “accurate” NHEJ in that it appears to be independent of the repair factors Ku70, Ku86, DNA-PKcs, XRCC4, Artemis, and DNA-Ligase IV (Roth 2003; Bentley et al. 2004).
Based on the microhomologies at the 5′ junctions of 5′-truncated standard L1 insertions, we propose a model for the generation of 5′-truncated L1 integrants that offers an alternative to the double-TPRT model: In contrast to double-TPRT, which relies exclusively on L1 proteins for synthesis and attachment of both L1 DNA strands (Supplemental Fig. 1), we suggest that joining of the cDNA to the genomic target DNA and subsequent second-strand synthesis are carried out by host-encoded factors that are involved in “error-prone” NHEJ (Fig. 3C). This proposed mechanism is closely related to twin priming since both models involve alternative NHEJ as a key part. While in the former model alternative NHEJ was proposed to be responsible for the attachment of the noninverted cDNA to the inverted cDNA (Fig. 3B), in the latter model the same DSB repair pathway facilitates the attachment of a noninverted cDNA to the genomic target (Fig. 3C). Our model is strongly supported by a recent study investigating the joining of unknown, complex DSBs (Odersky et al. 2002). This study reported repair junctions generated by NHEJ that displayed characteristics that were strikingly similar to those of 5′ junctions observed in 5′-truncated de novo L1 integrants, most notably the pronounced preference for microhomologies, but also the creation of deletions and extra nucleotide insertions at the junctions (Gilbert et al. 2002; Symer et al. 2002; N. Zingler and G.G. Schumann, unpubl.). While repair by alternative NHEJ can easily account for the generation of these structures at the 5′-ends of L1 insertions, they are hard to explain by the double-TPRT model. Also, Cost and coworkers demonstrated that unlike first-strand synthesis, second-strand cDNA synthesis by TPRT was quite inefficient in their L1 in vitro system, and none of the sequenced 5′ junctions of the second-strand DNA was reported to exhibit any microhomologies (Cost et al. 2002). As L1-ORF2p was the only protein present in their in vitro system, these observations suggest that additional factors, for example, L1-ORF1p (Martin and Bushman 2001; Martin et al. 2005) or host-encoded factors like NHEJ repair proteins, are necessary for efficient attachment of the first-strand cDNA to the target DNA and subsequent second-strand synthesis.
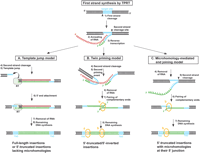
Alternative models for the attachment of the 3′-end of the L1 cDNA to the chromosomal target DNA. According to the TPRT model, L1-encoded EN nicks the bottom strand of the target DNA (1) and exposes a 3′-hydroxyl that primes reverse transcription of the element's full-length RNA (2, 3). (A) Template-jump model: After first-strand synthesis is completed, second-strand cleavage occurs (4), and the L1 RT jumps from the L1 RNA template onto the upstream target DNA (5). cDNA synthesis is continued using the genomic 3′ overhang as template, and the 3′-end of the cDNA is attached to the genomic DNA (6). Jumps from the 5′-end of full-length RNAs generate full-length L1 insertions. If the reverse transcriptase jumps from 5′-degraded L1 RNAs, 5′-truncated elements are generated without the need for microhomologies (Eickbush 2002; Bibillo and Eickbush 2004). (B) Twin priming model: L1 EN cleaves the second DNA strand (4) before reverse transcription of the first-strand cDNA has been completed (3), producing an additional 3′-hydroxyl and a stretch of single-stranded DNA, which serves as an internal primer. This primer anneals at complementing nucleotides in an internal region of the L1 RNA template, and primes reverse transcription upstream from the 3′-end of the L1 RNA (5), where the initial TPRT started. After the RNA is removed from the RNA/cDNA structure (6), the single-stranded cDNAs pair at a small region of limited complementarity (7), and the remaining DNA synthesis is completed (8) (Ostertag and Kazazian Jr. 2001b). In this model, patches of microhomology are a consequence of either RNA/DNA annealing at the 5′ junction (a) or DNA/DNA base-pairing at the inversion junction (b). (C) Microhomology-mediated end-joining model: After initiation of TPRT, the reverse transcriptase falls off the RNA template before it has reached the 5′-end of the RNA (3). Second-strand cleavage (5) occurs after reverse transcription has stopped and the L1 RNA has been degraded (4). The 3′ overhang of the chromosomal target site anneals to the 3′-end of the L1 cDNA at a region of limited complementarity (6), primes second-strand synthesis (7), and leads to the formation of 5′-truncated L1 elements with microhomologies at their 5′ junctions (b). In all three models, the second DNA strand is joined to the target site by the action of a cellular ligase after completion of second-strand synthesis.
Our genome-wide analysis also uncovered that, in contrast to 5′-truncated L1s, 5′ junctions of full-length L1 insertions as well as full-length and 5′-truncated Alu elements exhibit no or only a very weak preference for overlapping nucleotides. Neither the double-TPRT model nor our “microhomology-mediated end-joining” model can explain the lack of microhomologies at the junctions of 76% of endogenous full-length L1s, 38% of endogenous truncated L1 insertions, and the majority of Alu insertions analyzed (Fig. 2B,D,E,F). Some of these integrants might derive from alternative NHEJ events as it was reported that a minority of joining events mediated by this pathway lacks microhomologies (Kabotyanski et al. 1998; Pfeiffer et al. 2000). However, experimental data with R2Bm-encoded RT suggest a template-jump mechanism that could account for the lack of microhomologies at many 5′ junctions: In vitro studies indicate that the RT can simply switch templates from the RNA-template onto the upstream target DNA to finish the integration reaction without the need for sequence homology (Burke et al. 1999; Bibillo and Eickbush 2002a,b; Eickbush 2002). This mechanism could account for the majority of 5′ junctions of full-length L1 insertions (Fig. 3A). 5′-truncated insertions lacking overlapping nucleotides could also be generated by template-jumping, provided that the L1 template RNA is 5′-degraded, as the jumps occur preferentially from the 5′-end of the donor template (Bibillo and Eickbush 2004). However, the frequent occurrence of longer patches of microhomologies at the 5′ junctions cannot be explained by template jumping.
Adding our proposed microhomology-mediated end-joining mechanism to the previously described twin-priming model (Ostertag and Kazazian Jr. 2001b) and the template-jump model (Bibillo and Eickbush 2002b), we put forward a combination of mechanisms to account for the generation of full-length and 5′-truncated L1 integrations as well as for inversions (Fig. 3). We suggest that the following factors determine which pathway is chosen for L1 replication after the initial TPRT reaction: kinetics of second-strand cleavage, processivity of the RT, and the abundance of DSB repair proteins in the host cell. If the second strand is cleaved before reverse transcription is finished, twin-priming is likely to occur (Fig. 3B). If second-strand cleavage takes place while the RT is still bound to the (full-length or truncated) RNA, direct joining of the cDNA to the nicked target site is facilitated by template-jumping (Fig. 3A). However, if the second strand is cleaved after the RT has dissociated from the L1 cDNA and the RNA template has been degraded, microhomology-mediated end-joining might rescue and resolve these structures (Fig. 3C). The observed microhomologies covering up to 12 consecutive nucleotides could even imply “scanning” of the available L1 cDNA for complementary bases. This process would ensure a higher probability to come across homologous sequences than pairing of 3′-ends of fixed sequences.
The presented combination of models (Fig. 3) also provides an explanation for the characteristics observed in Alu insertions: The frequencies of microhomologies at the junctions of full-length Alus are almost identical to those of full-length L1s, suggesting that the 5′-ends of full-length Alus are preferentially attached by the same mechanism as discussed for full-length L1s. However, the minor bias for microhomologies found in 5′-truncated Alus is in striking contrast to that of 5′-truncated L1s, and could be a consequence of the short size of full-length Alu RNAs: As Alus are only ∼280 nt long, it is unlikely that 5′-truncated Alu cDNAs are generated frequently by premature dissociation of reverse transcriptase from the RNA template. This is also reflected by the fact that ∼90% of all identified Alus included in our refined data set are full-length. It is far more probable that most 5′-truncated Alu copies are derived from degraded full-length Alu RNAs that are subsequently reverse transcribed and processed like full-length RNAs.
So far, there is no direct evidence as to how, or even whether, the second DNA strand is synthesized by the LINE-1 machinery. However, the presented data imply that considerable assistance from the DNA repair machinery is involved in the attachment of the 5′-end of L1 to the chromosome. Interestingly, it was shown by Morrish and coworkers that the retrotransposition rate of EN-deficient L1s was significantly elevated in the absence of functional “accurate NHEJ” repair factors (Morrish et al. 2002). Additionally, recent data indicate that many transposable elements and retroviruses interact with host-encoded DSB repair factors (Moore and Haber 1996; Teng et al. 1996; Li et al. 2001; Fujimoto et al. 2004; Izsvák et al. 2004; Yu et al. 2004). It is clear at this stage that the effects of specific DSB factors in L1 retrotransposition need to be determined experimentally, in order to further our understanding of the insertional mechanisms responsible for the generation of more than 34% of our genome.
Methods
Identification of endogenous L1 and Alu sequences flanked by TSDs
Endogenous human L1 elements were identified by applying the program TSDfinder (Szak et al. 2002) on nonredundant human sequence contigs (NT_* records) assembled at NCBI (http://www.ncbi.nlm.nih.gov/genome/guide/human/). A DNA reference sequence of the human genome constituting ∼99% of the euchromatic genome (build 33 as of April 10, 2003) served as the data set, with the exception that the file “unplaced_contigs” was excluded from the study. For identification of L1 elements and their respective 5′- and 3′-flanking sequences, we used the method described by Szak et al. (2002) with minor modifications. Repeat elements were annotated using the RepeatMasker program (A.F.A. Smit, R. Hubley, and P. Green, 1996-2004. RepeatMasker version 20020713; http://repeatmasker.genome.washington.edu) and a custom library that contained only the L1.3 reference sequence (GenBank accession number L19088 with modifications as cited in Szak et al. 2002). TSDfinder (http://www.ncbi.nlm.nih.gov/CBBresearch/Landsman/TSDfinder/) was used to generate an output file with identification numbers, coordinates, and further information for all matching sequences (hits) of the RepeatMasker result. The search strategy reporting all elements with ≥90% identity to L1.3 sequences returned 30,265 hits in the RepeatMasker Output, and 10,031 of them were annotated to contain a TSD by the TSDfinder program. This data set was used for further studies (Supplemental material: Li_coord_ seq.txt). A similar search strategy was performed with the consensus sequence for AluY elements (Batzer and Deininger 2002) and the limitation to report only sequences with ≥90% identity. In the RepeatMasker search, 147,620 hits were identified, and 34,913 of these were reported to contain a TSD by the TSDfinder program. Further studies were carried out with this data set (Supplemental material: Alu_coord_seq.txt).
A Perl program was written to parse information from the TSDfinder output for each hit and join it with sequence information from the DNA reference sequence of the human genome. The program produced an output file in FASTA format reporting the TSDfinder identification numbers, GenBank accession numbers, and the position of the elements within the human sequence contigs. Additionally, it provided the L1 or Alu sequences as well as 150 nt of genomic sequence flanking the respective insertion. Another Perl program parsed the sequence information of the TSD as assigned by the program TSDfinder and the respective identification number for each element in a tabular fashion. Both programs are available on request. The data sets are listed as Supplemental material (L1_TSD.txt, Alu_TSD.txt).
Identification of microhomologies localized at the 5′ junctions of L1 and Alu insertions
We tested for reliability of the TSDfinder results by searching for TSD sequences in the output files. Only post-integration sites with the correct TSD at the 3′-end of each 5′-flanking 150-bp window included in the output sequences were used for further analysis. TSDs consisting exclusively of poly(A) stretches were discarded because of statistical uncertainties since the poly(A) sequences could be either real TSDs or fortuitous matches between the target site and the poly(A) tail. Next, it was examined whether the insertions carried inverted sequences or whether any nucleotides of unknown origin were inserted between the 5′-end of the L1 or Alu sequence and the TSD. For that purpose, the 30-bp region located 3′ of the TSD was aligned with the L1 or Alu consensus sequence, respectively, and all integration events that displayed <83% sequence identity with the L1 or Alu sequence in the best match were discarded. Moreover, a perfect match of the first 3 nt directly adjacent to the TSD was required. To exclude grossly rearranged L1 copies, we introduced a length criterion by demanding that the sum of L1 insertion length and corresponding 5′-truncation ranged between 6 and 7 kb. Similarly, we defined that Alu insertion length plus extension of the corresponding 5′-truncation should range between 282 and 312 bp, as a considerable number of hits consisted of two or more Alu sequences. After this preliminary selection process, microhomologies were searched for by comparing the 3′-end of the respective TSD with the sequence that lies 5′ of the truncation position in the L1 or Alu consensus. Identification numbers, start positions of the respective elements and numbers of complementary nucleotides at the junctions of L1 and Alu elements that met our criteria are listed as Supplemental material (L1_selection.xls, Alu_selection.xls).
In order to include as many full-length L1s in our analysis as possible, the entire procedure was repeated with two additional L1 consensus sequences, which differ from the original L1.3 sequence in a major polymorphism at the transcriptional start site, where a perfect match was required by our selection criteria. These two alternative sequences start with GAGGG and GGAGG instead of GGGGG. The data sets are available as Supplemental material (L1_selection.xls, Alu_selection.xls).
Statistical analyses
p-values were calculated to determine the significance of the differences between expected and observed distributions of microhomologies at the junctions between TSDs and 5′-ends of the retrotransposons. In the case of 5′-truncated L1 and Alu insertions, statistical assumptions were made according to Roth et al. (1985). The probability to observe a sequence of j homologies is computed as P(X = j)=(j +1) · pj · (1 - p)2, where p denotes the probability of random homology of a single nucleotide. When assuming an unbiased base composition of the target sequences, p was set to 0.25. However, to take into account the different base composition of L1 and Alu elements and the general A+T-bias of L1 target sequences, we also estimated p with the following formula: p = pA gen · pA te + pC gen · pC te + pG gen · pG te + pT gen · pT te (pN gen: probability of N in the genomic sequence, calculated as the actual base composition of the DNA sequences flanking each TSD in a 20-nt window; pN te: probability of N in transposable element L1 or Alu).
In the case of endogenous full-length L1 insertions, the 5′-end is defined by a G-rich purine stretch. Assuming the first nucleotides to be solely Gs, the probability to observe exactly j consecutive ties by chance is P(X = j)= pj · (1 - p), where j =0, 1, 2,... and p denotes the proportion of G in the target sequence. This means that the random variable X being defined as the number of ties until the first non-tie follows a geometric distribution with probability 1 - p. However, the assumption of the geometric distribution holds true only if the nucleotides at the 5′-end remain Gs. In order to consider the polymorphisms that we included in our analysis of full-length L1 elements (see above), we adjusted the formula to allow for the sporadic occurrence of As instead of Gs. The formula was broken down into a decision tree that takes into account all possible L1 start sequences and weights them by the observed occurrence of As at position 2 or 3 of the L1 consensus sequence. In full-length Alu insertions, the first 12 nt consist of Gs and Cs, so that the probabilities of only these two nucleotides were used in the formula.
To get an even more realistic assessment of the influence of the actual base composition directly at the TSD/L1 (or TSD/Alu) junction, we also determined the probability of microhomologies between each individual TSD sequence and any portion of its adjacent L1 (Alu) sequence. For full-length elements, the TSDs were sorted according to the sequence at their 3′-end, and overlaps between TSD and the first 5 nt of the respective consensus sequence (GRRGG for L1; GGCCG for Alu) were determined manually. For 5′-truncated insertions, a Perl program (available on request) was implemented that slides each TSD along its adjacent L1 sequence. The program determines and counts the number of longest, consecutive matches (suffices) between the 3′-end of the TSD and each position within the L1 sequence by exact pattern matching. The result is reported in a tabular fashion giving the length of the suffixes and number of hits obtained for each element. As microhomologies of the length j can be generated in j + 1 ways (see above and Roth et al. 1985), the number of hits of the length j was multiplied by j + 1 to obtain the correct expected distribution of microhomologies. The input data for the program were (1) TSD sequences as reported by the TSDfinder program (Szak et al. 2002). When TSDfinder reported mismatches between the 5′- and 3′-TSD sequence (∼10% of all cases), we used the 5′-TSD as search sequence in our program. (2) L1 or Alu sequences parsed from the original TSDfinder output by removing 150 bp of genomic sequence as well as the TSDs flanking each element. In the case of 3′-transduction events, the transduced sequences were included in the search. Likewise, the poly(A) tail was included since an attempt to remove putative poly(A) sequences from the 3′-end of L1 integrants yielded only minimal changes in the results (<0.1% difference).
For full-length as well as for 5′-truncated insertions, we tested whether the observed data could originate from the distribution specified under the assumption of random events. Therefore, we performed a Kolmogoroff-Smirnow Test computing p-values and 95% Clopper-Pearson confidence limits in a Monte Carlo simulation. This simulation consisted of 100,000 independent draws from the hypothesized distribution, and for each draw the maximum absolute distance (Dmax) of the observed and the theoretical cumulative distribution function was calculated. The proportion of draws that exceeded the analogous distance observed in our data is reported as an unbiased estimator of the true p-value. Simulations were performed with the software program S-Plus 4.5 (MathSoft Inc.). S-Plus programs are available as Supplemental material. Values for statistical significance (p), biological relevance (Dmax), and confidence intervals that were calculated for biased, unbiased, and “pattern-matched” distributions of microhomologies associated with extant human L1 and Alu insertions are listed in Supplemental Table 1.
Acknowledgments
We thank S.L. Martin for sharing unpublished information and S. Szak for helpful advice concerning the TSDfinder program. Special thanks to C. Stocking for crucial support and critical reading of the manuscript. This research was supported by grants Schu1014/2-1 and Schu1014/2-2 of the Deutsche Forschungsgemeinschaft and grant AZ.10.01.1.104 of the “Fritz-Thyssen Stiftung” to G.G.S. T.A.M. was supported, in part, by a grant from the NIH (GM60518).
Footnotes
-
[Supplemental material is available online at www.genome.org and at http://www.zbh.uni-hamburg.de/research/GI/projects. php. The following individual kindly provided reagents, samples, or unpublished information as indicated in the paper: S.L. Martin.]
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3421505. Freely available online through the Genome Research Immediate Open Access option.
-
↵5 Corresponding author. E-mail schgr{at}pei.de; fax 49-6103-771265.
-
- Accepted April 15, 2005.
- Received November 2, 2004.
- Cold Spring Harbor Laboratory Press








