
ORFeome Cloning and Systems Biology: Standardized Mass Production of the Parts From the Parts-List
Abstract
Together with metabolites, proteins and RNAs form complex biological systems through highly intricate networks of physical and functional interactions. Large-scale studies aimed at a molecular understanding of the structure, function, and dynamics of proteins and RNAs in the context of cellular networks require novel approaches and technologies. This Special Issue of Genome Research features strategies for the high-throughput construction and manipulation of complete sets of protein-encoding open reading frames (ORFeome), gene promoters (promoterome), and noncoding RNAs, as predicted from genome and transcriptome sequences. Here we discuss the use of a recombinational cloning system that allows efficiency, adaptability, and compatibility in the generation of ORFeome, promoterome, and other resources.
An important transition is taking place in biological research. The field of genome sequencing and annotation (Goffeau et al. 1996; Blattner et al. 1997; The C. elegans Sequencing Consortium 1998; Adams et al. 2000; Arabidopsis Genome Initiative 2000; Lander et al. 2001; Venter et al. 2001; Waterston et al. 2002; Gibbs et al. 2004) is now complemented by systems biology approaches that aim to decipher the biological networks in which cellular macromolecules function. Nearly complete lists of genes, the “parts-lists,” are available for several model organisms and for human. With such parts-lists in hand, it is possible to produce nearly complete collections of proteins, RNAs, or promoters, that is, to mass produce the parts from the parts-lists, and then to functionally characterize macromolecules in highly parallel assays, enabling global studies of the networks and systems in which macromolecules function (Vidal 2001).
Novel emerging methodologies and strategies enable this transition (Rual et al. 2004b). Particularly, new operating systems for mass cloning allow the generation of flexible, standardized clone collections that provide compatibility between resource collections, not only for a single organism, but across collections from different organisms. DNA cloning tools based on recombinational cloning (RC), such as the Gateway cloning technology (Hartley et al. 2000), allow the development of such resources for systems biology (Walhout et al. 2000a,b). Here we focus primarily on Gateway and Gateway-generated resources. Alternative RC methods (Liu et al. 1998; Paddison et al. 2004) are described and compared elsewhere in this Special Issue (Marsischky and LaBaer 2004; see also Table 1).
Summary of Recombinational Cloning Strategies in Terms of Demonstrated Efficiency, Adaptability, and Compatibility (See Text), Relative to Currently Available Full-Length cDNA Resources
Macromolecules as Components of Biological Networks
Proteins, RNAs, and DNA (promoters, e.g.) require intricate physical and functional interactions with other macromolecules, each with particular temporal and spatial aspects, to mediate their function. Understanding the complex networks formed by all these interactions is critical for a global perspective of biological systems (Barabasi and Oltvai 2004). For example, the incredible robustness exhibited by many cell types might relate to the overall topology of cellular networks (see below; Jeong et al. 2001). Thus, in addition to the reductionist characterization of macromolecules necessary for the detailed understanding of individual interactions (molecular biology), the complex networks they form by interacting with each other also need to be studied (systems biology; Fig. 1; Ideker et al. 2001; Vidal 2001).
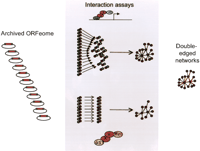
Modeling cellular networks requires efficient recombinational cloning “operating systems.” (A) Physical interactions. This example illustrates how the mapping of physical interactions between proteins can benefit from the use of two different assays (“Double-edged” networks). Here the yeast two-hybrid (Y2H) system is first used with individual DB-X baits and a pooled AD-ORFeome library (Reboul et al. 2003). Subsequently, positive Y2H interactions are retested using a different assay (Li et al. 2004): GST pull-down followed by anti-Myc Western-blot analysis. To express high numbers of proteins with the appropriate tags (DB, DNA binding domain; AD, activation domain; GST, glutathione-S-transferase), large collections of archived protein encoding open reading frames (ORFeomes) need to be available (efficiency), in ways that allow their subcloning in many different vectors (adaptability). The resulting “double-edged” networks are of greater overall quality.
Mapping Biological Networks
Modeling biological networks requires understanding of the structure, function, and dynamics of both the individual players and their effect on each other. Although some information is already contained in the scientific literature for a few thousand proteins, promoters, and RNAs studied individually in various organisms, these data cover only ∼10%-20% of the macromolecules encoded by those organisms (Costanzo et al. 2000). Thus, global studies of biological networks require the establishment of “maps” providing information simultaneously on hundreds, if not thousands, of proteins, RNAs, promoter sites, and other macromolecules (and often domains thereof).
Recent work in this emerging field of biological networks has focused on analyzing the structure of metabolic (Jeong et al. 2000), regulatory (Lee et al. 2002), and “interactome” networks (Uetz et al. 2000; Walhout et al. 2000a). Network maps are generally visualized as graphs composed of nodes and edges. Nodes represent the components of biological networks (metabolites, transcription factors or promoters, and proteins or RNAs in metabolic, regulatory, and interactome networks, respectively). Edges represent interactions between those components (enzymatic reactions in metabolic networks and physical interactions in regulatory and interactome networks).
Intriguing biological hypotheses have already emerged from early attempts at mapping the global structure of cellular networks. First, cellular networks appear scale-free, that is, they contain a small but significant proportion of nodes that are highly connected, whereas most nodes are sparsely connected (Jeong et al. 2000). The scale-free topology of cellular networks (Jeong et al. 2000; Lee et al. 2002; Li et al. 2004) might relate to cellular robustness (Jeong et al. 2001). Second, regulatory networks may contain subnetwork motifs, defined as logical subsystems linking small numbers of network components and potentially exhibiting semi-independent functions (Lee et al. 2002; Milo et al. 2002; Shen-Orr et al. 2002). Third, analyses of interactome networks suggest that biological processes are more interconnected molecularly than previously imagined (Walhout et al. 2000a; Walhout and Vidal 2001b). Together, these observations illustrate the need for further systematic mapping and modeling of cellular networks.
Efficiency and the Mass Production of Nodes
The challenges ahead lie in both the manipulation of hundreds of thousands of “biological nodes” (the components) and the determination of large numbers of “biological edges” (interactions) between them (Fig. 1). Network maps for half a dozen model organisms, humans, and key pathogens and parasites will be of value to both academic and pharmaceutical research. Ideally, nearly all proteins, noncoding RNAs, and promoters of these organisms should be available for network analyses.
Considering these challenges globally, the generation of nearly complete ORFeome, promoterome, and other “parts” resources for these different species will require the handling of hundreds of thousands, perhaps millions, of DNA segments. In the context of this enormous task, conventional cloning techniques have been increasingly replaced by recombinational cloning strategies.
Adaptability and the Mass Production of Edges
In concert with the production and manipulation of large numbers of biological nodes, hundreds of thousands of biological edges need to be mapped between them (Fig. 1).
Early attempts at mapping regulatory and interactome networks exist for Saccharomyces cerevisiae (Uetz et al. 2000; Ito et al. 2001; Gavin et al. 2002; Ho et al. 2002; Lee et al. 2002), Caenorhabditis elegans (Walhout et al. 2000a; Davy et al. 2001; Boulton et al. 2002; Walhout et al. 2002; Reboul et al. 2003; Li et al. 2004), and Drosophila melanogaster (Giot et al. 2003). Although highly informative, such projects need to be extended to greater proportions of the respective proteomes and promoteromes. Currently available interactome maps strongly suggest that a variety of methods will need to be applied to cross-validate the data quality of each edge (von Mering et al. 2002; Han et al. 2004; Li et al. 2004). False positives and false negatives inherent to high-throughput interaction assays are reduced to more acceptable levels when edges are tested in a variety of different assays (Han et al. 2004).
Although metabolic, regulatory, and interactome networks represent an important scaffold to comprehend the structure of cellular networks, other types of edges need consideration as well (Vidal 2001), such as protein modification networks in which nodes represent proteins and edges represent phosphorylation, acetylation, methylation, ubiquitination, or other posttranslational regulation relationships between them; and protein-RNA and RNA-RNA interaction networks.
Considering the many types of edges required, and that these edges need verification by different assays, ORFeome, promoterome, and other functional resources need to be produced in ways that are compatible with multiple different biological assay formats. For example, the production of proteins from ORFeome resources typically involves the handling of tens of thousands of ORFs and their incorporation into a myriad of expression vectors (Walhout et al. 2000b). Conventional cloning techniques that use restriction enzymes and ligase pose substantial challenges given such adaptability requirements of ORFeome projects. This is primarily because such conventional cloning strategies need to be redesigned for every new construct, both DNA segments and vectors.
Function of Biological Networks
Whereas the structure of biological networks is informative, studying the functional and dynamic features of biological networks is also crucial to understanding cellular biology. Ultimately, the functional consequence of interactions must be considered to appreciate the global role of nodes and edges within a network.
A global approach to assess the function of biological networks in vivo uses systematic node perturbations. In S. cerevisiae, gene knockouts are available for the whole ORFeome (Winzeler et al. 1999; Giaever et al. 2002). In multicellular organisms, “phenome” mapping analyses are now feasible with the development of RNA interference or RNAi (Fire et al. 1998). Genome-scale RNAi resources are available for C. elegans and D. melanogaster (Fraser et al. 2000; Gonczy et al. 2000; Piano et al. 2000; Maeda et al. 2001; Kamath et al. 2003; Lum et al. 2003; Simmer et al. 2003; Boutros et al. 2004). Similar resources are becoming available for human and mouse using small inhibiting RNAs (siRNAs) (Dorsett and Tuschl 2004) or small hairpin RNAs (shRNAs) (Berns et al. 2004; Paddison et al. 2004), and for Arabidopsis thaliana, using larger shRNAs (Hilson et al. 2004).
Here again we face the double challenge of efficiency and adaptability. Indeed, one of the advantages of RNAi techniques is provided by the potential to regulate silencing by spatial or temporal induction (Tavernarakis et al. 2000). Thus, constructs suitable for RNAi are needed in various expression vectors that include a diversity of tissue-specific or temporally specific promoters. For C. elegans, the ORFeome itself is effective as an RNAi resource, suggesting that genes can now be knocked down under many different conditions from various vectors (Rual et al. 2004a).
Lastly, crucial information can be gained by perturbing edges of biological networks by, for example, disrupting physical interactions between proteins. The reverse two-hybrid system can be used to genetically select “interaction-defective alleles” (Vidal et al. 1996; Endoh et al. 2002). Once selected, such alleles are characterized using in vitro biochemical assays and in vivo functional experiments (Yasugi et al. 1997; Endoh et al. 2002), and thus not only wild-type but also various mutant ORFs need to be cloned in flexible formats.
Dynamics of Biological Networks
Real-time evaluation of specific proteins and complexes, using the diverse collection of fluorescent proteins (FPs; Chalfie et al. 1994; Tsien 1998), provides a powerful approach to probe the dynamics of molecular networks. Experiments in which promoters direct expression of FPs inform whether, where (within a multicellular organism), and when (developmentally) promoters are activated or repressed. Similarly, proteins or protein domains fused to FPs report subcellular localization (Huh et al. 2003) or movement (e.g., translocation or trafficking; Simpson et al. 2000). Further developments, including fluorescence resonance electron transfer (FRET) techniques (Miyawaki and Tsien 2000), allow the in vivo detection of physical interactions and dissociations between gene products and metabolites or second messengers. Together FPs and FRET identify the where and when of large numbers of nodes and edges in vivo.
Homologous recombination was used to create a set of ∼6000 S. cerevisiae strains, each containing a GFP encoding sequence cloned precisely at the 3′-end of each predicted ORF in the S. cerevisiae genome sequence (Huh et al. 2003). This resource was used to generate an initial draft of the S. cerevisiae “localizome” map. The fortuitous availability of a highly efficient and specific operating system (homologous recombination) specific to yeast greatly facilitated these studies.
Unfortunately, precise homologous recombination strategies are not available for most multicellular organisms, at least not at ORFeome scale. For multicellular organisms, FP fusions typically are first generated in vitro, and then transferred into the relevant organisms by transgenesis. Large numbers of promoters and ORFs need to be molecularly manipulated and pieced back together for construction of the desired fusions. Once promoterome and ORFeome are cloned, they also need to be transferred together into many different expression vectors, such as those encoding different alleles of FPs. For example, promoters from a promoterome resource might need to be cloned upstream of ORFs from an ORFeome resource to generate large combinatorial collections of promoter::ORF::GFP fusions (Dupuy et al. 2004). Likewise, N-terminal and C-terminal FP fusions sometimes give rise to different results, and thus systematically testing both might be advantageous (Simpson et al. 2000). Thus, in addition to efficiency and adaptability, dynamic analyses also require compatibility.
Synthetic Biology
Biological networks can be organized into subnetworks comprising a discrete number of nodes and edges between them (Milo et al. 2002; Schuster et al. 2002). The logic of the wiring diagrams formed by the interacting components in subnetworks can be partly modeled from integration of complementary data on the structure, function, and dynamics of networks (Ge et al. 2003). Prototypical subnetwork motifs, such as the feedforward loop, were hypothesized from the analysis of regulatory network maps (Milo et al. 2002).
One goal of synthetic biology is to test models by the de novo reconstitution of entire subnetwork motifs exogenously. Synthetic reconstitutions of potential subnetwork motifs published so far include (1) the “synthetic genetic toggle switch,” which reconstitutes a model based on a positive feedback loop involving bacteriophage cro and cI proteins, each repressed by the product of the other (Gardner et al. 2000); and (2) the “repressilator,” a synthetic system that consists of three promoter-ORF pairs such that each promoter's gene product represses the next promoter in the cycle (Elowitz and Leibler 2000). In these synthetic biology experiments, many constructs needed to be generated with promoters and ORFs cloned adjacent to each other (Guet et al. 2002).
The ultimate goal of synthetic biology, still distant, is to engineer a self-sustaining, free-living cell. A prerequisite is the development of applicable cloning strategies. As with network biology, such synthetic biology will require cloning and assembling of large numbers of promoters, ORFs, and domains, as well as perhaps noncoding RNAs or 5′- or 3′-UTR regions, all with high efficiency, adaptability, and compatibility (Fig. 2).
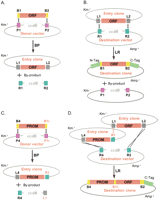
The Gateway recombinational cloning system. (A) ORFs flanked by recombinational sites B1 and B2 are directionally cloned into a P1::ccdB::P2 Donor vector using the BP reaction. The resulting Entry clones are selected using the kanamycin-resistance marker (Kmr). (B) ORFs flanked by L1 and L2 sites are directionally transferred from Entry clones into R1::ccdB::R2 Destination vectors using the LR reaction. The resulting Destination clones are selected using the ampicillin resistance marker (Ampr). (C) Promoters flanked by recombinational sites B4 and B1r are directionally cloned into a P4::ccdB::P1r Donor vector using the BP reaction. The resulting Entry clones are selected using the kanamycin-resistance marker (Kmr). (D) ORFs flanked by L1 and L2 sites and promoters flanked by L4 and R1 are directionally transferred right adjacent to each other from their respective Entry clones into R4::ccdB::R2 Destination vectors using the LR reaction. The resulting Destination clones are selected using the ampicillin-resistance marker (Ampr).
Mass Production: The Ford Model T Analogy
An intriguing analogy to the genome-wide and proteome-wide approaches described so far is the mass production of 15,000,000 Model T automobiles in the early 20th century. Genome and transcriptome sequencing followed by gene annotation projects have produced blueprints of the “parts” of biology. Before (re)placement of the parts in biological networks by reverse engineering, we need to learn how to mass-produce them from this parts list, and to standardize their assembly.
The Ford Motor Company established three principles in mass production: (1) continuous flow from a moving assembly line, (2) division of labor, and (3) precision manufacturing of standardized and interchangeable parts.
For molecular biology, the first two principles were implemented during the Human Genome Project. For example, the Genomatron and the Sequatron (Hawkins et al. 1997) introduced the notion of assembly lines and division of labor, reducing the unit cost of physical mapping and DNA sequencing to manageable levels. Likewise, many of the investigators represented in this Special Issue have converted their lab operations from manual pipetters and Eppendorf tubes to automated and integrated processes, including premium robotics devices, 96- or 384-well plates, and sophisticated database management with barcode tracking of reagents.
Producing and characterizing the parts from the parts-list also demands application of the third principle of mass production: standardization. Ideally, the cloning of ORFs, promoters, protein domains, noncoding RNAs, 5′- and 3′-UTRs, and other DNA segments of interest should be done analogously to the making of the Model T. Interchangeable parts meant that the individual pieces of the car were made the same way every time, so that any valve fit any engine, and any steering wheel any chassis: a revolutionary concept at the time. Previously, parts had been manufactured by hand, providing neither standardization nor interchangeability, severely limiting mass production. This shortcoming parallels the limitation of applying conventional DNA cloning techniques to generating genome-wide resource collections such as ORFeomes and promoteromes, and the great potentials of new RC methods.
Recombinational Cloning: In Vivo Gap Repair
Recombinational cloning techniques were first applied in S. cerevisiae by the “gap-repair” method (Orr-Weaver et al. 1983; Ma et al. 1987; Oldenburg et al. 1997). Gap repair allows the directional cloning of a DNA segment into a restricted “gapped” vector, provided that 30 to 50 nt identical to the vector's ends are added to the 5′- and 3′-ends of the segment to be cloned. In vivo homologous recombination in S. cerevisiae cells and to some extent in bacteria (Zhang et al. 1998), particularly those augmented with certain phage-derived proteins (Yu et al. 2000; Testa et al. 2003), allows robust transfer of such “tailed” segments into any vector of interest with relatively high efficiency.
Despite significant advantages, gap repair is limited by low adaptability and compatibility. Once DNA segments are cloned by gap repair into S. cerevisiae cells, the resulting clones cannot conveniently be extracted from these cells. Moreover, the construction of additional different vector backbones involves repeating the recombination reaction using portions of the PCR amplification mixture (or that from a repeat amplification). Given the potential of PCR-induced errors in amplification products, identity between the amplified DNA segments in the different vector backbones is not ensured, reducing the quality of a clone resource, and requiring extensive DNA sequence analysis to confirm the identity of each clone.
Recombinational Cloning: The Gateway System
The Gateway cloning system resolves all three challenges: efficiency, adaptability, and compatibility (Hartley et al. 2000; Walhout et al. 2000a,b; Cheo et al. 2004; Dupuy et al. 2004). Gateway provides ways to (1) directionally clone PCR products into a Donor vector (Fig. 2A,C); (2) transfer the resulting cloned segments into many Destination vectors in parallel (Fig. 2B); and (3) link DNA segments in a predefined order, orientation, and reading frame within Destination vectors (Fig. 2D). All this is made possible by the sophisticated knowledge of the biology of Escherichia coli bacteriophage λ (phage λ).
Phage λ grows either as a lytic phage or as a lysogen integrated into the genome of E. coli by a reversible recombination event. First, the integration reaction in which the phage attP site recombines with the bacterial attB site leads to an integrated prophage flanked by attL and attR sites. Second, the excision reaction in which the attL and attR sites recombine recreates the attP and attB sites in the phage and bacterial genome, respectively. The wild-type attP, attB, attL, and attR sites are made of 243, 25, 100, and 168 bp, respectively (Fig. 2). In Gateway, the in vitro attB × attP reaction is catalyzed by the “BP clonase,” comprised of the phage Integrase (Int) and the E. coli Integration Host Factor protein (IHF), whereas the attL × attR reaction is catalyzed by “LR clonase,” a mixture of phage λ Excisionase (Xis) together with Int and IHF (Landy 1989; Hartley et al. 2000).
In the Gateway system (Hartley et al. 2000), the modified att recombination sites render the excision reaction irreversible and more efficient, eliminate all stop codons, and, importantly, exist as two different specificities of recombination sites, referred to below as “1” and “2.” The “1” and “2” site specificities do not recombine with each other (Fig. 2), allowing directional cloning of DNA segments both from PCR products into Donor vectors, and from the resulting Entry clones into Destination vectors. An attB1 site only recombines with attP1, whereas the related but different attB2 site only recombines with attP2. Likewise, the corresponding attR1 site only reacts with attL1, whereas the attR2 site reacts only with attL2. Lastly, other introduced mutations minimize secondary structure formation in single-stranded forms of attB plasmids. All recombination reactions are conservative, meaning no net synthesis or loss of base pairs, ensuring maintenance of reading frame following recombination. For simplicity, we abbreviate the sites: (attB1) B1, (attB2) B2, (attP1) P1, (attP2) P2, (attL1) L1, (attL2) L2, (attR1) R1, and (attR2) R2.
In ORFeome cloning projects (Walhout et al. 2000b), ORFs flanked by B1 and B2 sites provided in the PCR primers (B1::ORF::B2) are directionally recombined into a Donor vector containing both P1 and P2 sites separated by a negative selectable marker such as ccdB (P1::ccdB::P2; Bernard 1996), and containing an antibiotic resistance marker (kanamycin or spectinomycin) that is different from the ampicillin resistance (AmpR) marker commonly present in expression vectors (Fig. 2). B1::ORF::B2 × P1::ccdB::P2 BP reactions lead to L1::ORF::L2 Entry clones recoverable in E. coli. In the second step, L1::ORF::L2 segments are transferred into various Destination vectors containing both R1 and R2 sites separated by ccdB (R1::ccdB::R2) and containing AmpR or other antibiotic resistance markers different from the Donor vector antibiotic resistance marker. L1::ORF::L2 × R1::ccdB::R2 LR reactions lead to B1::ORF::B2 Expression clones that survive in E. coli.
Expression clones can express either native or fusion proteins. For native (nonfusion) proteins, the coding sequence including the initiation and termination codons is placed by PCR between the B1 and B2 recombination sites (B1::ATG::ORF:: STOP::B2). The B1 and B2 sequences then reside in the 5′-UTR and 3′-UTR of the mRNA transcript, respectively. For N-terminal fusion proteins, however, the ATG codon is provided in the Destination vector and the 25-bp B1 site inserts an additional eight amino acids between the fusion Tag and the protein encoded by the ORF (ATG::Ntag::B1::ORF::STOP::B2). In C-terminal fusion proteins, the termination codon is provided in the Destination vector, and the 25-bp B2 site inserts another eight amino acids between the protein encoded by the ORF and the fusion Tag (B1::ATG::ORF::B2::Ctag::STOP). Finally, fusion proteins can be expressed with both N-terminal and C-terminal protein tags (ATG::Ntag::B1::ORF::B2::Ctag::STOP) (Walhout et al. 2000a). So far, there is no report available that suggests an impact on protein yield caused by the inserted B1 and B2 sequences.
In summary, the Gateway system provides both efficiency and adaptability, because it (1) has extraordinary fidelity, (2) is controllably reversible, and (3) uses small cis-acting sites that tolerate numerous modifications.
Multisite Gateway and Compatibility
To create Multisite Gateway (Cheo et al. 2004), the native attB, attP, attL, and attR cis-acting sites were systematically analyzed to identify nucleotide changes that increase site specificities without negatively affecting recombination efficiency. With that information, six new sets have now been engineered, designated “3” (B3, P3, L3, and R3), “4” (B4, P4, L4, and R4), and so on. With these additional sets of cis-acting recombination sites, it is possible to link together two or more DNA segments from different Entry clones into the same Destination vector, in a predefined order, orientation, and reading frame. For example, promoters can be cloned to generate a promoter Entry clone resource, and subsequently be mixed with ORF Entry clones and Destination vectors to link any promoter in the collection to any ORF in the second collection.
This feature of Gateway is unique among RC systems and allows optimal compatibility in projects that require the high-throughput cloning of two or more DNA sequences in many different vectors.
The Example of C. elegans
The C. elegans interactome project (Walhout et al. 1998) nicely illustrates the concepts outlined above. The Y2H-based map will eventually require the production of at least 38,000 proteins in yeast cells, because at least two hybrid proteins need to be made for each of the 19,000 predicted gene products, excluding splice variants (The C. elegans Sequencing Consortium 1998). After capture as an ORFeome resource (Reboul et al. 2003), C. elegans ORFs can be transferred into two Destination vectors: one containing a DNA-binding domain (DB) and the other containing an activation domain (AD; Walhout and Vidal 2001a). So far, 22,000 of the 38,000 clones have been generated and transformed into Y2H strains (J.-F. Rual, unpubl.). Concurrently, we transferred hundreds of Entry ORFs into other Destination vectors that allow expression of proteins fused to the glutathione-S-transferase (GST-tag) or the poly-histidine (His-tag) protein tags (Reboul et al. 2003; Li et al. 2004). These fusion proteins were used to verify, by coaffinity purification upon expression in mammalian cells, the overall quality of edges in the current C. elegans interactome network map (Li et al. 2004).
To initiate a comprehensive “phenome” analysis in C. elegans, we also transferred many worm ORFs into RNAi vectors (Rual et al. 2004a), an endeavor that nicely illustrates the adaptability of Gateway-cloned resources. Despite being unaware of the genome-wide potential of RNAi when designing the worm ORFeome project, the conversion of an RNAi-by-feeding vector into a Gateway Destination vector took about a week, then a few weeks more were needed to shuffle 11,000 worm Entry ORFs into the new RNAi vector.
To map various dynamic features of the C. elegans proteome (Hope et al. 1996, 2004; Dupuy et al. 2004), we have started a promoterome cloning project that takes advantage of the compatibility provided by the Multisite Gateway reaction (Cheo et al. 2004). Many C. elegans prom::ORF::GFP fusions have already been generated, and other variations are envisioned (Dupuy et al. 2004). Both ORFeome and promoterome Entry clone resources are being used in yet another new project: the mapping of the regulatory network of C. elegans using the yeast one-hybrid (Y1H) system (Deplancke et al. 2004). For this, ∼600 transcription factors will be produced from the ORFeome resource, and thousands of promoters will be manipulated from the promoterome resource. Although this project was designed after the start of the ORFeome project, “the engineered parts were sufficiently standardized and interchangeable,” so that available resources could be applied to this new endeavor.
Standardization and Uniformity
Uniformity follows standardization. Ford's engineers standardized the design of the Model T, a simple and sturdy car with no options, not even color. The Model T retained the same design for all 15,000,000 cars produced. Unfortunately, uniformity is not always wise for biology. Therefore, although ORFeome resources need to be designed to maximize future uses, they are not likely to be suitable for every conceivable application.
Given the relatively high costs of primer synthesis in ORFeome projects, we initially followed, to reduce cost, a single design that includes the absence of a termination codon for each cloned ORF (Walhout et al. 2000a,b). This design allows optimal adaptability and compatibility and maximal uses of ORFeome resources, that is, the expression of both N-terminal and C-terminal fusion proteins. However, this design precludes protein expression systems in which the C-terminal end of proteins needs to be native, such as signaling by receptor tyrosine kinases, which might be problematic with the B2-encoded peptide present. Such research would entail redesign of the Entry cloned ORFs.
Another limitation is that full-length protein-encoding ORFs have been used so far downstream from ORFeome projects. Some assays benefit from having functional domains of proteins expressed, rather than full-length proteins. For example, Y2H screens show increased sensitivity when libraries of protein domains are used, rather than full-length ORF constructs (Rain et al. 2001).
Solutions to those hindrances can be imagined and will likely come with time. For example, the removal of the C-terminal B2 site would be straightforward using existing Entry clones, a modified 3′-primer that includes a stop codon, and a short five-cycle amplification regimen. Nevertheless, the advantages of standardized resources remain apparent.
Utility and Resource Composition
Entry clones are typically generated from PCR products. Given the mutation rate of conventional PCR typically required to achieve the robustness necessary in large-scale projects, the population of Entry clones generated actually represents a library of sequences related to the original template sequence, the diversity of which reflects the fidelity of the amplification. Two approaches are possible: either a minipool of resulting clones is captured, a solution designated “Version 1.1,” or individual clones are isolated, each of which is sequenced to eventually archive one as the “wild-type” representative (“Version 2.1”; Reboul et al. 2003).
Depending on the size of the resource to be constructed, the budget, the available templates (cDNA libraries or isolated full-length cDNAs or genomic DNA), and the time frame for completion, v1.1 may be favored over v2.1 or vice versa. Our experience with C. elegans strongly argues that, at least for large resources in the thousands, pools of clones should be used. First, it is prohibitively expensive and time-consuming (clone isolation, template preparation, sequence analysis, bioinformatics, clone tracking, reanalysis, etc.) to recover single clones devoid of misincorporation for each target. Second, gene annotation, describing the sequence and intron/exon structure of each target (e.g., ORFs), is often inadequate, particularly for higher eukaryotes such as human. For tens of thousands of genes, the “wild-type” ORF sequences are as yet unknown and hence selecting any particular allele as representative of “wild type” could be erroneous. Additionally, for multicellular organisms, the incomplete understanding of differential splicing represents an enormous challenge. Finally, resources in which each Entry clone comprises a minipool of (mostly “wild-type”) individuals are perfectly suitable (perhaps more so) for screening experiments, particularly for functional proteomics. When individual sequence-validated isolates are required for detailed single-gene studies, it is straightforward to generate the appropriate clone from the corresponding pools.
To address fidelity, while simultaneously aiming for improved data quality and coverage for ORFeome and promoterome cloning projects, we adopted a system of evolving versions. Version 1.1 of an ORFeome captures minipools of clones for each template (e.g., ORFs). This approach minimizes errors of PCR-induced misincorporation while maximizing amplification fidelity, and maximizes the recovery of large portions of differentially spliced variants when a cDNA library is used as template for ORF PCR reactions (Reboul et al. 2003). This strategy provides at any moment a snapshot of the whole ORFeome of an organism, and does not attempt to clone a few hundred ORFs perfectly before moving to the next batch. Two practical examples highlight the tradeoffs between fidelity and coverage.
Version 1.1 of the C. elegans ORFeome was obtained in reference to the worm genome sequence annotation (WS9) of August 1999 (Vaglio et al. 2003). Approximately 11,000 ORFs of the 19,000 predicted ORFs were recovered as PCR products with a worm cDNA library as template and cloned by the Gateway system as minipools of in-frame Entry clones. Most of the 8000 missing ORFs correspond to genuine genes for which the exact exon structure is unknown (Reboul et al. 2001). This explains why the original primers did not generate any product in the first pass of the C. elegans ORFeome project (Reboul et al. 2003). Despite the absent ORFs, Version 1.1 has already been used to express thousands of proteins for structural proteomics, proteome-wide Y2H mapping, genome-wide RNAi analyses, and more (Chance et al. 2002; Reboul et al. 2003; Xu et al. 2003; Li et al. 2004; Luan et al. 2004; Rual et al. 2004a; Tewari et al. 2004). Version 1.1 of the C. elegans ORFeome is developing into Version 2.1, in which single isolated clones corresponding to different splice products are captured from ORF minipools (Reboul et al. 2003).
Incorporating four years of improved gene annotations, the WS100-derived version of the C. elegans predicted ORFeome provides corrections for ∼3000 ORFs that were not obtained in Version 1.1. By redesigning new PCR primers for these repredicted ORFs, we were able to Gateway-clone ∼2000 of them (Lamesch et al. 2004), generating Version 3.1 that now contains ∼13,000 ORFs ready for use. Future genome reannotations, incorporating (1) comparative genomics with related nematode species (Chen et al. 2004), (2) improved PCR-amplification methods (Rual et al. 2004c), and (3) more complex cDNA libraries (Hirozane-Kishikawa et al. 2003), will contribute to ever improving versions of the C. elegans ORFeome. We estimate that we should eventually be able to clone ∼17,000 ORFs (Reboul et al. 2001), representing 85%-90% of the worm ORFeome.
The Human ORFeome
Cloning and expressing the human ORFeome is an important goal in the development of systems approaches to biology. The complete human ORFeome, again defined as the complete set of protein-encoding ORFs, including all splice variants present in all cells at all stages of development and in all environmental conditions, is clearly an impossible task at the moment. However, the methods, strategies, and concepts described here and elsewhere in this Special Issue are now mature enough to initiate the recovery of the human ORFeome using one or more RC methods. From our experience with the C. elegans project, we propose to organize this effort as a set of ever improving versions.
An original attempt is described in this issue (Rual et al. 2004c). It consists of a first version of Entry constructs recovered as minipools from the currently available Mammalian Gene Collection (MGC; Strausberg et al. 2002) of ∼10,000 full-length cDNA clones (hORFeome v1.1). As new cDNA collections such as those created by RIKEN (Okazaki et al. 2002) are added to MGC and new genome annotation versions become available, the scientific community will be able to further develop the human ORFeome resource and thus characterize the cellular networks likely responsible for the great complexity of human biology.
Conclusions
The Gateway system was first conceived to address the problems of adaptability and limited efficiency of standard cloning approaches. One of us faced, almost a decade ago, the most frustrating problem in gene cloning. After working for more than a year on the design and cloning of a complex DNA construct, involving dozens of restriction and ligation steps, the final plasmid obtained would not express the expected protein product. The Gateway system, which provides a convenient way to transfer any DNA segment into many different expression vectors, once it has been created or captured, was born of that frustration.
With the great advances in genome sequencing leading to prediction of tens of thousands of novel genes, new opportunities emerged to study biology as molecular networks (Walhout et al. 1998). It soon became clear that new types of resources were needed, such as cloned ORFeomes, to support the automated production and subsequent analysis of a nearly full proteome, under many conditions and in many different functional assays (Walhout et al. 1998; Vidal 2001). This, in turn, created new needs for efficiency and compatibility in DNA cloning, in addition to that for adaptability.
The beautiful utility of phage λ integration provided the necessary scaffold for the development and application of a system that combines solutions for all three challenges: efficiency, adaptability, and compatibility.
Acknowledgments
We thank D. Dupuy, D. Hill, P. Hilson, and an anonymous reviewer for useful comments. We also thank M. Cusick for careful editing of the manuscript. The C. elegans ORFeome and interactome projects are supported by grants from NCI, NHGRI, and NIGMS awarded to M.V.
Footnotes
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.2769804.
-
↵4 Corresponding author. E-MAIL marc_vidal{at}dfci.harvard.edu; FAX (617) 632-5739.
- Cold Spring Harbor Laboratory Press








