
Fish genomics and biology
Abstract
The last common ancestor between fish and mammals dates back to the very origin of the vertebrate lineage and today, half of modern vertebrates are fish. It is thus not surprising that several fish species have played important roles in recent years to advance our understanding of vertebrate genome evolution, to inform us on the structure of human genes, and, somewhat more unexpectedly, to provide leads to understanding the function of genes involved in human diseases. Genome sequence comparisons between such distantly related organisms are highly informative due to the accumulation of neutral mutations in nonfunctional regions. Yet humans and fishes share many developmental pathways, organ systems, and physiological mechanisms, making conclusions relevant to human biology. The respective advantages of zebrafish, medaka, Tetraodon, or Takifugu have been well exploited so far with bioinformatics analyses and molecular biology techniques. However the full potential of fish genomics is about to be unleashed with the integration of more traditional disciplines such as biochemistry and physiology, with the study of additional species such as carp, trout, or tilapia and a broadening of its applications to environmental genomics or aquaculture.
“There the nets brought up beautiful specimens of fish: Some with azure fins and tails like gold, the flesh of which is unrivalled; some nearly destitute of scales, but of exquisite flavour; others, with bony jaws, and yellow-tinged gills, as good as bonitos; all fish that would be of use to us.” While the gastronomic qualities of fish did not escape Jules Verne in his 1870 20,000 Leagues Under the Sea, fish are no less put to good use in twenty-first century biology. In this new context, one could easily replace fin color and flesh quality by genome size and embryo transparency in a similar enumeration of the advantages of these animals for biology in general and molecular genetics in particular. If Captain Nemo was in a position to offer such variety on his menu, it is partly because fish comprise more than 25,000 species, by far the most successful vertebrate group. Indeed few aquatic ecosystems have eluded colonization by at least some fish species, from Tibetan streams to the abyss of the oceans via sub-zero Antarctic seas (Nelson 1994). Of these species, many have long been used as models in different disciplines of biology (Fig. 1) because of this very diversity: The atrophy or exaggeration of important anatomical or physiological functions occur with sufficient frequency to have attracted biologists to fish models (Epstein and Epstein 2005). This includes molecular genetics and genome research, for which fish also possess interesting and outstanding features, if not all-time records, among vertebrates.
About 30 years ago, a popular tropical aquarium fish named Danio rerio (zebrafish) was already seen as endowed with many advantages for genetic analysis: a short generation time (about 3 mo), large egg clutches all year round, easy maintenance, and external development of a transparent embryo (Streisinger et al. 1981). Combined with large-scale mutagenesis screens initiated in the early 1990s (Haffter et al. 1996; Stainier et al. 1996), zebrafish filled a gaping hole in vertebrate developmental biology: the ability to study genes via their mutant phenotypes on a large scale as in Drosophila melanogaster or Caenorhabditis elegans. Since then, capital discoveries for our understanding of vertebrate development and human disease have already emerged from zebrafish studies. However if one considers genome analysis a question of DNA sequence acquisition and “mining,” then fishes really became a major player in 1993 when Sydney Brenner suggested a new species as a genome model, the marine pufferfish Takifugu rubripes (fugu) (Brenner et al. 1993). Aside from its gastronomic delicacy status in Japan and China, fugu possess one of the smallest vertebrate genomes. This feature, already recorded for its freshwater relative Tetraodon nigroviridis in 1968 (Hinegardner 1968), is a major advantage to rapidly gain access to a large catalog of genes in a vertebrate at a cost comparatively smaller than for the much larger genome of a mammalian species. However, both pufferfish are species for which we know little in terms of physiology, reproduction, or life cycle. Since biology is still a science largely driven by the quality and depth of the experimental data and our ability to extract meaning from it, the outcome of both pufferfish genome programs has until recently been confined to discoveries on the structure and evolution of genes and genomes, with few connections to development, cell biology, or physiology.
However this is about to change, with the emergence of networks of scientists and cross-disciplinary platforms where precise biological questions are examined with an array of tools and resources that include genome sequences and genomic techniques. Here we review the impact of the genome sequence for those fish species for which it is already available, and we examine how the combination of genomics with more traditional disciplines might pave the way for a much wider impact on biology.
Angling in the genomic aquarium
The sequence of the human genome was still a distant goal in 1993, but the project of sequencing the entire genome of a multicellular eukaryote, that of the nematode worm C. elegans, was well on its track (Sulston et al. 1992). As it happens, many important eukaryotic model organisms that were already being studied with molecular biology approaches had genome sizes within the reach of sequencing technologies of the time: Saccharomyces cerevisiae (14 Mb), D. melanogaster (180 Mb), C. elegans (100 Mb), and Arabidopsis thaliana (125 Mb). But none of these were vertebrates, a situation which motivated a pilot project to evaluate the usefulness of fugu as a model vertebrate genome (Brenner et al. 1993). This influential analysis showed that the genome was indeed about 400 Mb, or eight times smaller than the human or mouse genomes. Just as important, exon–intron boundaries seemed conserved, suggesting that gene structures were likely to be very similar, albeit in a much more compact sequence. Rapidly, other studies strengthened the notion that pufferfish DNA could help identify and better understand the structure and sometimes the function of mammalian sequences.
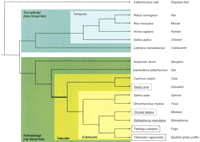
Consensus phylogenetic relationships between fish (simplified from Nelson 1994; Inoue et al. 2003) and tetraopods, including fish considered as biological models (underlined), and those for which a sequence has been published (boxed, continous line) or is underway (boxed, dashed line). The thick branch indicates the most likely position of the whole-genome duplication at the root of the teleosts, based on Hoegg et al. (2004).
In one such early attempt, a fugu sequence next to the Hoxb4a gene and conserved with mouse, showed enhancer activity in transgenic mice (Aparicio et al. 1995). The power of comparative genomics in vertebrates was also soon illustrated when the complete Huntington disease (HD) gene from fugu was sequenced and compared to its human ortholog (Baxendale et al. 1995). The analysis showed that the fugu HD gene possesses a four-glutamine repeat, whereas mouse has seven and healthy humans a minimal of eight. Because a tract of four glutamines is unlikely to form a functional site in itself (such as a polar zipper-,Perutz et al. 1994), the fugu protein supported the view that it is only after expanding to over 37 residues in HD patients that it somehow gained a new pathogenic function. This initial sequencing project in fugu was followed by many more that generally showed limited long-range conservation of gene order, rarely more than four genes per conserved synteny block, thus dampening down the initial hope that this compact genome was a valuable tool to accelerate the mapping of human genes, then a priority in the Human Genome Project (Gilley et al. 1997). However there were still major reasons to establish complete sequences of fish genomes: (1) these early studies had shown the usefulness of comparative sequencing when constructing gene models on the human genome, (2) the initiation of large scale mutagenesis projects on zebrafish was calling for a global genome effort already in preparation with the construction of genetic maps (Shimoda et al. 1999; Kelly et al. 2000) and radiation hybrid maps (Geisler et al. 1999; Hukriede et al. 2001).
Trawling for whole genomes
Reevaluating the number of human genes
Fugu is a relatively large marine fish that contains elevated doses of tetrodotoxin causing live specimen or frozen samples to be the subject of restrictive importation laws in most countries outside of Asia and thus posing practical problems for genomic analyses. A different pufferfish, Tetraodon nigroviridis (Green spotted puffer, sometimes confused in the aquarium fish market with Tetraodon fluviatilis, which is a different species) was proposed (Crnogorac-Jurcevic et al. 1997) that alleviates this restriction: Tetraodon also possesses a small compact genome (Hinegardner 1968) but it is a popular aquarium fish that can live in freshwater (Ebert 2001). In contrast to fugu, few specific Tetraodon loci were sequenced and studied in comparison to their homologs in other species. From the beginning instead, Tetraodon genomic DNA was exploited in large-scale comparisons between different vertebrate genomes. The initial rationale behind this second pufferfish project was to assist in the annotation of human genes, a slow and fastidious task when performed by humans, often unreliable when performed by automatic approaches, and yet one of the primary goals of the Human Genome Project. On the basis of an initial sampling of random sequences from the Tetraodon genome (about 30%), a tool named Exofish that was based on BLAST sequence alignments was developed to identify conserved regions in human genomic DNA that correspond to coding exons, rapidly and with high specificity (Roest Crollius et al. 2000). A surprising outcome of this first example of a global sequence comparison between large samples of two vertebrate genomes is that the number of conserved sequences identified by Exofish in the human genome was not compatible with the 60,000 to 150,000 genes that it was thought to possess at the time. Indeed comparisons with the Tetraodon sequence sample indicated that the entire human genome would contain about 88,000 evolutionary conserved regions (termed ecores) corresponding to human exons, while known human genes possessed on average between 2.6 and 3.2 ecores. A simple ratio between these figures yields a total of about 30,000 human genes. This new estimate, confirmed later by the initial analysis of the human genome sequence (Lander et al. 2001), challenged the notion that the complexity of genetic information contained in a genome is a function of the number of protein-coding genes.
Puffer fish genome features in draft sequences
The fugu genome, the first vertebrate genome to be sequenced after human, was obtained using the whole-genome shotgun method (Aparicio et al. 2002). This sequence draft enabled a number of interesting observations, such as differences in specific protein families between human and fugu. The Tetraodon genome sequence was subsequently produced (Jaillon et al. 2004), also with the whole-genome shotgun method albeit with a higher redundancy in sequence reads (8.3 vs. 5.6). Both pufferfish possess about 70 different families of transposable elements against only 20 for human or mouse, but in pufferfish they comprise two to three orders of magnitude fewer copies. Interestingly in Tetraodon, SINE and LINE families are distributed in opposite regions of the genome compared to human or mouse: SINEs are more abundant in G + C-rich sequences in mammals, and in A + T-rich regions in Tetraodon, and vice versa for LINE elements. More surprisingly, these initial studies of Tetraodon and fugu showed a number of differences in their genomes. For instance a G + C-rich region present in both Tetraodon and mammal genomes is absent in fugu. Also some gene families such as type I cytokines and their receptors, present in all vertebrates studied so far, were notably difficult to find in fugu, while over 30 members of the family could be identified in Tetraodon. These discrepancies are most likely attributable to biases in clone libraries or differences in methodologies, and hopefully should be resolved as the genomes reach completion. When comparing fish and mammal gene catalogs, surprisingly few major differences could be documented when using the Gene Ontology (Harris et al. 2004) classification system. More striking differences could be seen using protein domain comparisons: Proteins involved in sodium transport are more abundant in fish, which also contain an allantoin pathway for purine degradation that is absent in humans. Neutral nucleotidic sequence evolution per year was found to be twice as fast in pufferfish as between human and mouse, and protein evolution also appears to proceed at a faster rate in fish, although the reasons for this are still unclear. It should be noted that these results depend on the dating of the divergence between Tetraodon and fugu (18–30 Mya) (Crnogorac-Jurcevic et al. 1997).
Insights into vertebrate genome evolution
Perhaps one of the major differences in the two pufferfish draft sequences resides in the fact that the fugu genome sequence was assembled purely by the whole-genome shotgun method with no physical mapping, whereas most (64%) of the Tetraodon genomic sequence is anchored on each of the chromosomes, providing a long-range view of gene organization in the genome. This added information primarily resolved a long-standing issue on the occurrence of a whole-genome duplication in the fish lineage.
Remarkably, the idea that an increase in chromosome numbers may be a source of phenotypic novelty is nearly a century old. In 1911, Kuwada already observed that some varieties of maize were tetraploid and suggested that this may be the source of “innumerable races” (Taylor and Raes 2004). During the following 60 years, the occurrences and consequences of gene and genome duplications continued to be discussed (Taylor and Raes 2004), with for instance the proposal by Stephens in 1951 that increasing the number of genetic loci was the only path to “evolutionary progress” and his suggestion that genome duplication could be one way of achieving this (Stephens 1951). These theories reached a high point in 1970 with the publication of Susumo Ohno's book (Ohno 1970) that stated several landmark notions: (1) without duplicated genes, the emergence of metazoans, vertebrates, and mammals from unicellular organisms would have been impossible, (2) this process required the creation of new loci with previously nonexistent functions, and (3) he postulated that at least one whole-genome duplication facilitated the evolution of vertebrates. While these ideas were met with mild enthusiasm at the time (Lewin 1971; Spofford 1972), it is today a widely accepted notion that to create functional novelty, gene duplications are at least as important as point mutations in individual loci. Moreover when the duplication affects the entire genome at once, this potential for novelty is theoretically amplified by allowing the duplication and retention of partial or complete metabolic pathways.
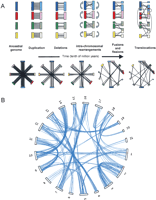
(A) A schematic model of whole-genome duplication with four chromosomes, followed by massive gene loss, chromosome fusions and fissions, inter- and intrachromosomal rearrangements. Each colored rectangle is a chromosome, and lines are drawn between duplicate copies of genes present on sister chromosomes or chromosome segments. The top panel starts with the sister chromosomes facing each other and illustrates the changes induced by chromosome rearrangements, while the lower panel shows the situation in a circular representation, which assumes one does not know the relationships between chromosomes a priori. After several million years of evolution, the distribution of the few duplicate genes that remain do not bear a trace of the ancient duplication event. (B) The same representation as in the lower panel in (A), but with real data from the Tetraodon genome: Despite more than 300 million years since the duplication, the distribution of about 2% of Tetraodon genes that remain strictly in two copies (joined by blue lines) in the genome shows a striking pattern where chromosomes are associated in pairs (e.g., chromosome 9 and 11, or 10 and 14), or sometimes in triplets (e.g., chromosomes 5, 13, and 19). The former suggests that no interchromosomal rearrangements have occurred on these chromosome pairs since the duplication, while the latter is reminiscent of a chromosome fusion or fission.
However the fate of most duplicate copies of genes, over tens or hundreds of million years, is to be eliminated from the genome in a global process called diploidization (Wolfe 2001; Jaillon et al. 2004; Kellis et al. 2004) (Fig. 2). The fact that most duplicate copies of genes have long disappeared from anciently duplicated genomes is one reason why providing proof of the duplication is often difficult. In the case of fish, a strong indication in support of the duplication came from the revelation that zebrafish possess seven HOX clusters on seven different chromosomes, instead of the four clusters found in mammals (Amores et al. 1998). This observation immediately suggested that these four chromosomes at least —but most likely the entire genome— duplicated once in an ancient teleost, followed by the loss of one HOX cluster in the zebrafish lineage. This conclusion was sustained by examples in other fish species (Meyer and Schartl 1999) but stronger support came from comparative analyses in zebrafish using many more gene loci (Postlethwait et al. 2000) as well as results from Expressed Sequence Tags (ESTs) positioned on the zebrafish genetic linkage map (Woods et al. 2000), where it was clear that this genome contained large duplicated segments that did not exist in human or mouse. However, the possibility that the frequent occurrences of duplicated genes in fish originated from a high level of segmental or local duplications could not be entirely ruled out (Robinson-Rechavi et al. 2001). These studies were followed by several attempts at dating the emergence of duplicate gene copies in fugu (Christoffels et al. 2004; Vandepoele et al. 2004) but with a relatively large uncertainty.
The ultimate demonstration rested on the long-range continuity of the Tetraodon sequence assembly covering parts of all chromosomes (Jaillon et al. 2004). Although the paralogs (pairs of genes that appear by duplication of an ancestral gene) that were identified using very conservative criteria represent less than 2% of the current set of genes, their distribution in the genome clearly associates chromosomes in pairs or in triplets, a situation expected if a single tetraploidization event took place at some point in the past, followed by a few chromosome fissions or fusions (Fig. 2). A second and even more telling signature of the duplication was found in the alternating pattern of Tetraodon syntenic groups along human chromosomes. Indeed, the return to a diploid state after the whole-genome duplication mostly affects genes, not necessarily chromosomes. While the two duplicated sister chromosomes remain an integral part of the genome, they each gradually loose about 50% of their genes, in small clusters that alternate between the two (Fig. 3A). To observe the result of this process, one needs to compare the position of genes on a chromosome of a species that did not duplicate during the same period of time, with orthologous genes of the species that did duplicate. The characteristic pattern that results seems quite universal, from single-cell eukaryotes (Kellis et al. 2004) to vertebrates (Jaillon et al. 2004), and is illustrated in Figure 3B. The deletion process takes place because the supernumerary gene copies are not under selection and they thus rapidly acquire deleterious mutations. The choice of which copy will be deleted is driven by the first mutation to occur in one of the two copies, a process that must be random since ultimately each sister chromosome inherits about half of the initial gene complement.
However some duplicate copies are not deleted, in this case implying either that both copies are immediately placed under selection, that a deleterious mutation partially obliterates the function of one copy making the other gene essential, or that the first mutation is not deleterious but favorable. Because it is a genome-wide process of gene selection affecting all functional classes at the same time, it is of interest to investigate, in the case of fish, which classes have emerged as advantageous compared to genomes that did not duplicate. Ideally, the question would be best addressed using one of the living fish species that diverged just before the whole-genome duplication, such as sturgeons (acipenseriformes) or gar (semionotiformes) (Hoegg et al. 2004) (Fig. 1). Until the genome sequence of such a species becomes available, a comparison with the gene catalogs of much more distant species such as mammals using Gene Ontology annotations (Harris et al. 2004) and phylogenetic classifications reveals significant differences: Development, cell differentiation and cell communication classes are enriched in gene duplicates (F.G. Brunet, H. Roest Crollius, M. Paris, J.M. Aury, P. Gibert, O. Jaillon, V. Laudet, and M. Robinson-Rechavi, in prep.). This result is interesting in the light of the teleost radiation that took place approximately at the time of the genome duplication. Also strikingly and as previously shown in yeast and nematode (Davis and Petrov 2004), fish genes that evolve slowly prior to the duplication seem to be preferentially retained in two copies after the duplication: but what of the rate of evolution between the two retained copies? Whereas the subfunctionalization model (Force et al. 1999) predicts that the two copies will share the ancestral function and thus evolve under the same constraints, the neofunctionalization model (Ohno 1970) proposes that the emergence of a new function in one copy occurs under positive selection, i.e., one copy will evolve faster than the other. Interestingly, in the above-mentioned study, Tetraodon gene duplicates recurrently show a markedly accelerated evolution of one of the two copies, in agreement with the latter model and supporting the view that genome (and hence gene) duplication is a driving force behind the emergence of functional novelty. The ancestral teleost genome duplication provided its owner with a powerful toolkit to adapt and diversify: twice as many genes as any other emerging vertebrate. It is tempting to propose that a consequence of this genome doubling is to be found in the rich diversity of extant fish species, unparalleled today among vertebrates (Amores et al. 1998; Meyer and Schartl 1999). Ultimately, the availability of more fish genome sequences will help distinguish between teleost-wide and lineage-specific strategies for the retention of beneficial duplicate functional classes. But for now, the teleost genome duplication provides a direct entry point into another exciting theme: the reconstruction of the ancestral genome prior to the duplication, which would closely resemble that of the ancestral bony vertebrate genome. Studies in zebrafish (Postlethwait et al. 2000), medaka (Naruse et al. 2004), and Tetraodon (Jaillon et al. 2004) have already delineated the probable 12 proto-chromosomes with increasing precision.
Zebrafish and medaka: Biological models come back
The zebrafish as a model system has accompanied the development of molecular biology from the 1960s to the genomic revolution of the 1990s (Grunwald and Eisen 2002). Following the publication of the results from two large-scale genetic screens in 1996 in a special issue of Development (1996;123:1–461), zebrafish was propelled at the forefront of developmental biology research. While such screens are currently being expanded to tackle a wider range of mutations in embryonic and adult stages (see below), many researchers are also studying zebrafish models of human diseases (Dooley and Zon 2000) including blood diseases (de Jong and Zon 2005), heart disorders (MacRae and Fishman 2002), and cancer (Amatruda et al. 2002). As mentioned above, genome research on zebrafish started in parallel to these developments. Early efforts to map genes and genetic markers served to identify the loci of specific mutant genes, and to compare the organization of genes with other vertebrates (Barbazuk et al. 2000; Woods et al. 2000). Microarray technology has also been applied, in combination with single nucleotide polymorphism (SNP) identification, to accelerate the linking of mutant phenotypes to their genotypes (Stickney et al. 2002).The zebrafish genome is about 1700 Mb, and that of the Tübingen strain is now being sequenced at the Sanger Institute using a mixed strategy where both large insert clones and the entire genome are submitted to the whole-genome shotgun method. Today the zebrafish community worldwide has expanded tremendously with over 3500 scientists registered in the ZFIN community database (zfin.org). The availability of the genome sequence will provide a unifying reference to integrate the wealth of functional data accumulated so far.
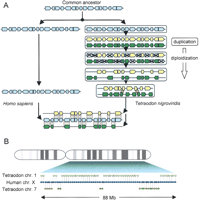
(A) Duplication leads to double-conserved synteny. After speciation, a chromosome or chromosome segment from an ancestral species is duplicated in one lineage but not in the other. In the former (e.g., Tetraodon), supernumerary copies of genes are progressively deleted from each of the duplicated segments in approximately equal proportion (diploidization). Ultimately the two duplicated chromosomes only contain 50% of the initial gene complement and are thus very different from each other. The difficulty of finding the original pair of sister chromosomes can be alleviated by a comparison with a genome that originates from the same pre-duplication ancestor, but that did not duplicate (e.g., human). The nonduplicated chromosome segment should contain genes with orthologs alternating between the two duplicated chromosomes (adapted from Kellis et al. 2004). (B) Example of double-conserved synteny. An 88-Mb region covering the majority of the long arm of human chromosome X contains 65 genes with orthologs in the Tetraodon genome, alternating between chromosome 1 and chromosome 7. Genes are represented by small arrows that indicate the orientation of transcription.
The Medaka (Oryza latipes) has also long been used as a model in genetics, dating back to the beginning of last century (Wittbrodt et al. 2002). While in many respects it matches zebrafish in its advantages as a laboratory model, Medaka possesses several characteristics of its own as a useful biological model such as the existence of several fertile inbred strains and embryonic stem cells that can be stably cultured long enough to enable genetic manipulation (Hong et al. 1996), although their use is currently limited by the fact that they do not contribute to the germ line. The complementarity between medaka and zebrafish is obvious when considering the numerous mutations of the same locus that affect each fish differently or even uniquely, thus enabling the deciphering of species-specific patterns for these mutations (Furutani-Seiki and Wittbrodt 2004). Interestingly, Medaka is the only fish so far for which a single genetic locus, DMY, has been found to be responsible for sex determination, as in mammals (Matsuda et al. 2002; Nanda et al. 2002). While DMY is a recent invention in the Medaka lineage (Lutfalla et al. 2003; Volff et al. 2003) and thus was not found in other model fish species (Kondo et al. 2003), it provides the first opportunity to study the molecular basis of sex determination in fish. Many resources for genome research have been developed for Medaka, including genetic maps (Naruse et al. 2004), EST sequences (Kimura et al. 2004), and physical maps (Khorasani et al. 2004). The 800-Mb genome of Medaka has now been sequenced and assembled, and the analysis that is under way should reveal exciting insights into vertebrate genome evolution.
The future of fish genomics
The future of fish genomics is bright, and this prediction is sustained below by three examples where specific characteristics of fish species were successfully exploited to gain penetrating insights in a broad range of subjects.
Fish, as a highly diversified group of the vertebrate family, experience an astonishing range of environmental conditions to which their physiologies, body shapes, and lifestyles have adapted. However, a common denominator of all fish species is their aquatic habitat, meaning that water is in direct contact with several tissues and internal compartments of the animal, potentially inducing a high sensitivity to water-borne parameters such as temperature, oxygen levels, salinity, and sometimes toxic chemicals. This intimate relationship between an organism and a wide range of different environments has recently prompted the view that fish could be used as models for “environmental genomics” (Cossins and Crawford 2005), in other words the study of the interface between an organism and its environment using genomic approaches. This concept had recently been illustrated by an elegant study involving the exposure of carp to increasing levels of cold, from 30°C to 10°C and measuring the change in the level of expression of several thousand genes using microarray techniques (Gracey et al. 2004). Carp, as most fish, is poikilothermic (cold-blooded), which implies that its body temperature follows that of the water it is immersed in. In the seven tissues that were monitored, the decrease in temperature was correlated with a graded increase in expression of a core set of 252 genes predominantly from transcriptional regulation, RNA splicing and translation systems, while very few genes common to all tissues showed a decrease in expression. Conversely, tissue specific responses showed that brain modifies its glycolytic activity while a switch to lipid metabolism is observed in liver. Interestingly this study was performed in a nonmodel species, exemplifying the use of a specific species of fish with a specific feature of interest, in this case the carp which tolerates a wide range of temperature, to investigate a whole body physiological adaptation to a change in environment. While the results are useful to better understand the molecular basis of the response to cold in carp, fish share so many aspects of their developmental pathways, physiological mechanisms and organ systems with mammals that these results are also relevant to human physiology.
The second illustration of the strength of future genomic research using fish species is its recognition by international funding agencies. One such recent example is the major impetus to zebrafish genomics provided by the European Union Framework Programme for Research. By funding the ZF-MODEL consortium (www.zf-models.org), an Integrated Project devoted to the study of zebrafish models for human development and disease, the project links about 30 groups from seven European countries (Bradbury 2004). The research plan is to take advantage of the zebrafish model in combination with an array of large-scale techniques to identify zebrafish genes that replicate to some degree human pathologies, or are involved in developmental pathways similar to ours. The scale of this endeavor can be measured by the thousands of mutants already generated by the consortium members using either forward genetics (for instance with chemical mutagenesis screens), or reverse genetics (for instance using the TILLING technique; McCallum et al. 2000) and the scope of the screens used to analyze them. Designed to identify mutants that resemble human disorders, these screens are also carried out on adult fish and encompass for instance bone malformation, skin development, eye movement or addiction behavior. Combined with enhancer detection techniques using Green Fluorescent Protein (GFP) transgenic lines and expression profiling with microarray analysis, the consortium will integrate a broad range of expertise to tackle the function of the mutated genes, which hopefully will lead to a better understanding of corresponding human disorders.
In addition to counting several member species elevated to the status of genomic models such as zebrafish or pufferfish, fish also represent a major source of food for humans. The European Union thus also recognized the need for improving aquaculture research by funding the AQUAFIRST consortium to identify genes associated with stress and disease resistance in sea bream, sea bass, and rainbow trout in order to provide a physiological and genetic basis for marker-assisted selective breeding. Genomic techniques and sequence comparisons with genomic models are cornerstones of this project, which illustrates how fish genomics may grow in coming years outside of fundamental research labs toward more applied objectives nonetheless essential to human welfare.
A final example of the use of fish genomes that may expand in the near future rests on the phylogenetic position of fish species in comparison to mammals (Fig. 1). Today a great deal of attention is focused on the part of eukaryote genomes that do not code for proteins but are nevertheless functional. Elements that may carry out specific functions in these regions include non-coding RNA genes and regulatory control regions. One of the most powerful techniques to identify such elements is to align genomic sequences of distantly related organisms and look for regions that have remained similar during evolution, thus suggesting that a functional constraint is acting to preserve the sequence from mutations. The advantage of fish in this context is the long evolutionary distance, approximately 450 million years, since their last ancestor with mammals. Neutral mutations have since saturated the genome to a point where any conserved region between for instance human and pufferfish is indicative of a functional constraint. This comparative approach was first applied on a genome scale to identify coding exons (Roest Crollius et al. 2000), and more recently to identify ultra-conserved regions (UCRs) of unknown function (Sandelin et al. 2004; Woolfe et al. 2004). However an additional assumption that can be deduced from the discovery of UCRs conserved between such distant organisms is their fundamental importance across vertebrates. In line with this, UCRs found in this way lie in clusters around genes involved in the regulation of development. Indeed when the orthologous regions were assayed using GFP reporter constructs in zebrafish embryos, most showed significant enhancer activity in one or more tissues (Woolfe et al. 2004). So fish–mammal sequence alignments not only provide the means to identify functional elements, they also act as a screen to select those elements essential to vertebrates. With the production of new fish genome sequences as well as new mammalian sequences, such comparative studies are likely to play an important role in guiding the identification of functional noncoding elements, and in deciphering the subtle sequence variations that might lead to phenotypic changes (Ahituv et al. 2004; Boffelli et al. 2004).
Many fish species are routinely being studied at the molecular level and even at the genomic level and have not been cited here. For instance thousands of EST sequences are available for carp, catfish, salmon, trout, killifish, stickleback, or tilapia and large insert BAC libraries are also available for several of these species, further illustrating the widespread interests in using fish for genomic research. Four fish genome sequences are or will be available soon: fugu, Tetraodon, medaka, and zebrafish. The stickleback genome (Gasterosteus aculeatus) is also well advanced (Table 1), but to our knowledge no other is currently ongoing beyond these, although obviously several interest groups are actively working towards promoting certain species. Research on salmon and trout for instance would greatly benefit from the availability of the genome sequence. Indeed more is known about the physiology and biology of rainbow trout than any other fish species, although genomic sequencing could be complicated by an additional genome duplication in the salmonid lineage some 25–100 Mya (Allendorf and Thorgaard 1984). The sarcopterygian fish coelacanth is the nearest living relative of tetrapods (Gorr et al. 1991) (Fig. 1) and thus escaped the whole-genome duplication that affected the teleosts. This may be a serious advantage since it might provide access to a genome that resembles the early tetrapod genome, unaffected by the consequence of massive gene duplications such as gene conversion. Altogether the coelacanth would indeed be an excellent candidate for genome sequencing as it would provide a reference genome for tetrapods while allowing the identification of genomic features that differentiate them from teleosts (Noonan et al. 2004). For similar reasons but across a wider evolutionary scale, the elephant fish (Callorhinchus milii, a cartilaginous fish; Fig. 1) has recently been proposed as a good model to study the genome structure and gene content of a basal jawed vertebrate, and provide a common reference for tetrapods and ray-fined fishes (Venkatesh et al. 2005).
Summary of current genomic resources on fish species
Up to now fish genomics has been able to draw on the similarities and the differences between mammalian and fish genomes to gain profound insights into the evolution of vertebrate genomes in general, and into the function of individual genes often associated with human disorders in particular. Captain Nemo's fishing exploits with the Nautilus may be hard to match but the net cast by genome scientists has also reeled in some unexpected surprises, and the end of this story is certainly a long way off.
Footnotes
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3735805.
-
↵3 Corresponding author. E-mail hrc{at}ens.fr; fax 33-1-44-32-39-41.
- Cold Spring Harbor Laboratory Press








