
Abstract
We have developed GFScan (GeneFamily Scan), a tool that identifies members of a gene family by searching genomic DNA sequences with genomic DNA motifs (or matrices) that are representative of the family. We have tested GFScan on four human gene families including the neurotransmitter-gated ion-channels (NGIC) family, the carbonic anhydrases (CA) family, the Dbl homology (DH) domain family, and the ETS-domain family. All known members of these families with motifs mapped to sequenced genomic DNA regions were found, whereas some novel genomic locations were also found to match the motifs, which may indicate new members in these families. Compared with other methods,GFScan recognized all true positives with much fewer false positives. We also showed that motifs constructed based on human genes could be used to search the mouse genome to identify orthologous family members in mouse. This program is available athttp://www.cshl.org/mzhanglab/.
[The following individuals and institutions kindly provided reagents, samples or unpublished information as indicated in the paper: J. Maddock and Celera Genomics.]
With the advances of several whole-genome sequencing projects, including human, mouse,Drosophila, and so on, more and more genomic DNA sequences have become available. These projects make it possible to analyze gene families in one species systematically. One of the well-known strategies for gene family analysis is to detect all the gene models first in one genome with some gene prediction methods, such asGenscan (Burge and Karlin 1997), Genie (Kulp et al. 1996), or FGENES (Solovyev and Salamov 1997); translate these genes into proteins; then try to find gene families at the protein level using similarity search or protein motif databases, such as BLOCKS+ (Henikoff et al. 1999), Pfam (Bateman et al. 1999), ProDom (Corpet et al. 1999), PRINTS (Attwood et al. 1999), PROSITE (Hofmann et al. 1999), IntroPro (http://www.ebi.ac.uk/interpro/). Additionally, mRNAs can also be used to find gene family members byBLAST or FASTA searches (Pearson and Lipman 1988; Altschul et al. 1990, 1997). Recently, Henikoff (Henikoff and Henikoff 2000) had tried to use protein fragments in the BLOCKS+ database to search the Drosophila genomic sequence usingBLAST.
Our method seeks to find all members of a gene family by searching the whole genome with the representative genomic DNA motif of this family. Motif search at the protein level is a reliable method to find protein family members based on known proteins. However, protein motifs can only be used to search the known proteins, and some proteins remained undiscovered by existing experimental or theoretical methods. On the other hand, TBLASTN, a program of the BLASTpackage, can align protein sequences with genomic DNA sequence directly to find matched regions that may code new members of the gene family. However, as shown in the Results and Discussion sections, programs in the BLAST family are general sequence-alignment programs and find many false positives. To circumvent this problem, we developedGFScan (Gene FamilyScan), which uses a representative DNA motif of a gene family to search genomic DNA sequence directly to identify new members of the gene family. The representative genomic DNA motif is constructed based on protein motifs in PROSITE (release 16.0, updates up to September 2000) and the genomic structure of known members of the family. As more and more mRNA and protein sequences are submitted to the public databases, and as each genome becomes more complete,GFScan will be increasingly effective to find new members of a gene family.
RESULTS
GFScan was developed in C++ language. To show the usefulness of this program, we applied it to four gene families, searching for new members of the family in the whole human genome (Genome Sequencing Consortium 2001) in GoldenPath (April 2001 freeze and August 2001 freeze; http://genome.ucsc.edu/) and mouse genome in the Celera Genomics Company's database.
Neurotransmitter-Gated Ion-Channels (NGIC) Family
The human neurotransmitter-gated ion-channels family is a large family, whose members include GABA (gamma-aminobutyricacid) A receptors, glycine receptors, acetylcholine receptors, and 5-hydroxytryptamine-3 receptor. All members of the family have a common protein motif, called NEUROTR_ION_CHANNEL in the PROSITE database (ID: PS00236). Using the known 37 human genes of this family in the public database and the protein motif in PROSITE, a 45-bp intronless genomic DNA motif was constructed. We also found that one family member, CHRNB1, has an intron in the motif-matching genomic region, and the intron separates the 45-bp motif into two parts. An intron-containing genomic DNA motif was then constructed (see Methods). Both genomic DNA motifs were used to search the whole human genome. Of 37 known motif regions, 29 were found by GFScan. For the missed eight genes, all the genomic regions corresponding to the motifs fell into the gaps of the genome. Moreover, nine additional genomic regions were found. Three of them were duplications of the known genes. Among the remaining six novel genomic regions, one is located in the repeat region, and the other five were likely to be members of this gene family that are previously unidentified. Based on the human genome annotation in GoldenPath (http://genome.ucsc.edu/), these five regions were reported to be similar to mouse glycine receptor subunit α1, rat GABA A receptor subunit γ1, rat ρ3, mouse GABA A receptor subunit α3, and Gallus nicotinic acetylcholine subunit α8, respectively. With the exception of GABA A receptor α3, no mRNA or protein sequence has been known for the other four genes (see Table1).
Results on Neurotransmitter-Gated Ion-Channels Family
| No. | Chromosome | Strand | Motif position | Description | |
| 1 | X | + | 13920432 | 13920477 | GLRA2 |
| 2 | X | − | 104174460 | 104174415 | (Similar to mouse Glra1)New |
| 3 | X | − | 152091851 | 152091806 | GABRE |
| 4 | X | − | 152454684 | 152454639 | GABRA3 |
| 5 | 1 | − | 1478204 | 1478159 | GABRD |
| 6 | 2 | − | 178143495 | 178143450 | CHRNA1 |
| 7 | 2 | + | 237873656 | 237873701 | CHRND |
| 8 | 2 | + | 237886519 | 237886564 | CHRNG |
| 9 | 3 | − | 104298942 | 104298897 | (Similar to Rat Gabrr3)New |
| 10 | 4 | − | 50087519 | 50085574 | (Similar to Rat Gabrg1)New |
| 11 | 4 | − | 50330244 | 50330199 | GABRA2 |
| 12 | 4 | + | 50782012 | 50782057 | GABRA4 |
| 13 | 4 | − | 51222098 | 51222053 | GABRB1 |
| 14 | 4 | + | 166964539 | 166964584 | GLRB |
| 15 | 4 | − | 184669299 | 184669254 | GLRA3 |
| 16 | 5 | − | 174399338 | 174399293 | GABRB2 |
| 17 | 5 | + | 174946361 | 174946406 | GABRG2 |
| 18 | 5 | + | 183945827 | 183945872 | GABRP |
| 19 | 6 | − | 98310008 | 98309963 | GABRR2 |
| 20 | 8 | − | 29298237 | 29298192 | CHRNA2 |
| 21 | 8 | + | 45473102 | 45473147 | CHRNB3 |
| 22 | 8 | − | 45498244 | 45498199 | CHRNA6 |
| 23 | 10 | − | 58232334 | 58232289 | (REPEAT region)New |
| 24 | 10 | + | 96875579 | 96875624 | (Similar to mouse Gabra3)New |
| 25 | 11 | − | 2590955 | 2590910 | CHRNA10 |
| 26 | 11 | − | 125455260 | 125455215 | (HTR3A duplication)New |
| 27 | 11 | − | 125490375 | 125490330 | HTR3A |
| 28 | 15 | + | 22459117 | 22459162 | GABRB3 |
| 29 | 15 | + | 24261884 | 24261929 | (Similar to Gallus Chrna8)New |
| 30 | 15 | + | 24323276 | 24323321 | (CHRNA7 duplication)New |
| 31 | 15 | + | 26565917 | 26565962 | CHRNA7 |
| 32 | 15 | + | 76709488 | 76709533 | CHRNA5 |
| 33 | 15 | − | 76721764 | 76721719 | CHRNA3 |
| 34 | 15 | − | 76752832 | 76752787 | CHRNB4 |
| 35 | 17 | − | 5014125 | 5014080 | CHRNE |
| 36 | 17 | − | 5277247 | 5277202 | (CHRNE duplication)New |
| 37 | 17 | − | 8070655 | 8070258 | CHRNB1 |
| 38 | 20 | − | 63883819 | 63883774 | CHRNA4 |
[i] Missed known genes in this family: GABRA6 (NM_000811), GABRQ (NM_018558), CHRNA9 (NM_017581), GLRA1 (NM_000171), GABRA1 (NM_00806), GABRA5 (NM_000810), GABRR1 (NM_002042), CHRNB2 (NM_000748).
Carbonic Anhydrases (CA) Family
Human carbonic anhydrases (CA) are zinc metalloenzymes that catalyze the reversible hydration of carbon dioxide. There are 14 known members in the family. From the mRNAs of the known members, we first constructed a 57-bp cDNA motif based on the PROSITE protein motif (ID: PS00162). All of the genomic sequence regions corresponding to this cDNA motif contain one intron. The splice locations of the introns are identical among all members, but the lengths of the introns are different. We next constructed a genomic DNA motif from the cDNA motif incorporating information on the intron. By searching the whole human genome with the genomic DNA motif, 12 of 14 known genes were found, and the two genes that were missed had their motif-matching genomic region falling into the genomic gaps. Moreover, we found two additional genomic regions that match the motif: One was related to a non-CA family gene, PTPRG (protein tyrosine phosphatase, receptor type G) in Chromosome 3; the other was found in Chromosome 8, whose closest homologous gene was the mouse Car13 gene. It is worth noticing that the human CA13 gene has not been found before, and our finding may have shed light on this new member of the family (see Table 2).
Results on Carbonic Anhydrases (CA) Family
| No. | Chromosome | Strand | Motif position | Description | |
| 1 | 1 | − | 230527424 | 230527143 | (CA14 duplication)New |
| 2 | 1 | + | 230570250 | 230570531 | CA14 |
| 3 | 1 | + | 9063801 | 9065482 | CA6 |
| 4 | 3 | + | 68787364 | 68790131 | (PTPRG)New |
| 5 | 4 | + | 138352740 | 138353600 | (CA7 duplication)New |
| 6 | 8 | + | 89796759 | 89803951 | (Similar to mouse Car13)New |
| 7 | 8 | − | 89874524 | 89871143 | CA1 |
| 8 | 8 | + | 89982297 | 89984196 | CA3 |
| 9 | 8 | + | 90013921 | 90014490 | CA2 |
| 10 | 9 | + | 38809920 | 38810067 | CA9 |
| 11 | 15 | + | 60482427 | 60485863 | CA12 |
| 12 | 16 | − | 23050079 | 23047764 | CA5 |
| 13 | 16 | − | 33403805 | 33401499 | (CA5 duplication)New |
| 14 | 16 | − | 33988707 | 33986402 | (CA5 duplication)New |
| 15 | 16 | + | 77218848 | 77219708 | CA7 |
| 16 | 16 | + | 77672109 | 77672969 | (CA7 duplication)New |
| 17 | 17 | − | 55621766 | 55527814 | LOC56934 |
| 18 | 17 | + | 64513194 | 64513368 | CA4 |
| 19 | 19 | − | 57550194 | 57549926 | CA11 |
[i] Missed known genes in this family: CA8 (NM_004056), CA5B (NM_007220).
Dbl Homology (DH) Domain Family
The Dbl homology (DH) domain is responsible for the guanine nucleotide exchange factor (GEF) catalytic activity (Zhu et al. 2001). Eight human genes belong to this family, and some of these genes are oncogenes, including DBL, Break Cluster Region(BCR) oncogene, VAV, VAV2, andVAV3. The protein sequences of all eight members share the DH domain (PROSITE ID: PS00741). From their mRNA sequences, a 78-bp cDNA motif was constructed. In the genomic regions corresponding to the motif, no intron was found for one of the family members,TIAM; two introns were found for ABR andBCR; and one intron was found for the remaining five members of the family. Based on above information on gene structure, we next constructed three genomic DNA motifs of this domain from the cDNA motif. Searching the whole human genome with the genomic DNA motifs revealed nine genomic regions that significantly match the motifs. Among the nine regions, seven contain known genes, one of the two new locations was the VAV gene's genomic DNA sequence duplication, and the other overlapped with the known VAV2's motif region (see Table 3). VAV3was the only known member of the family that was missed by the search, and this is because the genomic region matching the motif region was not available in the April 2001 Goldenpath freeze (it was found in the August 2001 freeze).
Results on DH-Domain Family
| No. | Chromosome | Strand | Motif position | Description | |
| 1 | X | − | 141522542 | 141523440 | DBL |
| 2 | 9 | − | 137956245 | 137957015 | VAV2 |
| 3 | 9 | − | 137956245 | 137964371 | (VAV2 pseudo-site)New |
| 4 | 13 | + | 117682791 | 117683814 | DBS |
| 5 | 17 | − | 631694 | 639380 | ABR |
| 6 | 19 | + | 28364356 | 28364681 | (VAV duplication)New |
| 7 | 19 | − | 7283797 | 7284122 | VAV |
| 8 | 21 | − | 29371499 | 29371577 | TIAM |
| 9 | 22 | + | 20261116 | 20264414 | BCR |
[i] Missed known genes in this family: VAV3 (NM_006113) (found in chr1 125174019-125178912 in Goldenpath Aug 2001).
ETS-Domain Family
The ETS-domain gene family includes a group of proteins that function as transcription factors under physiologic conditions and, if aberrantly expressed, can cause cellular transformation (Karim et al. 1990). These proteins share a conserved domain, the ETS domain, which is involved in DNA binding. From the mRNAs of the 19 known members and a protein motif in the PROSITE database (ID: PS00346), a 48-bp cDNA motif was constructed. Four of these 19 genes have one intron in their genomic regions matching the motif, and the splice location of the intron is the same. Therefore, we constructed an intron-containing genomic DNA motif, and it is used to search the human genome together with the cDNA motif. Twenty-six genomic regions were found to match the motifs, which include 18 of the 19 known genes. ETV5's genomic DNA motif region was missed because the genomic DNA sequence around the motif-matching region was uncompleted. Out of the eight additional motif-matching regions, three were duplications of three known genes (i.e., GABP, ETV6, and ERF). The other five were related to unknown genes in human: one was in the FEVgene region, two were similar to mouse Ets-protein Spi-C (GenBank accession no. AF098863), and the last two were located in two genes predicted by Genscan and Ensembl. Both FEV and Spi-C are ETS-domain family members (Bemark et al. 1999). FEV was not listed in the PROSITE database because of the database-updating problem, and human Spi-C has not been found. Likely, these new motif-matching regions will provide experimental scientists with useful guidance to identify new members of the ETS-domain family in the human genome (see Table 4).
Results on ETS-Domain Family
| No. | Chromosome | Strand | Motif position | Description | |
| 1 | X | + | 46781345 | 46781393 | ELK1 |
| 2 | 1 | + | 177575228 | 177576887 | ETV3/PEP1 |
| 3 | 1 | + | 177611558 | 177611851 | (Genscan predicted gene)New |
| 4 | 1 | − | 229691250 | 229691298 | ELK4 |
| 5 | 2 | − | 223797915 | 223797963 | (FEV gene)New |
| 6 | 6 | − | 40661831 | 40661879 | TEL2 |
| 7 | 7 | − | 13211815 | 13211863 | ETV1 |
| 8 | 7 | + | 63416487 | 63416535 | (GABP duplication)New |
| 9 | 11 | − | 142926355 | 142926403 | ETS1 |
| 10 | 11 | + | 143364213 | 143364261 | FLI1 |
| 11 | 11 | − | 34770889 | 34770937 | (Similar to Mus.AF098863)New |
| 12 | 11 | − | 48738245 | 48738293 | SPI1 |
| 13 | 12 | + | 105487761 | 105487809 | ELK3 |
| 14 | 12 | + | 110940578 | 110940626 | (Similar to Mus.AF098863)New |
| 15 | 12 | + | 110951587 | 110951635 | (Similar to Mus.AF098863)New |
| 16 | 12 | + | 13676162 | 13676210 | ETV6 |
| 17 | 13 | − | 40208762 | 40210606 | ELF1 |
| 18 | 17 | − | 45424547 | 45424595 | ETV4 |
| 19 | 19 | − | 41606511 | 41606559 | ETV2/ER71 |
| 20 | 19 | − | 50620314 | 50620753 | (Ensembl predicted gene)New |
| 21 | 19 | + | 51122030 | 51122466 | (ERF duplication)New |
| 22 | 19 | − | 51228203 | 51228639 | ERF |
| 23 | 19 | + | 62153841 | 62153889 | SPIB |
| 24 | 21 | + | 23995905 | 24000169 | GABP |
| 25 | 21 | − | 36613278 | 36613326 | ERG |
| 26 | 21 | + | 37052185 | 37052233 | ETS2 |
[i] Missed known genes in this family: ETV5 (NM_004454).
Comparison with the BLAST Results
The other common method to search for new members of a gene family is to run the BLAST program against the whole genome using known members' sequences as queries. We compared BLASTand GFScan on all four families. We searched the protein sequence of each known member of a given family in human genome usingTBLASTN. We also used the motif region of the mRNA sequence of each known member to search the human genome usingBLASTN. The results are listed in Table5.
Comparison Results with BLAST
| GFScan | TBLASTN | BLASTN | |||||
| E<E m | E<1e− 4 | E<10 | E<10 | ||||
| A. NGIC Family (E m = 9e − 6)[i] | |||||||
| Known member | 37 | 37 | 37 | 37 | 37 | ||
| Location found | 38 | 45 | 48 | 59 | 33 | ||
| Known location found | 29 | 29 | 29 | 29 | 28 | ||
| Potential candidates[ii] | 8 | 8 | 8 | 8 | 5 | ||
| False positives[iii] | 1 | 8 | 11 | 22 | 0 | ||
| Known location missed | 8 | 8 | 8 | 8 | 9 | ||
| B. CA Family (E m = 9e − 10) | |||||||
| Known member | 14 | 14 | 14 | 14 | 14 | ||
| Location found | 19 | 19 | 23 | 38 | 16 | ||
| Known location found | 12 | 12 | 12 | 12 | 11 | ||
| Potential candidates[ii] | 6 | 6 | 6 | 6 | 5 | ||
| False positives[iii] | 1 | 1 | 5 | 20 | 0 | ||
| Known location missed | 2 | 2 | 2 | 2 | 3 | ||
| C. DH-Domain Family (E m = 1c − 8) | |||||||
| Known member | 8 | 8 | 8 | 8 | 8 | ||
| Location found | 9 | 11 | 16 | 44 | 5 | ||
| Known location found | 7 | 7 | 7 | 7 | 5 | ||
| Potential candidates[ii] | 1 | 1 | 1 | 1 | 0 | ||
| False positives[iii] | 1 | 3 | 8 | 36 | 0 | ||
| Known location missed | 1 | 1 | 1 | 1 | 3 | ||
| D. ETS-Domain Family (E m = 1c − 10) | |||||||
| Known member | 19 | 19 | 19 | 19 | 19 | ||
| Location found | 26 | 34 | 37 | 58 | 15 | ||
| Known location found | 18 | 18 | 18 | 18 | 14 | ||
| Potential candidates[ii] | 8 | 8 | 8 | 8 | 1 | ||
| False positives[iii] | 0 | 8 | 11 | 32 | 0 | ||
| Known location missed | 1 | 1 | 1 | 1 | 5 | ||
[i] E m: The minimum E-value used to find all known members by TBLASTN.
[ii] Genomic location that is not related to known members. The translated protein could match regular expression pattern of the gene family in the PROSITE database.
[iii] Genomic location where no gene family member locates (see detail in Methods).
Table 5 indicates that GFScan had less false positives than TBLASTN (except for the CA family under a lowE-value threshold, but the false positives ofTBLASTN were increased when the E-value threshold was increased). In the BLASTN search, even with a very high E-value threshold (e.g., E = 10), some known genes were still not found, especially the ones whose motifs contain introns. For those genes, the match of the motif region to the genomic sequence is rather poor. Meanwhile, very few new genomic regions were found in this case. In short, compared with BLAST,GFScan offers both higher sensitivity and higher specificity, especially in intron-containing cases.
Mouse Genome Searching with Two Human DNA Motifs
We searched Celera's mouse genome using the motif constructed from human genes. For the neurotransmitter-gated ion-channels family, 23 of 24 known mouse members in the NCBI LocusLink Database (http://www.ncbi.nlm.nih.gov/LocusLink/) were found byGFScan. For the one that was missed (NM_017369: 1824–1868), the genomic DNA sequence of this gene was incomplete in the database. At the same time, 13 new motif-matching genomic locations were found, which may correspond to 13 novel mouse members of this family.
The result was different for the CA family. For 13 known mouse CA members in the LocusLink Database, 11 had the genomic DNA sequence matches. Using GFScan and the motif constructed by human genes, we could only find five loci. The reason for missing the other six was that the motif segments in these mouse genes are different from the motif in human genes (Fig.1). Three of these six genes cannot even match the motif in human (NM_030558, mouse Car15; NM_009802, mouse Car6; NM_007608, mouse Car5a) at the protein level. However, two new genomic locations matching the human motif were still found, which may correspond to novel members in mouse.
The mRNA regions of 11 members of the mouse CA family identified by searching the Celera Mouse genome using the motif constructed based on CA family members in human. The first five lines were regions found byGFScan as they matched the human motif in the middle lines (R = A or G, Y = T or C, K = G or T, M = A or C, S = G or C, W = A or T, B = G or T or C, D = G or A or T, H = A or C or T, V = G or C or A, N = A or T or G or C). The bottom six lines show the other regions of the gene family that were missed byGFScan. The unmatched sites are in bold fonts. The underlined lower-case triplet represents the amino acid code that did not even match the protein motif of this family.

In summary, GFScan is capable of identifying all the true members of a family with very few false positives and requiring no gene prediction. It performs especially well with intron-containing motifs where most BLAST-based tools may fail. One should be cautioned when using GFScan for cross-species search, however, as the results may depend on the divergence among members of the family, as well as the evolutionary distance between the two species. By adding more mRNAs from different species or modifying a genomic motif to allow species-specific codon usages, further improvement on performance can be achieved. GFScan is implemented in a way that such customizations can be easily made (see Methods for more detail).
DISCUSSION
Same Species versus Cross-Species
As DNA sequences are usually less conserved than protein sequences in evolution, we recommend constructing motifs using known mRNAs in one species and then using the motif to search the genome of the same species. This will reduce false positives. For cross-species searches, this method sometimes worked well, as in neurotransmitter-gated ion-channels family; at other times it missed many true positives, as in the case of the CA family described above. As the program allows users to reconstruct motifs by adding more mRNAs from other species, it is easy to extend the search to the cross-species cases. One could also redefine the motif by relaxing on codon usage when searching related species or adding other conserved information into the motif.
Regular Expression Pattern Search and Weight Matrix Search
From the mRNA sequences and protein motifs of the known members of a given gene family, we could construct both a regular expression pattern and weight matrix for later searching. GFScan can use either of them to search the genomic DNA. Based on the matrix constructed, the scores of all known motif regions were calculated. When we chose the minimum score of the known motif regions as the threshold of matrix search to minimize false positives, we found that the genomic locations whose scores were higher than the threshold could all be found by a regular expression pattern search (Table6), whereas the latter saved a lot of CPU time, because searching with regular expressions is almost 15–20 times faster than searching with matrices. However, because matrix search has higher sensitivity (at the expense of specificity and CPU time), the genomic locations missed by a regular expression pattern search may be recovered by a matrix search, especially in the cross-species cases.
Genomic Locations with Highest Score Identified by Matrix Search
| Chromosome | Motif location | Strand | Score | Index in pattern search | |
| A. NGLC Family (S min = 24.027[i]) | |||||
| 20 | 63883774 | 63883819 | − | 29.4595 | 38 |
| 8 | 29298192 | 29298237 | − | 29.4595 | 20 |
| X | 104174415 | 104174460 | − | 29.0811 | 2 |
| 8 | 45473102 | 45473147 | + | 29.0541 | 21 |
| X | 13920432 | 13920477 | + | 28.8649 | 1 |
| 15 | 76752787 | 76752832 | − | 29.7568 | 34 |
| 17 | 8070258 | 8070655 | − | 28.5135 | 37 |
| 6 | 98309963 | 98310008 | − | 28.4324 | 19 |
| 17 | 5014080 | 5014125 | − | 28.4054 | 35 |
| 17 | 5277202 | 5277247 | − | 28.4054 | 36 |
| 2 | 237873656 | 237873701 | + | 28.3514 | 7 |
| 15 | 22459117 | 22459162 | + | 28.0811 | 28 |
| 1 | 1478159 | 1478204 | − | 28.0541 | 5 |
| 11 | 125455215 | 125455260 | − | 27.973 | 26 |
| 11 | 125490330 | 125490375 | − | 27.973 | 27 |
| 15 | 76721719 | 76721764 | − | 27.8919 | 33 |
| 4 | 184669254 | 184669299 | − | 27.6487 | 15 |
| 2 | 237886519 | 237886564 | + | 27.5946 | 8 |
| 15 | 76709488 | 76709533 | + | 27.5676 | 32 |
| 8 | 45498199 | 45498244 | − | 27.4324 | 22 |
| X | 152454639 | 152454684 | − | 27.4324 | 4 |
| 2 | 178143450 | 178143495 | − | 27.3514 | 6 |
| 15 | 24323276 | 24323321 | + | 27.1081 | 30 |
| 15 | 26565917 | 26565962 | + | 27.1081 | 31 |
| 5 | 183945827 | 183945872 | + | 27.0811 | 18 |
| 15 | 24261884 | 24261929 | + | 27.0541 | 29 |
| 5 | 174399293 | 174399338 | − | 26.7027 | 16 |
| 11 | 2590910 | 2590955 | − | 26.5946 | 25 |
| 4 | 51222053 | 51222098 | − | 26.5135 | 13 |
| 10 | 96875579 | 96875624 | + | 26.2432 | 24 |
| 4 | 50330199 | 50330244 | − | 26.2432 | 11 |
| 4 | 166964539 | 166964584 | + | 25.7027 | 14 |
| 5 | 174946361 | 174946406 | + | 25.6757 | 17 |
| 4 | 50782012 | 50782057 | + | 25.5135 | 12 |
| 3 | 104298897 | 104298942 | − | 25.1351 | 9 |
| 4 | 50085574 | 50085619 | − | 24.3243 | 10 |
| X | 152091806 | 152091851 | − | 24.027 | 3 |
| B. CA Family (S min = 32.6429) | |||||
| 16 | 77218848 | 77219708 | + | 39.2143 | 15 |
| 16 | 77672109 | 77672969 | + | 39.2143 | 16 |
| 4 | 138352740 | 138353600 | + | 39.2143 | 5 |
| 16 | 23047764 | 23050079 | − | 39.0714 | 12 |
| 16 | 33401499 | 33403805 | − | 39.0714 | 13 |
| 16 | 33986402 | 33988707 | − | 39.0714 | 14 |
| 8 | 89982297 | 90014490 | + | 38.8571 | 8 |
| 15 | 60482427 | 60485863 | + | 38.4286 | 11 |
| 9 | 38809920 | 38819255 | + | 37 | 10 |
| 8 | 89796759 | 89803951 | + | 36.9286 | 6 |
| 8 | 90013921 | 90014490 | + | 36.8572 | 9 |
| 17 | 64513194 | 64513368 | + | 36.4286 | 18 |
| 8 | 89871143 | 89874524 | − | 36.4286 | 7 |
| 1 | 9063801 | 9065482 | + | 35.8571 | 3 |
| 17 | 55604726 | 55621766 | − | 35.3571 | 17 |
| 3 | 68787364 | 68790131 | + | 34.5 | 4 |
| 19 | 57549926 | 57550194 | − | 34 | 19 |
| 1 | 230527143 | 230527424 | − | 32.6429 | 1 |
| 1 | 230570250 | 230570531 | + | 32.6429 | 2 |
[i] S min is the minimum score of the motifs from known family members.
Motifs
In the present program, the motif length is taken as a constant; in other words, all the motif regions in the family should have the same length. For those families whose protein motifs have variable lengths, it is difficult to construct the DNA motif, and allowing gaps in the motif can be very CPU-expensive. We will address these issues in future work.
Although GFScan constructs the genomic motif automatically, it also accepts user-defined motifs as its input. This makes GFScan a very flexible tool for gene family analysis at the genomic level. In conjunction with gene prediction tools, it can be used for gene finding and gene structure prediction as well.
METHODS
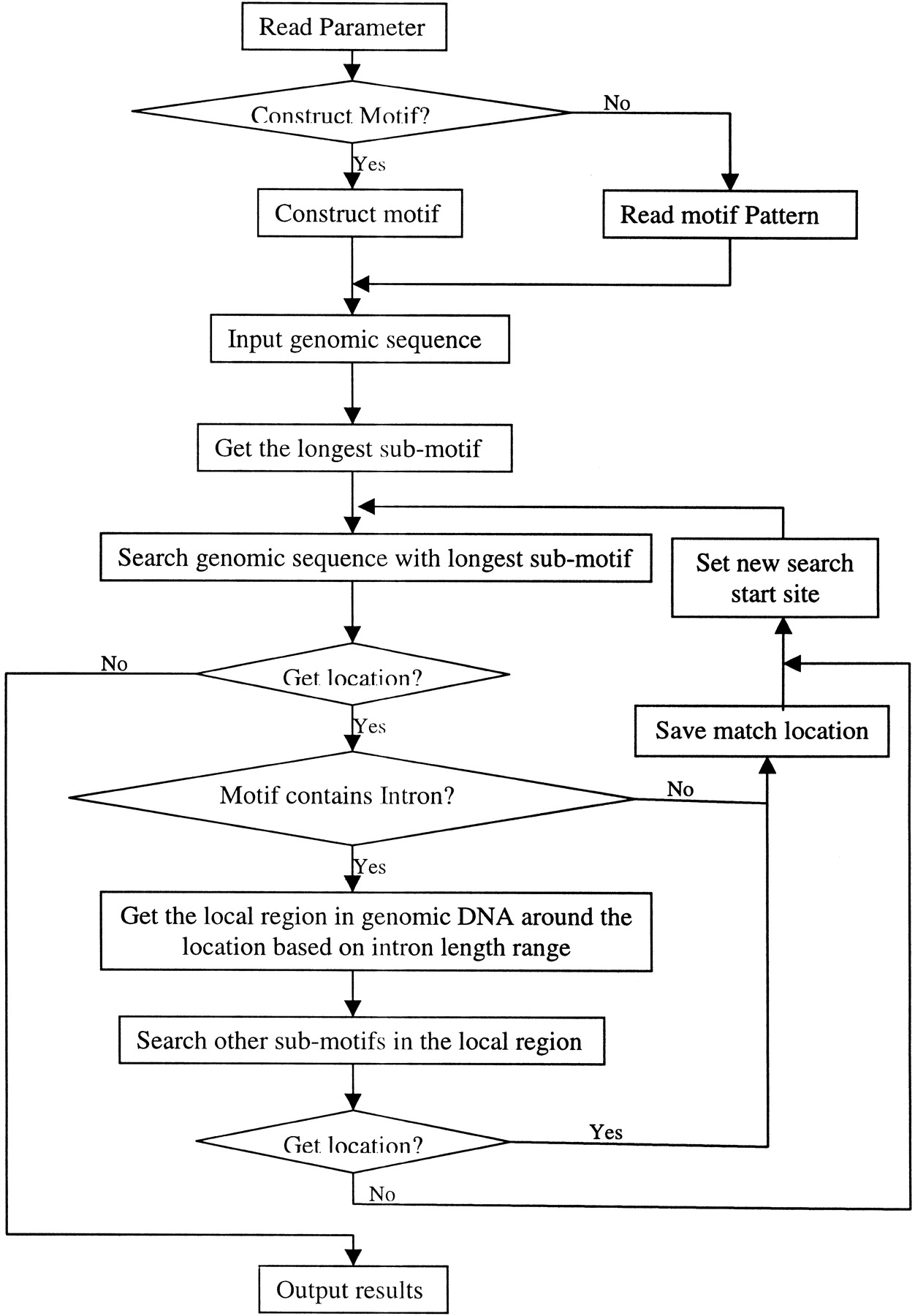
For a protein or a gene family, we collected protein, mRNA, and genomic DNA sequences of all known members, as well as the PROSITE entry. Using the protein motif in PROSITE, we extracted the protein motif fragments and their corresponding mRNA fragments. Based on the protein motif, these mRNA fragments were aligned, and the consensus pattern was created. Each site in the consensus pattern was determined from all the corresponding sites in the known mRNA sequences. In other words, each site in the protein motif was converted into three sites in the cDNA motif based on all existing codons in known mRNAs. UsingSIM4 (Florea et al. 1998) to align mRNAs with genomic DNAs, we find the potential intron position and its length range within the genomic regions that matches the motif regions. This intron information was incorporated into the cDNA motif as the genomic DNA motif of this family was constructed (see Fig.2). For each genomic DNA motif, if there were introns inside, the motif was divided into several submotifs, and the longest submotif would be used first to find the potential match location, then the other submotifs were used to search the sequences around this location (see Fig. 3). Each genomic DNA region matching the motif would be translated into a protein sequence, and this protein fragment was tested by the protein motif to identify the false-positive results.
(A) Method to construct genomic DNA motif. Three steps were taken to construct the genomic DNA motif for a given family from known protein, mRNA, and genomic DNA sequences of the family. First, based on the locations of the protein motif in protein sequences, the corresponding mRNA regions were extracted and aligned to reveal the consensus pattern. Each site in the consensus pattern would include all nucleotides existing in the mRNAs at the site. Second, gene structures were obtained by aligning mRNA with genomic DNA sequences, and the intron information was collected. Third, the intron information was incorporated into the cDNA consensus pattern to generate the final genomic DNA motifs. (B) Motif construction example in the CA family. Conservative sites in DNA motifs are in bold font. Donors and acceptors of introns are in small letters. The number in the brackets in the DNA sequence alignments is the intron length in each gene. In the final Genomic DNA Motif, the two numbers separated by a comma in the parentheses (34 , 105467) are the minimum and maximum lengths of the intron in this position. Each pair of brackets in the DNA motif represents one site in the sequence, and the bases within each pair of brackets represent all possible nucleotides at that site.

Flowchart of the motif search algorithm. Rectangle boxes repesent steps, diamond boxes represent decision switches, and arrows show steps' order. For an intron-containing motif, the genomic DNA motif was separated into several submotifs, and the longest one was used to search the genome first. If a genomic region matches the longest submotif, this region is extracted based on intron information and the other submotifs would be only searched within this region.
The weight matrix can be created while constructing the consensus regular expression pattern. In this algorithm, we simply used the nucleotide occupation frequencies at each site of the motif as the weights. For the intron-containing motif, we used the same strategy as we did in pattern search, namely, the longest submatrix was used first to find a candidate genomic location, and the local region around this location would be searched by the other submatrices.
We used protein sequences of all known members to search the human genome by TBLASTN, and we used the motif region of known members' mRNA sequences to search the human genome byBLASTN. As the exact number of the real members in a given gene family is unknown, we regarded the locations found byGFScan or BLAST false positives if the DNA fragment in these locations could not be translated into protein sequences without a stop codon, or the translated protein sequences did not match the motif pattern of the gene family. If the location is overlapped by one gene that is obviously not a member of the gene family by knowledge, the location would also be regarded as false positive. At the same time, those locations that do not code the known proteins listed in one PROSITE entry and are not false positive will be regarded as potential candidates. In TBLASTN search, only genomic DNA regions that could match the protein motif region partially or completely were considered as the locations of gene family members. The other genomic regions where the matches between genomic DNA sequence and protein sequence were outside of the motif were not considered. In BLASTN search, because the query sequences were so short that the significance of matches was low, only those genomic DNA match regions that could be aligned completely with the query sequence were regarded as the gene member's locations to avoid many short, partial, and random matches. The Expect-value (E-value) was used as the threshold to filter the most significant match in BLAST. In our comparison, we chose different E-values as thresholds in TBLASTNsearches and used the default setting in BLASTN(E-value < 10) searches. To compare the specificity withGFScan meaningfully, we chose the smallestE-value that could find all known gene members as the threshold for TBLASTN, then compared the new motif match locations number with that obtained from GFScan.
Availability
The program GFScan is available athttp://www.cshl.org/mzhanglab/.
WEB SITE REFERENCES
http://genome.ucsc.edu/; GoldenPath.
http://www.cshl.org/mzhanglab/; GFScan program.
http://www.ebi.ac.uk/interpro/; IntroPro.
http://www.ncbi.nlm.nih.gov/LocusLink/; NCBI LocusLink Database.
We thank Theresa Zhang for revising the English text. We also thank Celera Genomics for the mouse genome database. The C++ Boost library was kindly supplied by Dr. John Maddock. This work is supported by grant CA81152 from NIH.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Notes
[9] Corresponding author.
Notes
[10] E-MAIL [email protected]; FAX (516) 367-8461.
[11] Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.220102. Article published online before print in June 2002.
REFERENCES
- ↵S.F. AltschulW. GishW. MillerE.W. MyersD.J. Lipman(1990) Basic local alignment search tool. J. Mol. Biol. 215:403–410.
- ↵S.F. AltschulT.L. MaddenA.A. SchafferJ. ZhangZ. ZhangW. MillerD.J. Lipman(1997) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402.
- ↵T.K. AttwoodD.R. FlowerA.P. LewisJ.E. MabeyS.R. MorganP. ScordisJ.N. SelleyW. Wright(1999) PRINTS prepares for the new millennium. Nucleic Acids Res. 27:220–225.
- ↵A. BatemanE. BirneyR. DurbinS.R. EddyR.D. FinnE.L.L. Sonnhammer(1999) Pfam 3.1: 1313 multiple alignments and profile HMMs match the majority of proteins. Nucleic Acids Res. 27:260–262.
- ↵M. BemarkA. MartenssonD. LibergT. Leanderson(1999) Spi-C, a novel Ets protein that is temporally regulated during B lymphocyte development. J. Biol. Chem. 274:10259–10267.
- ↵C. BurgeS. Karlin(1997) Prediction of complete gene structure in human genomic DNA. J. Mol. Biol. 268:78–94.
- ↵F. CorpetJ. GouzyD. Kahn(1999) Recent improvements of the ProDom database of protein domain families. Nucleic Acids Res. 27:263–267.
- ↵L. FloreaG. HartzellZ. ZhangG.M. RubinW. Miller(1998) A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 8:967–974.
- ↵Genome Sequencing Consortium (2001) Initial sequencing and analysis of human genome. Nature 409:860–921.
- ↵J.G. HenikoffS. Henikoff(2000) Drosophila genomic sequence annotation using the BLOCKS+ database. Genome Res. 10:543–546.
- ↵S. HenikoffJ.G. HenikoffS. Pietrokovski(1999) BLOCKS+: A non-redundant database of protein alignment blocks derived from multiple compilations. Bioinformatics 15:471–479.
- ↵K. HofmannP. BucherL. FalquetA. Bairoch(1999) The PROSITE database, its status in 1999. Nucleic Acids Res. 27:215–219.
- ↵F.D. KarimL.D. UrnessC.S. ThummelM.J. KlemszS.R. McKercherA. CeladaC. Van BeverenR.A. MakiC.V. GuntherJ.A. Nye(1990) The ETS-domain: A new DNA-binding motif that recognizes a purine-rich core DNA sequence. Genes & Dev. 4:1451–1453.
- ↵D. KulpD. HausslerM.G. ReeseF.H. Eeckman(1996) A generalized hidden Markov model for the recognition of human genes in DNA. in Proceedings of the Fourth International Conference on Intelligent Systems for Molecular Biology, ed D. States(AAAI Press, Menlo Park, CA), pp 134–142.
- ↵W.R. PearsonD.J. Lipman(1988) Improved tools for biological sequence comparison. Proc. Natl. Acad. Sci. 85:2444–2448.
- ↵V.V. SolovyevA.A. Salamov(1997) The Gene-Finder computer tools for analysis of human and model organisms genome sequences. Intell. Syst. Mol. Biol. 5:294–302.
- ↵K.J. ZhuB. DebreceniF. BiY. Zheng(2001) Oligomerization of DH domain is essential for Dbl-induced transformation. Mol. Cell. Biol. 21:425–437.