
Nonrandom Tripeptide Sequence Distributions at Protein Carboxyl Termini
Abstract
The availability of complete genome sequences enables the statistical analysis of sequence features without significant database-imposed bias. The carboxyl termini of proteins often contain regions associated with protein targeting and enhanced translational termination. We analyzed the frequency of occurrence of C-terminal tripeptides in representative archaeal, bacterial, and eukaryotic genomes. The sequence distribution in prokaryotic genomes nearly matches that generated by the randomization of the observed tripeptide set. In contrast, eukaryotic genomes contain large numbers of overrepresented sequences. Some of these correspond to highly repeated sequences from either duplicated endogenous genes or transposon open reading frames. Gratifyingly, others represent previously known targeting signals or sequences associated with an increase in translational termination efficiency. However, a number of overrepresented tripeptides have not been previously noted and may represent novel functional sequences. For example, the sequence XSS may enhance translational termination efficiency in plants, whereas FWC may be a targeting or processing signal for certain amino acid permeases in yeast.
The complete genomic sequence of an organism provides a unique opportunity for insight into the mechanisms of biological processes. The generation of these data has proceeded at a remarkable rate. Initially performed on prokaryotes, complete genome sequencing efforts now have been successfully completed on a diverse set of eukaryotes as well, including the recent landmark achievement of a substantially complete human genome sequence (Lander et al. 2001;Venter et al. 2001). Once an entire genomic DNA sequence has been elucidated, it is possible to predict the location and identity of the entire complement of open reading frames (ORFs) and, hence, the sequences of essentially all the proteins for a given organism. Importantly, this set of ORFs contains little bias as to the nature of the protein product. One application for this information is the determination of patterns and correlations within protein sequences. Because of the large number of ORFs within a given genome, the utilization of statistical methods can be particularly powerful, enabling the distinction between chance occurrences and the results of biological selection. Moreover, one can compare the results of these analyses across a number of species and generate testable hypotheses about the evolutionary history of these sequence distributions.
One example of the potential utility of complete genome analysis which does not appear to have been extensively explored is the study of amino acid sequences located at the ends of the polypeptide chain. These residues often are of functional significance, as they can serve as determinants for interactions with other proteins (Chung et al. 2002). Binding sites located at the termini of proteins have particular advantages over internal sites. For example, they are commonly solvent-exposed and, hence, easily accessible by their appropriate binding partners. Moreover, these regions can evolve readily, as they usually do not require compensatory adjustments at some distant position along the polypeptide chain. In addition, a sequence that must be terminally located adds information content to the signal: The sequence alone is not sufficient, its position relative to either the amino or carboxyl terminus is also critical. From a computational perspective, terminal signal identification within a genomic sequence database is simplified considerably, assuming that the translation initiation and termination codons have been correctly identified.
One prominent example of recognition sites at the termini of proteins includes certain organelle targeting signals. A protein targeted to a particular organelle typically contains a short signal sequence that is sufficient to direct that protein to its appropriate compartment (Blobel and Dobberstein 1975). In some cases, these targeting signals are located at sequence termini. For example, the signal for many proteins to be retained within the lumen of the endoplasmic reticulum (ER) is the C-terminal tetrapeptide -Lys-Asp-Glu-Leu-COO− (KDEL; Munro and Pelham 1987). Additionally, many proteins destined to reside within the lumen of the peroxisome contain the peroxisomal targeting signal-1 (PTS1), a C-terminal tripeptide with the consensus sequence -Ser-Lys-Leu-COO− (SKL; Gould et al. 1989). Also, several known targeting signals are found at the N-termini of proteins, including the signal sequence for proteins entering the secretory pathway and the mitochondrial targeting sequence (Schatz and Dobberstein 1996).
In this paper, we present the use of complete genome information to study patterns of C-terminal tripeptide sequences within the entire ORF complement of a set of representative organisms. Genomes of the following species were studied: Methanococcus jannaschii (an archaeon), Escherichia coli (a bacterium), Saccharomyces cerevisiae (a yeast), Arabidopsis thaliana (a plant),Caenorhabditis elegans (a nematode), and Homo sapiens(humans). These genomes were analyzed by determining, for a given genome, how many occurrences of a tripeptide sequence can be defined as overrepresented beyond that expected by chance alone. To achieve this goal, the collection of C-terminal tripeptides from a given genome was randomized multiple times, then compared to the original set.
As might have been anticipated, analysis of the archaeal and bacterial genomes revealed little deviation from the randomized results. Many of the overrepresented sequences within the E. coli genome can be explained by transposon ORFs, repeated sequences, or possible effects of certain codons on translation termination efficiency. However, analysis of the eukaryotic genomes yielded a collection of sequences that appear distinctly overrepresented, even after ruling out homologous ORFs. Some of these sequences can be attributed to known targeting signals or binding sites, but others remain less well characterized. Possibilities for selected sequences will be discussed.
RESULTS
Sequence distributions were analyzed using two approaches. The first analysis method determines sequence significance by estimating how many times a sequence must occur to be considered unusually abundant. To accomplish this task, a tally was generated of the number of occurrences for each of the 8000 possible C-terminal tripeptides within a given genome. Then, a “dummy” tripeptide database was created in which the sets of amino acids used at positions −1, −2, and −3 across all ORFs were independently jumbled, then rejoined to form a new set of tripeptides. Typically, 1000 iterations of this jumbling procedure were performed. The sequence tallies from these randomized “dummy” sets were then averaged and compared with the observed tally. The importance of performing this analysis in a position-dependent fashion for each organism is highlighted in Table1. There is considerable variation in the amino acid usage profile not only from organism to organism, but also from the terminal residues to the rest of the protein sequence within a given genome. The scrambling of an existing set of amino acids in a position-dependent manner thus takes into account this unequal distribution of amino acid frequencies. Moreover, the repetitive nature of this analysis allows for the calculation of the distributions for the expected number of sequences that occur a particular number of times.
Position-Specific Amino Acid Frequencies (Expressed as a Percentage) at the Three C-Terminal Positions for Each of the Genomes Studied
A sampling of the results from the genome jumbling method is shown in graphic form in Figure 1, in which the number of sequences is plotted as a function of the number of occurrences for each sequence. For a prokaryote such as the archaeonMethanococcus jannaschii (Fig. 1A), the observed number of sequence occurrences generally falls within one standard deviation of the randomized sets. These data contrast sharply with the results from the eukaryote Saccharomyces cerevisiae (Fig. 1B). For this organism, there is a collection of sequences that appear in the genome more times than predicted by chance alone. Table2 lists the nonrandomly distributed sequences observed in all the genomes studied. The expected and observed number of sequences with a given number of occurrences is shown. Where possible, a likely reason why that sequence might be highly repeated is indicated (see below).
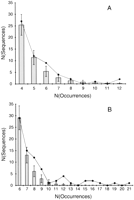
Comparison of observed vs. expected sequence occurrences inMethanococcus jannaschii (A) and Saccharomyces cerevisiae (B). The number of tripeptide sequences is plotted as a function of the number of times that that sequence occurs in the genome. The line indicates the genomic data, and the bars show the results from 1000 iterations of the jumbling procedure. Error bars are drawn at one standard deviation.
Most Frequent Tripeptide Sequences Observed Within the Genomes Studied
This method identifies a set of tripeptides that can be considered overrepresented based on the number of times these sequences
appear within a given genome. To further highlight the importance of a particular sequence, the expected number of occurrences
for each individual tripeptide was determined based on positional amino acid frequencies. If f(a,b) is the frequency of amino acid a at position b, then the expected number of occurrences of the C-terminal tripeptide XYZ,NExp
(XYZ), can be expressed as the product: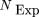 where NORF
is the total number of ORFs within the genome of the organism. Comparison of NExp
(XYZ) with NObs
(XYZ), the observed number of occurrences of XYZ, can give an indication of the significance of that sequence. Although simple
to calculate, the results from this frequency method must be interpreted carefully. Sequences that have a relatively low number
of occurrences but contain the less frequent amino acids (such as cysteine and tryptophan) tend to show inflatedNObs
(XYZ)/NExp
(XYZ) ratios. The results from the frequency method for each sequence, expressed as theNObs
(XYZ)/NExp
(XYZ) ratio, are also presented in Table 2. The application of these ratios to the sequences identified in the jumbling analysis serves to emphasize particular sequences
over others.
where NORF
is the total number of ORFs within the genome of the organism. Comparison of NExp
(XYZ) with NObs
(XYZ), the observed number of occurrences of XYZ, can give an indication of the significance of that sequence. Although simple
to calculate, the results from this frequency method must be interpreted carefully. Sequences that have a relatively low number
of occurrences but contain the less frequent amino acids (such as cysteine and tryptophan) tend to show inflatedNObs
(XYZ)/NExp
(XYZ) ratios. The results from the frequency method for each sequence, expressed as theNObs
(XYZ)/NExp
(XYZ) ratio, are also presented in Table 2. The application of these ratios to the sequences identified in the jumbling analysis serves to emphasize particular sequences
over others.
DISCUSSION
Reasons for High-Frequency Tripeptides
Although a major goal of this study was to identify previously unrecognized binding sites and signals, there are additional expected causes for overrepresented C-terminal tripeptides. First, many organisms possess highly conserved proteins, the genes for which have been duplicated many times within their genomes. For example, the human genome contains multiple copies of each of the histone-encoding genes. Thus, although the sequence KGK is observed 17 times, 11 of them can be attributed to isoforms of the histone protein H2A. In other cases, the actual protein product itself is less well understood. In A. thaliana, there exists a large novel protein family named AtPCMP (Aubourg et al. 2000). The members of this family appear to be unique to plants, and their function is not known. Of this group, 57 ORFs contain the “H motif” at their C-terminus. Consequently, they end with the same tripeptide, DYW.
A related reason for finding highly repeated C-terminal sequences is the result of multiple copies of transposon proteins within the genome. In S. cerevisiae, the sequence WIH is found 16 times. This tripeptide corresponds to the final three amino acids of the transposon Ty1 gag-pol protein product. All 16 ORFs from this genome ending in WIH encode such proteins. Similarly, all 10 occurrences of RSH and seven of 10 occurrences of RSR from E. coli can be explained by transposon ORFs (IS5 and IS1, respectively).
An additional cause of overrepresentation in C-terminal tripeptides may be a consequence of the synthesis of the polypeptide chains themselves. In both E. coli (Mottagui-Tabar et al. 1994; Björnsson et al. 1996; Mottagui-Tabar and Isaksson 1997) and S. cerevisiae(Mottagui-Tabar et al. 1998), it has been shown that certain amino acids at the last two positions of the polypeptide chain can affect the efficiency of translational termination. Using the analysis presented here, an overrepresentation of ORF sequences that end in XKK is found in all species except A. thaliana. For example, in the C. elegans genome, the tripeptide KKK is observed 70 times, GKK 32 times, SKK 28 times, AKK and LKK 24 times each, and EKK 22 times. In contrast, the expected number of occurrences of these sequences calculated using position-specific amino acid frequencies are: KKK, 15 times; GKK, 7 times; SKK, 15 times; AKK, 9 times; LKK, 15 times; and EKK, 10 times. These data are consistent with the reported results that in E. coli, lysine codons at the −1 (Björnsson et al. 1996) and −2 (Mottagui-Tabar et al. 1994; Mottagui-Tabar and Isaksson 1997) positions can enhance the efficiency of translational termination. Moreover, Arkov et al. (1995) noted that the 5′ contexts of stop codons are similar in both E. coli and humans, suggesting that effects on termination efficiency may be related. However, it should be noted that the observation of XKK inS. cerevisiae in this study is not consistent with previously observed translational termination effects in this species, which apparently do not depend on lysine (Mottagui-Tabar et al. 1998).
Since the genome jumbling analysis already takes into account the amino acid frequencies at each position, our data not only reflect the previous observation that lysine is found preferentially at the −1 position of many proteins (Berezovsky et al. 1997, 1999), but suggest that the correlated appearance of the terminal dipeptide KK may also be important. Interestingly, the XKK pattern is not found in the A. thaliana genome. Instead, an overabundance of XSS sequences is observed. We speculate that in this plant species, the serine residue or its corresponding codons may have a similar effect on translation efficiency as lysine in the other species studied.
Identification of Known and Potential Interaction Motifs
Not surprisingly, in addition to the reasons listed above, this study identified a set of tripeptide sequences that are known to serve as targeting signals or other recognition motifs in eukaryotes. The last three amino acids of the tetrapeptide signal for retention of proteins within the ER (Munro and Pelham 1987), DEL, were identified as being overrepresented in all eukaryotic genomes. In addition, the known variant EEL (Mazzarella et al. 1990) was found in humans. Similarly, the PTS1 (Gould et al. 1989), SKL, was also found in all eukaryotic genomes, as well as the variant AKL in humans and C. elegans. Moreover, an additional known C-terminal binding site was found. The sequence EVD occurs eight times in S. cerevisiae and 12 times in humans. This sequence corresponds to the last three amino acids of the tetrapeptide recognition motif, EEVD, found in the hsp70/hsp90 protein family, recognized by such proteins as Hop, FKBP52, and PP5 (Young et al. 1998; Buchner 1999; Scheufler et al. 2000).
Finally, previously unidentified recognition motifs may emerge from the results of this analysis. For example, the tripeptide FWC was found nine times in the S. cerevisiae genome. This particular sequence has several characteristics which make it potentially intriguing as a candidate recognition site: (1) All nine proteins that contain FWC belong to the family of amino acid permeases, and (2) the absolute sequence conservation extends only across the last three residues; upstream residues, although similar, are not identical (Fig.2). Furthermore, previous work has suggested that amino acid permeases in yeast utilize a unique set of proteins for proper trafficking (Ljungdahl et al. 1992; Kuehn et al. 1996; Gilstring et al. 1999), and that the C-terminal portion of these permeases is important for turnover (Helliwell et al. 2001; Omura et al. 2001). Of the remaining genomes presented in Table 2, we did not identify FWC as an overrepresented tripeptide. These results, along with the data from this study, implicate the C-terminal tripeptide FWC as potentially important for proper localization of the yeast amino acid permeases. Interestingly, in our preliminary analysis of the genome from the yeast Candida albicans, we noted that the FWC tripeptide does appear five times: once in the ORF for the amino acid permease Gap1p, and in four additional ORFs with high sequence similarity to Gap1p and other amino acid permeases, as identified through a BLAST search (Altschul et al. 1997). However, we were unable to identify any occurrences of the FWC sequence in the fission yeastSchizosaccharomyces pombe, a species more distantly related toS. cerevisiae than C. albicans, despite the fact that this genome does contain ORFs encoding amino acid permeases with significant similarity to those of S. cerevisiae.

Alignment of the amino acid sequences of the nine amino acid permeases from S. cerevisiae that end in the tripeptide FWC (highlighted) across the C-terminal 30 residues. Note that there are no other absolutely conserved residues within this region besides the terminal tripeptide.
Conclusions
The complete sequence of the genome of an organism can serve as a powerful tool for the analysis of sequence patterns and distributions. Since this information comprises a nearly complete array of a large number of data points, statistical methods can be applied to distinguish between chance occurrences and the results of selection. In the work presented here, C-terminal tripeptide sequences from a given genome were analyzed to determine which sequences can be considered overrepresented. Through this study, a collection of nonrandomly distributed tripeptide sequences was identified in eukaryotes. In addition to known targeting signals and binding motifs, several sequences could be explained by ORF homology or effects of certain codons on translational termination efficiency.
For many of these remaining sequences, however, distinguishing features are not readily apparent without more detailed analysis. This was, in part, due to the fact that many of these tripeptides belong to ORFs whose functions have not yet been determined. Hence, it is not yet possible to correlate those C-terminal sequences with a particular function or subcellular localization. Moreover, the difficulty of a comprehensive analysis of these data is enhanced by the large number of overrepresented sequences and the myriad of functions in which their corresponding proteins participate. By making these results available to the scientific community, we hope to enable the identification of additional common themes.
METHODS
Databases of the protein sequences of predicted ORFs from an entire genome were downloaded from the Web sites of the following institutions: The Institute for Genomic Research (M. jannaschii, A. thaliana; Web site: http://www.tigr.org[Bult et al. 1996; The Arabidopsis Genome Initiative 2000]), The E. coli Genome Project at the University of Wisconsin-Madison (E. coli K-12, Web site:http://www.genome.wisc.edu/k12.htm [Blattner et al. 1997]), TheSaccharomyces Genome Database at Stanford University (S. cerevisiae, Web site: http://genome-www.stanford.edu/Saccharomyces/[Goffeau et al. 1996]), The Sanger Centre (C. elegans, Web site: http://www.sanger.ac.uk/Projects/C_elegans/ [The C. elegans Sequencing Consortium 1998]), and the National Center for Biotechnology Information (H. sapiens, Web site:http://www.ncbi.nlm.nih.gov/genome/guide/human/ [Lander et al. 2001;Venter et al. 2001]). At the time of this study, the annotated array of predicted ORFs from the human genome was comprised of 14,760 entries.
Programs used for both the frequency method and the genome jumbling method were written in Perl.
WEB SITE REFERENCES
http://www.tigr.org; The Institute for Genomic Research (source ofM. jannaschii and A. thaliana ORF sequences).
http://www.genome.wisc.edu/k12.htm; The E. coli Genome Project at the University of Wisconsin-Madison (source of E. coli ORF sequences).
http://genome-www.stanford.edu/Saccharomyces/; TheSaccharomyces Genome Database at Stanford University (source of S. cerevisiae ORF sequences).
http://www.sanger.ac.uk/Projects/C_elegans/; The Sanger Centre (source of C. elegans ORF sequences).
http://www.ncbi.nlm.nih.gov/genome/guide/human/; The National Center for Biotechnology Information (source of H. sapiens ORF sequences).
Acknowledgments
G.J.G. was supported in part by a grant through the Medical Scientist Training Program. We thank the NIH for support.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵1 Corresponding author.
-
E-MAIL jberg{at}jhmi.edu; FAX (410) 502-6910.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.667603.
-
- Received July 29, 2002.
- Accepted January 28, 2003.
- Cold Spring Harbor Laboratory Press








