
Analysis of Sequence Variations in Several Human Genes Using Phosphoramidite Bond DNA Fragmentation and Chip-Based MALDI-TOF
Abstract
The challenge in the postgenome era is to measure sequence variations over large genomic regions in numerous patient samples. This massive amount of work can only be completed if more accurate, cost-effective, and high-throughput solutions become available. Here we describe a novel DNA fragmentation approach for single nucleotide polymorphism (SNP) discovery and sequence validation. The base-specific cleavage is achieved by creating primer extension products, in which acid-labile phosphoramidite (P-N) bonds replace the 5′ phosphodiester bonds of newly incorporated pyrimidine nucleotides. Sequence variations are detected by hydrolysis of this acid-labile bond and MALDI-TOF analysis of the resulting fragments. In this study, we developed a robust protocol for P-N-bond fragmentation and investigated additional ways to improve its sensitivity and reproducibility. We also present the analysis of several human genomic targets ranging from 100-450 bp in length. By using a semiautomated sample processing protocol, we investigated an array of SNPs within a 240-bp segment of the NFKBIA gene in 48 human DNA samples. We identified and measured frequencies for the two common SNPs in the 3′UTR of NFKBIA (separated by 123 bp) and then confirmed these values in an independent genotyping experiment. The calculated allele frequencies in white and African American groups differed significantly, yet both fit Hardy-Weinberg expectations. This demonstrates the utility and effectiveness of PN-bond DNA fragmentation and subsequent MALDI-TOF MS analysis for the high-throughput discovery and measurement of sequence variations in fragments up to 0.5 kb in length in multiple human blood DNA samples.
The ultimate goal of the Human Genome Project (Lander et al. 2001; Venter et al. 2001) is to provide the fundamental information necessary for better understanding the genetic causes of human disease. The draft of the human genome sequence and the publicly available single nucleotide polymorphism (SNP) map produced by The SNP Consortium open the way for an intensive search for novel disease targets. However, the challenge of this task is enormous because the genetic background of complex traits is multifactorial and poorly understood. In addition, the probability that a given polymorphism is implicated in a given disease may be low. To establish statistically valid gene/disease associations, one will need to measure multiple sequence variations over large genomic fragments in numerous patient samples (Altshuler et al. 2000; Roses 2000). Thus large-scale SNP discovery and sequence validation (resequencing) will be important tasks. This volume of work can only be completed if more accurate and cost-effective high-throughput solutions become available.
Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) allows highly accurate measurements of label-free diagnostic DNA fragments with an average molecular-weight resolution of 1 to 2 D (Tang et al. 1999). Cocrystallization of matrix and DNA, followed by soft ionization, reduces in situ photocleavage of analyte molecules. Combined with robotic sample handling and on-chip dispensing, this approach has been successfully applied to the genome-wide analysis of gene-related SNP patterns (Little et al. 1997; Buetow et al. 2001). We and others recently introduced the use of baseselective nucleic acid cleavage followed by MALDI-TOF MS to retrieve the sequence information (Shchepinov et al. 2001; Rodi et al. 2002; von Wintzingerode et al. 2002; Böcker 2003; Hartmer et al. 2003; Krebs et al. 2003). The DNA fragmentation-based approach has a number of advantages over traditional techniques for DNA analysis and can be successfully used for SNP discovery, resequencing, and sequence validation.
We earlier developed an approach to study sequence variations in targets averaging 100 nucleotides in length (Shchepinov et al. 2001). This approach was based on the simultaneous MALDI-TOF MS analysis of a number of short DNA species resulting from site-selective phosphoramidite bond cleavage of a larger fragment. This reduction in size is necessary to overcome existing MS size limitations, which prevent the practical application of this technology for DNA sequencing. Although the separation of fragments as large as 500 bases has been reported (Tang et al. 1994; Henry 1997), routine MS of nucleic acids has a short read-length capability (<50 to 100 bases). The use of nucleic acid molecules <30 bases in length is generally preferred. We previously demonstrated that the base-specific cleavage of a large DNA fragment into shorter species resulted in suitable signal intensity of the resulting fragment set while retaining useful diagnostic characteristics of the whole fragment (Shchepinov et al. 2001). Most sequence changes in the region under interrogation would result in the appearance of additional cleavage products or the disappearance of expected products. The cleavage selectivity was achieved by creating primer extension products, in which acidlabile phosphoramidite bonds replaced 5′ phosphodiester bonds of newly incorporated pyrimidine nucleotides. The cleavage itself was performed by using slightly acidified MALDI-TOF 3-hydroxypicolinic acid (3-HPA) matrix or by the addition of a short acid hydrolysis step.
We originally used a manual reaction set-up for proof-of-concept and analyzed short PCR fragments up to 174 bp. We observed an undesirable degree of acid depurination during the course of PN-bond cleavage. Thus, this original procedure was not suitable for the large-scale analysis of human samples. In this work, by using an optimized incorporation and cleavage protocol, we present validation of a method and a comprehensive analysis of several human genomic targets ranging from 100-450 bp in length. By using an automated sample processing protocol, we investigated an array of SNPs within a 240-bp segment of the NFKBIA gene (including the 3′-UTR) in 48 human DNA samples belonging to two different ethnic groups. We successfully identified and measured frequencies for the two common SNPs in this region (separated by 123 bp), then confirmed this finding by an independent genotyping experiment. The calculated allele frequencies in white and African American donor groups differed significantly, yet both fit Hardy-Weinberg expectations. This demonstrates the utility and effectiveness of phosphoramidite bond-specific DNA fragmentation and subsequent MALDI-TOF MS for high-throughput discovery and measurement of single nucleotide sequence variations in fragments ranging up to 0.5 kb in length.
RESULTS
Optimization of P-N Fragmentation Protocol
A P-N fragmentation procedure was optimized for large-scale analysis of DNA samples. Site-specific cleavage of P-N bond-containing DNA is accomplished via hydrolysis in acidic conditions. To make hydrolysis products compatible with MALDI-TOF MS, volatile acids such as trifluoroacetic acid (TFA) or hydrochloric acid (HCl) have been used (Shchepinov et al. 2001). The general process scheme is shown in Figure 1. The double-stranded target DNA fragment is generated by performing PCR with one 5′-biotinylated primer. A single-stranded template is obtained by immobilizing the PCR product on streptavidin-coated magnetic beads followed by removal of the nonbiotinylated strand. Primers are extended by using P-N modified dTTP or dCTP while the template is still attached to the beads. Extension products are isolated, and P-N bonds are hydrolyzed in acidic conditions. The resulting mixture of short DNA fragments is then analyzed by MS. To hydrolyze the P-N bonds, we had originally used 3-HPA matrix, acidified by the addition of 1.5% TFA (Shchepinov et al. 2001). In the present study, we have optimized a number of cleavage parameters to obtain a reproducible and automation-ready protocol. The concentration of P-N deoxynucleotide triphosphates in the extension reaction was optimized, as were the conditions of extension product purification (see Methods). We currently incorporate 7-deaza-purine deoxynucleotide triphosphates to compensate for the decreased stability of purine bases in acidic environment (Siegert et al. 1996). We include a separate acid exposure step prior to matrix addition because this greatly improves assay reproducibility by decreasing the number of incompletely cleaved fragments.
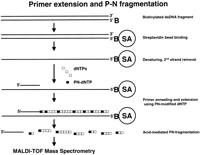
Scheme of P-N-bond fragmentation assay. The experimental protocol is described in Methods.
Figure 2 demonstrates the experimental design and results of a typical fragmentation reaction. The target, a 265-bp fragment of the APOB gene, was amplified by PCR using a biotinylated forward primer and then immobilized on streptavidin beads. A 20-nt extension primer was designed in the reverse orientation for sequence analysis (Fig. 2A). We typically place the extension primers so that they overlap the PCR primers by five to 10 nucleotides. Extension was performed with Sequenase DNA polymerase using P-N modified dTTP. The expected length of the extension product is 255 nucleotides, including a one nucleotide overhang that may be produced by sequenase. Note that in the previous study we did not observe the most distal fragmentation product and explained this by the negative effect of the biotinstreptavidin complex on the DNA polymerase (Fig. 1; Shchepinov et al. 2001). Therefore, the actual length of the extension product may be slightly <255 bases.
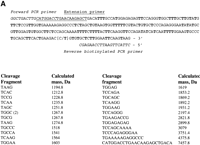
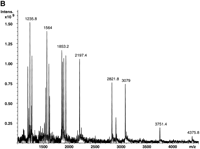
Fragmentation analysis of an APOB target by using an optimized cleavage protocol. (A) Sequence of a 265-bp APOB amplicon and the list of fragments resulting from a P-N dT-specific cleavage of an extension product. Locations of PCR and extension primers are labeled. Potential cleavage sites (dT) are indicated in bold. (B) Experimentally obtained fragmentation mass spectrum of a 265-bp product. Only measured peak m/z values are shown.
Figure 2A lists all fragments with four or more nucleotides that would result from the complete cleavage of all P-N bonds in the extension product. Fragments smaller than four bases (1100 m/z) are not included because of their redundancy. In addition, we often observe increased chemical noise in this size range, which may mask signals from mono- to trinucleotide DNA molecules (Shchepinov et al. 2001). The list of calculated fragments was then compared to the experimentally derived peaks (Fig. 2B) to determine if there was any deviation from the expected spectrum. All but one of the expected fragments were confirmed. We could not detect the largest and most proximal fragment (calculated mass, 7457.8 D), most likely because it is composed of the entire sequence of the extension primer and an additional GACA tetramer. The synthetic extension primer does not contain deaza purines and therefore is susceptible to acid depurination. This will lead to the random loss of adenine and/or guanine bases from the proximal fragment and will result in a distribution of resulting peaks with drastically reduced intensities. The difficulties in detecting the most proximal and distal cleavage products are some of the limitations of the present method.
The mass profile seen in Figure 2B was reproduced in several independent reactions and by performing 48 replica reactions in a 96-well microtiter plate. Comparison of expected peaks and raw spectra showed that all expected peaks were unambiguously and consistently detected, thus providing complete T fragmentation sequence information except for mono- to trinucleotides. Any missing information can usually be obtained by running the C fragmentation reaction in the same direction, or the T/C reactions in the reverse direction.
In the next experiment, we analyzed a longer target. A 450-bp fragment of the NFKBIA gene was generated by using primers described in Methods. The results of this experiment are presented as online Supplemental material available at www.genome.org. After a standard fragmentation reaction, we were able to identify 53 peaks corresponding to fragments larger than three to four bases. Only three peaks were missing out of 56 expected. One missing peak corresponded to the most proximal cleavage fragment. The ability to analyze longer fragments significantly improves the scope of this technique. However, increased amplicon size has certain limitations because the possibility of overlapping peaks with the same mass increases. If polymorphic peaks overlap with nonpolymorphic peaks, it becomes difficult to identify missing and additional signals.
Sequence Validation and Molecular Genotyping of Common SNPs in CETP and UCP2
We next tested the application of P-N bond-based fragmentation for sequence verification and genotype determination in human DNA samples. Initially, we chose the cholesteryl ester transfer protein (CETP) gene due to a number of well-documented cardiovascular disease-related polymorphisms ascribed to this target (Yamashita et al. 2000). We focused on exons 12-14 (GenBank accession no. M32997) described by Agellon et al. (1990) and designed the experiment to detect an exonic WIAF-10949 A/G SNP (rs5882) located at nucleotide position 1301 in the sequence M32997. Analysis of the P-N fragmentation pattern in the region surrounding this site showed results conflicting with the expected pattern (Fig. 3). In the P-N dTTP incorporation reaction, the expected peak at 3439 D was absent, whereas an additional peak at 3766.1 m/z was observed (Fig. 3A,B). Similarly, in the P-N dCTP reaction, the calculated peak at 3109 D was not seen, whereas an additional peak at 3437.2 m/z was observed. Reverse incorporations also produced unexpected cleavage profiles (data not shown). The mass difference between obtained and expected signals in both forward reactions was exactly 329 D for both analogs. The only possible sequence change that could lead to the observed profile shift was an intronic insertion of a G (329.4 D). Based on the appearance of measured masses 3766.1 and 3437.2 m/z (Fig. 3C), we unambiguously determined that the insertion was located between nucleotides 1281 and 1282 and not at any other location.
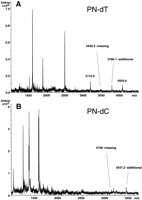
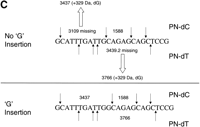
Identification of a single base insertion by fragmentation. A CETP target was analyzed as described in the text. (A, B) Mass spectra of P-N dTTP and P-N dCTP incorporation reactions, respectively. (C) Scheme showing cleavage sites in both reactions revealing a single dG insertion.
To verify this finding, we designed a simple genotyping assay by using the MassEXTEND method (Tang et al. 1999; Buetow et al. 2001) and confirmed this sequence change in 48 randomly chosen human DNA samples (data not shown). Finally, the correct sequence was found in the contig NT_024766, as well as in the Ensembl database. This invalidates the sequence listed in M32997, as well as the 5′ flanking fragment in SNP rs5882 submitted record at http://www.ncbi.nlm.nih.gov/SNP/snp_retrieve.cgi?subsnp_id=7484. Thus the P-N cleavage-based approach works for validation of known sequences, provided that the cleavage pattern allows sufficient interpretation. The probability that at least one cleavage pattern out of four possible patterns (P-N dTTP and P-N dCTP, forward and reverse direction) will indicate a sequence change is extremely high, whereas the chance that the location of this change may be uniquely mapped to the fragment is lower.
Next, we analyzed coding regions of two human genomic targets: the shorter CETP fragment surrounding codon 422 (rs5882, Val422Ile), and a fragment of the Uncoupling Protein-2 (UCP2) gene surrounding codon 55 (rs660339, Val55Ala). The aim of these experiments was to prove that accurate genotypes of the two known SNPs could be confirmed by the P-N fragmentation approach. The genomic sequences, peak lists, and representative mass spectra can be seen as online Supplemental material. All three different genotypes for each target were investigated. DNA samples that we used were previously genotyped by the MassEXTEND approach (Tang et al. 1999; Buetow et al. 2001). By using P-N fragmentation, we were able to unambiguously detect and confirm genotypes of all samples. Heterozygous samples were characterized by decreased intensity of additional peaks. This could aid in distinguishing between homozygotes for a minor allele and heterozygotes. The characteristic sequence changes for the minor alleles of both targets were detected by running just a single P-N dTTP fragmentation reaction in one direction. This shows the great potential of this approach for the analysis of sequence variations in multiple samples.
Analysis of Sequence Variations in the 3′ Untranslated Region of the NFKBIA Gene Using Multiple Human DNA Samples
A semiautomated high-throughput procedure was established by using a Multimek 96 automated pipetting station (Beckman). PCR was set up manually in 96-well microtiter plates, followed by automated primer extension and acid hydrolysis. Onchip nanodispensing with a SpectroJET (Sequenom) and MALDI-TOF analysis were also performed in operator-free mode. Forty-eight samples (in duplicates) were analyzed. Post-PCR processing time was ∼4 h 30 min. We targeted a 240-bp genomic fragment of nuclear factor-κB inhibitor α (NFKBIA), encompassing parts of intron 5, exon 6, and the 3′UTR. Specifically, we set out to map the two common SNPs in this area (rs696 and rs8904, positions +2 and +126 from the stop codon), as well as measure the frequencies of alleles in two different ethnic populations. A total of 24 unrelated white and 24 unrelated African American individuals were investigated. The target sequence, primer locations, and typical reaction spectra are shown in Figure 4. The PN-dTTP analog was used for incorporation. The reaction conditions allowed for unambiguous genotyping of two common SNPs and one low-frequency SNP in a single fragmentation reaction. The results obtained reflect a situation in which a homozygous sample for each of three alleles was characterized by one signature fragmentation peak. Therefore, a heterozygous sample for all three alleles should show six signature peaks; this is clearly seen in Figure 4E. It is important to realize that the resolution limit for MS is ∼1 D. Therefore, the mass difference between the closest peaks in this spectrum, #3A (3698.9 m/z) and #3G (3714.2 m/z) falls well above the limit. This triple-heterozygous genotype only occurred in one white sample out of 48 total individuals (96 chromosomes). This indicates a frequency ∼1% for this combination. The cumulative SNP scoring results and allele frequencies for the two common SNPs are presented as online Supplemental material. The results revealed high levels of heterozygosity for the common variations rs696 and rs8904. These variants were in Hardy-Weinberg equilibrium with their respective reference alleles. Frequencies of C and T alleles for rs8904 were completely opposite in the two populations and differed by ∼27%. Interestingly, frequencies of G and A alleles for rs696 also differed by 27%. This indicates significant variation between the white and African American samples for these markers. The newly discovered SNP at the position +18 (18 nucleotides downstream of the stop codon) was not confirmed as a polymorphic marker (i.e., having a frequency >10%) in our samples. To validate these findings, we analyzed all samples in an independent genotyping experiment. Genotyping data obtained by the MS (data not shown) and P-N fragmentation methods (Supplemental Table) were identical. These results show that the P-N bond-based fragmentation approach achieves a high level of accuracy and is capable of providing novel sequence information. SNPs can even be analyzed without the knowledge of the exact sequence around the polymorphism. By comparing the additional and missing signals between samples, one can identify the signals that represent polymorphic regions. It is possible to automate all the steps of P-N fragmentation microplate processing, thus making it an operator-free procedure.
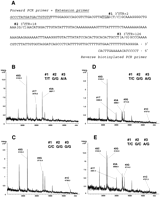
Automated fragmentation analysis of NFKBIA. (A) Analyzed fragment of the NFKBIA gene. PCR and extension primers, three interrogated polymorphisms, and a stop codon (boxed) are shown. (B-E) Representative mass spectra of observed genotypes. Note that the genotype exemplified in E was measured in a single sample only.
DISCUSSION
The main goal of this study was to prove that, upon optimization, a P-N bond-based DNA fragmentation could be successfully used for semiautomated, high-throughput analysis of sequence variations in human DNA samples. Base-selective cleavage has several apparent advantages for SNP identification. While this manuscript was in preparation, several studies describing cleavage-based MS approaches were published (Rodi et al. 2002; von Wintzingerode et al. 2002; Böcker 2003; Hartmer et al. 2003; Krebs et al. 2003). Most of these studies used RNA polymerasemediated incorporation of RNase-cleavable ribonucleotides in a transcription reaction. The advantage of RNA-based fragmentation is an option to run all stepwise reactions in a homogeneous format. The major disadvantages include a number of contaminating adduct peaks and a necessity to work with a ribonuclease reagent limiting the attractiveness of this procedure for molecular biology laboratories. P-N bond fragmentation features a minimal number of chemical adduct species and a remarkable clarity of specific mass peaks.
How can the base-specific fragmentation procedure be compared with traditional sequence analysis techniques such as capillary gel electrophoresis (CGE) in terms of SNP discovery power, throughput, and costs? A very recent investigation directly compared results of SNP discovery in 30 non-overlapping PCR amplicons averaging 500 bp performed by CGE or by a site cleavage-based MS approach (Böcker 2003). This study showed that MS method yielded 50 new SNPs versus 47 SNPs delivered by CGE. In addition, the CGE method missed four SNPs confirmed by MS, whereas MS missed only 1 SNP identified by CGE. In our experience, both RNase-mediated and P-N bond-based cleavage reactions had similar SNP discrimination success rates; therefore, Böcker's conclusions may be extended to P-N fragmentation. These results clearly underscore the power of base-specific cleavage in combination with MS.
It is also useful to assess the throughput and costs of the acid cleavage procedure. We estimate that the total costs of a P-N fragmentation reaction are well below $2, thus making this method very competitive to CGE. The retail price of the DNA synthesis enzyme that we currently use, Sequenase, makes up ∼75% of this estimate. The ability to use alternative, less expensive DNA polymerases will significantly reduce these already low costs. The synthesis of P-N modified bases is achieved in a simple single-step reaction by using inexpensive reagents (Shchepinov et al. 2001). These modified analogs can be stored frozen for >1 year without decrease in the incorporation efficiency. The total cost of P-N dNTPs per reaction is just a fraction of a cent. It takes <5 h for a single operator to complete a 96- or 384-well plate post-PCR reaction, including data analysis. Moreover, the final MS part of the current approach using miniaturized MALDI chips is a fully automated, walk-away procedure (Buetow et al. 2001).
By using the fragmentation results for human amplicons described in this study (ApoB, NFKBIA [450 bp], CETP, and UCP2), we attempted to estimate the accuracy of P-N cleavage procedure from the number of missing peaks versus the total number of expected peaks. Only eight peaks were missing out of 125 expected. Among these, six were either most proximal or most distal fragments (see below), one could contain a potential SNP (data not shown), and one could be a true MS failure. Thus, the accuracy of P-N fragmentation approach is very high. Note that a significant advantage of a site-selective cleavage over CGE is that a single sequence change results in multiple MS changes. For example, if the detected homozygous sequence change is a cleavage-blocking event, the resulting disappearance of a peak will coincide with the appearance of an additional signal of higher mass (Fig. 4). This helps to validate newly discovered SNPs in the same cleavage reaction. The aggregate of four independent fragmentation reactions (e.g., dT and dC cleavages on both strands) provides a multilayer validation tool. Such approach is routinely used for analysis of RNase-mediated fragmentation (D. van den Boom, pers. comm.). A large-scale P-N fragmentation study may provide better assessment of the accuracy of SNP identification. The failure rate for P-N fragmentation is extremely low. Out of >50 fragmented amplicons analyzed, we failed to fragment only one amplicon. The reason for this single failure may be the individual features of a template sequence, making it less amenable to Sequenase-mediated polymerization.
Overall, the P-N fragmentation procedure is simple and straightforward, yet robust and fully compatible with highly accurate MALDI-TOF MS. Hydrolysis of P-N bonds at low pH yields only one type of cleavage product, and therefore, the mass spectrum is not overloaded with adduct species. We have easily overcome problems with acid-induced depurination by incorporating 7-deazapurine triphosphates. Long sequence coverage is another advantage of a fragmentation approach. Validation of SNPs in highly polymorphic regions may face difficulties when primer extension- or hybridization-based methods are used. If the sequence chosen for the genotyping primer also contains SNP(s), proper primer annealing and extension may be difficult or impossible. The fragmentation approach presented here has fewer sequence restrictions because both the fragment length and the location of extension primer may be varied. Here we demonstrate that fragments of up to 450 bp in length may be analyzed without loss of sensitivity. Using magnetic streptavidin beads with higher binding capacity for large DNA molecules will help to measure even longer fragments. Amplicons may be chosen in a flexible way and shifted easily for better sequence coverage. This flexibility helps to resolve the problem of most proximal and distal cleavage products by designing overlapping amplicons. In addition, the fragmentation-based approach should be useful in genotyping difficult sequences. In the MassEXTEND method, primer duplex/hairpin formation and other sequence effects can lead to poor primer extension or uneven allele peaks in a heterozygous sample (Buetow et al. 2001). Redesigning the extension primer for the opposite strand can sometimes help, but not in all cases. Genotyping by fragmentation is not limited by the sequence surrounding the SNP and may therefore assist in resolving challenging sequences. The genotyping application of this method, however, bears a higher unit cost than does the conventional MassEXTEND method and should only be used for problematic sequences.
The fragmentation/MS approach is also well suited for the discovery of SNPs in regions with known reference or draft sequence (von Wintzingerode et al. 2002; Böcker 2003; Hartmer et al. 2003; Krebs et al. 2003). When a change in a certain fragment peak is noticed, the exact mass shift can be measured, and it is often possible to deduce the sequence change responsible for this mass shift (Böcker 2003). The analysis is greatly facilitated when the disappearance of an expected fragment coincides with the appearance of an additional fragment (Fig. 4). The base composition but not the sequence may be deduced for any unexpected (additional) fragment. We call such base compositions “compomers.” For example, the compomer A(x)G(y)C(z) refers to the molecular ion composed of x deoxyadenosines, y deoxyguanosines, z deoxycytosines, and no deoxythymidines. The inhouse SpectroCLEAVE software program has been created for the analysis of MALDI-TOF MS fragmentation spectra (Böcker 2003). This software reports additional (and missing) peak signals and the corresponding compomers. Accurate sequence mapping of the polymorphism is achieved by comparing two or more appropriate fragmentation reactions, and, if necessary, by confirming a base change in a MassEXTEND assay. The SpectroCLEAVE software program has been described in detail (Böcker 2003).
By using the P-N fragmentation method, we obtained good signals for fragments longer than three bases. Another level of discrimination may be reached when a cleavable base is incorporated in only a fraction of possible positions, and consequently, a partial (or incomplete) base-selective cleavage is performed. Because the phosphoramidite bond is much more susceptible to acid cleavage than are natural phosphodiester bonds, we added normal dTTP in addition to PN-dTTP to create a distribution of uncleavable thymidine nucleotides. Figure 5 shows a model experiment when, together with 5 mM P-N dTTP, a lower concentration (10 μM) of natural dTTP was incorporated in the APOB fragment. This pattern was obtained reproducibly in several experiments. When comparing this spectrum with Figure 2B, one can notice a number of extra “partial cleavage” peaks, each carrying additional sequence information.
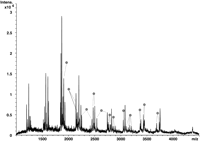
Incomplete (partial) P-N-bond cleavage. APOB extension product (265 bp) was generated as in Figure 2B with the exception that 10 μM natural dTTP was incorporated together with 5 mM P-N dTTP. Note the increased total number of fragmentation peaks compared with Figure 2B. Representative incomplete cleavage peaks are labeled by a crossed circle.
The incomplete cleavage scheme allowed us to shift the smaller fragments into the larger mass range by linking adjacent sequence fragments. In practice, this allows the original one, two, and three nucleotide fragments to be seen in a higher mass range. The number of dTs incorporated relative to PN-dTs depends on the kinetic properties of the polymerase and the ratio of blocking to cleavable nucleotides. By using the relative incorporation rate of the polymerase, a Poisson distribution can be used to approximate the relative number of fragments with blocking nucleotides. In this case, only one or two partial cleavage reactions may reveal the full sequence context of a given amplicon.
METHODS
PCR and extension primers were obtained from IDT. 5′-PN-triphosphates of deoxythymidine and deoxycytidine were synthesized as described (Shchepinov et al. 2001). 7-Deaza-dGTP and 7-deaza-dATP were purchased from TriLink. All other chemicals were from Sigma/Aldrich.
Human DNA Samples
Human genomic DNA was from a previously published study (Buetow et al. 2001). Blood samples were obtained at two California locations from healthy, anonymous American volunteer participants according to collection protocols approved by the Loma Linda University Institutional Review Board (IRB) (#48057) and by the Sacramento Medical Foundation IRB (#99-015). Genomic DNA was isolated and quantitated as described (Buetow et al. 2001).
Amplification of SNP-Containing Genomic Fragments
PCR was performed in a 20 μL reaction volume by using a HotStar (Qiagen) DNA polymerase and a standard thermal cycling protocol (Shchepinov et al. 2001). The following PCR primers were used for amplification of CETP and UCP2 targets, respectively: CETP-US4-F, 5′-CCCAGTCACGACGTTGTAAAACGTCCAGGGAGGACTCACCATG; CETP-US5-R, 5′-AGCGGATAACAATTTCACACAGGTGACTGCAGGAAGCTCTGG; UCP2-US4-F, 5′-CCCAGTCACGACGTTGTAAAACGTCTTGGCCTTGCAGATCCAAG; and UCP2-US5-R, 5′-AGCGGATAACAATTTCACACAGGCCATCACACCGCGGTACTG. PCR and extension primers for APOB and NFKBIA (240-bp PCR product) are shown in Figures 2A and 5A, respectively. A 450-bp PCR product of the NFKBIA gene was obtained using primers (F) 5′-CCCAGTC ACGACGTTGTAAAACGGGCCAGCTGACACTAGAAAA and (R) 5′-AGCGGATAACAATTTCACACAGGGTTCTTTCAGCCCCTTTG.
P-N Primer Extension and Fragmentation
Single-stranded DNA templates for extension reactions were generated by using a standard MassEXTEND (PROBE) procedure (Buetow et al. 2001). Extension primers TCTGGGCTATGAGATCA and TGGTCATCGTGGCCATCGCC were used on the single-stranded templates, CETP and UCP2, respectively. The extension primer for the 450-bp fragment of NFKBIA was 5′-GGCCAGCTGACACTAGAAAA. PCR product (20 μL) was bound to streptavidin-coated magnetic beads (Dynal) by room temperature incubation for 15 min. DNA was denatured by incubation with 0.1 M NaOH for 5 min, and the beads were washed three times with 10 mM Tris-HCl (pH 9.5). Extension primer (20 pmole) was annealed to target fragment for 5 min at 70°C and then at 37°C for 10 min. The extension mix (25mM Tris-HCl at pH 9.5, 200μM dCTP or dTTP, 200 μM deaza-dA, 200 μM deazadG, 5 mM P-N dT or P-N dC, 20 mM MgCl2, 50 mM NaCl, 5 mM DTT, and 1 to 2 U Sequenase version 2.0) was added, and samples were incubated at 37°C for 30 min. The beads were washed three times with 10 mM Tris-HCl (pH 9.5), and DNA was eluted by heating for 10 min at 90°C. The samples were acidified by adding acid to a final concentration of 0.33% TFA or 0.1% HCl and then incubated for 90 min at 45°C.
For semiautomated sample processing, a Multimek multisample pipettor (Beckman) program was created. This program allowed the processing of all bead manipulation steps. All reaction mixtures were also added with the Multimek. Samples were processed manually after the extension product was eluted from the magnetic beads. Fragmented extension products were robotically dispensed onto a silicon chip (SpectroCHIP, Sequenom) by using a piezoelectric dispensing device (SpectroJET, Sequenom). The samples were then analyzed on a Bruker Biflex mass spectrometer (Bruker Daltonics) by using parameter settings that were optimized for low-mass DNA fragments.
Identification of MS peaks in this work was performed visually by an operator. It should be noted, however, that in the course of this manuscript preparation, proprietary software has become available that is fully capable of resolving the P-N fragmentation peaks (Böcker 2003). The algorithm is based on comparison of experimentally derived spectra with those predicted from analysis of the in silico information. It is described in detail (Böcker 2003).
Acknowledgments
We wish to thank M. Shchepinov for synthesis of P-N deoxytriphosphates and C. Rodi and D. van den Boom for helpful discussions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
[Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to National Center for Biotechnology Information, dbSNP, under accession nos. ss12704301-ss12704303.]
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.1653504.
-
↵1 Corresponding author. E-MAIL mdenissenko{at}sequenom.com; FAX (858) 202-9001.
-
- Accepted November 4, 2003.
- Received June 12, 2003.
- Cold Spring Harbor Laboratory Press








