
Abstract
Alu repetitive elements are found in ∼1.4 million copies in the human genome, comprising more than one-tenth of it. Numerous studies describe exonizations of Alu elements, that is, splicing-mediated insertions of parts of Alu sequences into mature mRNAs. To study the connection between the exonization ofAlu elements and alternative splicing, we used a database of ESTs and cDNAs aligned to the human genome. We compiled two exon sets, one of 1176 alternatively spliced internal exons, and another of 4151 constitutively spliced internal exons. Sixty one alternatively spliced internal exons (5.2%) had a significant BLAST hit to an Alu sequence, but none of the constitutively spliced internal exons had such a hit. The vast majority (84%) of theAlu-containing exons that appeared within the coding region of mRNAs caused a frame-shift or a premature termination codon.Alu-containing exons were included in transcripts at lower frequencies than alternatively spliced exons that do not contain anAlu sequence. These results indicate that internal exons that contain an Alu sequence are predominantly, if not exclusively, alternatively spliced. Presumably, evolutionary events that cause a constitutive insertion of an Alu sequence into an mRNA are deleterious and selected against.
Alu elements are short interspersed elements (SINEs), typically 300 nucleotides long, which account for >10% of the human genome (International Human Genome Sequencing Consortium 2001; Li et al. 2001). Despite their being genetically functionless, Alu elements have been suggested to have broad evolutionary impacts (Mighell et al. 1997; Szmulewicz et al. 1998; Hamdi et al. 1999; International Human Genome Sequencing Consortium 2001). Alus are found in all primates (including prosimians), but in no other organism (Kapitonov and Jurka 1996; Schmid 1996). Therefore, it is tempting to suggest that they have played a role in the evolution of primates. However, the nature of this role is still under debate.
It has been shown in numerous studies that fragments of Alusequences may appear in mature mRNAs, sometimes in the protein-coding region (Makalowski et al. 1994; Yulug et al. 1995; Nekrutenko and Li 2001). Some Alu insertions were found to be translated in vivo. For example, translated splice variants of the biliary glycoprotein containing an Alu fragment were identified by Western immunoblot analysis (Barnett et al. 1993). Another example is that of the human decay-acceleration factor (DAF), in which 10% of its transcripts contain an Alu fragment. There are indications that the Alu-containing DAF mRNA is translated to create a peptide that differs from the common DAF by a hydrophilic carboxy terminus, which inhibits the migration of DAF into the cell membrane (Caras et al. 1987).
A recent study reports that transposable elements are found in the protein-coding regions of ∼4% of human genes, and that Aluelements account for about one-third of these insertions (Nekrutenko and Li 2001). Under the assumption of 30,000 genes in the human genome, there should be ∼400 genes that contain fragments of Aluelements in their protein-coding regions. The insertion of anAlu sequence into a mature mRNA may cause a genetic disease, but an Alu insertion may also contribute to protein variability and versatility (Makalowski et al. 1994).
The vast majority of the insertions of Alu sequences into mature mRNAs are splicing mediated (Makalowski et al. 1994; Nekrutenko and Li 2001). This is possible because both strands of Alusequences contain motifs that resemble consensus splice sites (Makalowski et al. 1994). Mutations within intronic Alusequences may yield active splice sites, that is, part of the intronicAlu sequence will be exonized.
In theory, an insertion of an Alu sequence into a mature mRNA, especially if it is in the protein-coding region, should be deleterious to the organism. Therefore, there must be a mechanism that allows such a large number of Alu insertions into the human transcriptome, keeping it yet unharmed. Using genomically aligned cDNAs and ESTs, we scanned the genome to locate Alu-derived internal exons. We show that all Alu-derived exons found in our study are alternatively spliced. Thus, from an evolutionary point of view, exonized Alu sequences increase the coding and regulatory versatility of the transcriptome, and at the same time, maintain the intactness of the genomic repertoire.
RESULTS
To obtain the intron-exon structures of human genes, we used the output of the LEADS software platform (Shoshan et al. 2001) that was run on the December 2000 draft human genome, and the cDNAs and ESTs from GenBank version 121. The software cleans the expressed sequences from repeats, vectors, and immunoglobulins. It then aligns the expressed sequences to genome, taking alternative splicing into account and clusters overlapping expressed sequences into clusters that represent genes or partial genes (see Methods for a detailed description of the process).
Our search focused on internal exons, that is, exons that are flanked by at least one exon on the 5′ side and one on the 3′ side. We chose to work with internal exons because the prediction of terminal exons using EST alignments is problematic. We searched the LEADSoutput for cases of exon skipping, that is, internal exons that are skipped in some of the splice variants of a certain gene (alternatively spliced internal exons). We also created a set of constitutively spliced internal exons, for example, internal exons that are found in all detected splice variants of the gene. For these compilations, we first selected clusters containing four or more expressed sequences, in which at least one sequence was a cDNA (13,097 clusters). In this set of clusters, we searched for substructures of the cluster containing three exons separated by two introns. We took only those cases in which both introns agreed with the GT/AG, GC/AG, or AT/AC rules, and were not covered by expressed sequences. An internal exon was defined as an exon embedded between the two introns. An internal exon was classified as an alternative internal exon if there was at least one sequence that contained the three exons, and one sequence that contained both flanking exons, but skipped the middle one. A constitutive internal exon was defined as an internal exon supported by at least four sequences for which no alternative splicing was observed (Fig.1). We limited our search to exons shorter than 400 bases, because the length of internal exons only rarely exceeds a few hundred bases (Deutsch and Long 1999).
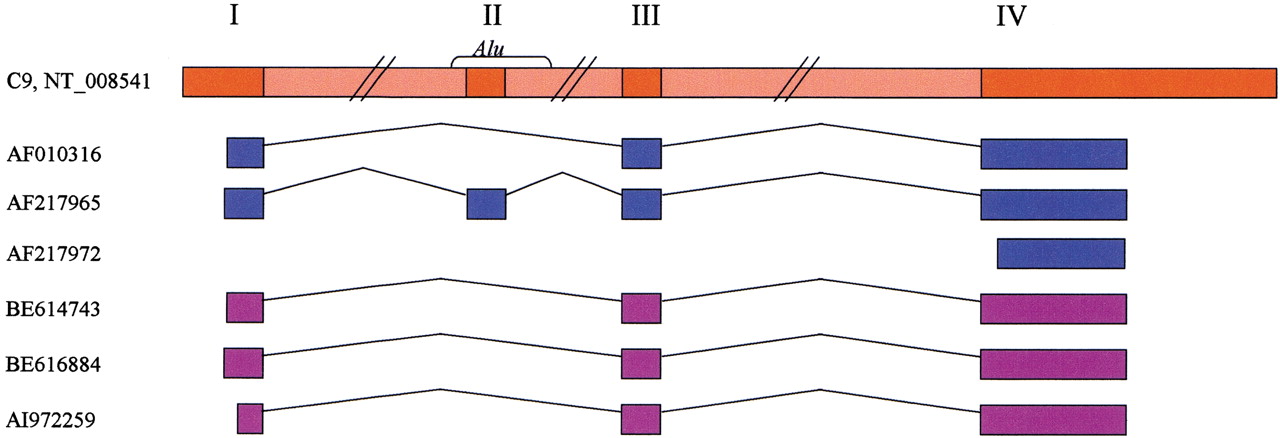
Schematic representation of the multiple alignment of the mRNAs of a microsomal glutathione transferase homolog gene with the genomic sequence. Three GenBank mRNAs (blue) align to the same genomic locus on chromosome 9, NT_008541 (red). Three ESTs that map to this locus are presented (purple), 38 other ESTs that align to the locus are not displayed to save space. Gaps in the alignment of mRNAs represent introns in the DNA. Four exons (marked I, II, III, and IV) are inferred from the presented alignment. Exon II is an alternative internal exon, contained entirely within an Alu repeat. Exon III is a constitutive internal exon, found in all detected splice variants and supported by seven expressed sequences (only five are shown). TheLEADS output was searched for internal exons. A total of 1176 alternatively spliced internal exons were found, 61 of them (5.2%) contained an Alu fragment. A total of 4151 constitutive internal exons were found; none of them contained anAlu fragment.
Under the rules defined above, we obtained 4151 constitutively spliced internal exons (coming from 1662 clusters) and 1176 alternatively spliced internal exons (coming from 1042 clusters). These sets represent, of course, only a fraction of the real number of internal exons in the genome. There are several reasons for not identifying all internal exons. First, a large number of ESTs that represent intron contamination align to places in the genome that are normally introns. Because we searched only for exons flanked by introns that are not covered by expressed sequences, we may have missed introns masked by the contaminated ESTs. Second, for the set of constitutively spliced internal exons, we chose only exons supported by four sequences or more, namely from relatively highly expressed genes. This condition may have led to the exclusion of exons from genes poorly represented in the EST database (dbEST). And finally, we searched only the subset of genes for which a cDNA sequence had been deposited in GenBank.
A BLASTn search of the alternatively spliced internal exons against the NCBI Alu database (Claverie and Makalowski 1994) yielded 61 exons (5.2%) hitting an Alu sequence with an E score lower than 10−10 (Table1). These exons were declared Alu-containing exons. A second search of the database with the 4151 constitutive exons has failed to identify even oneAlu-like sequence. These results indicate that internal exons that contain an Alu sequence are predominantly, if not exclusively, alternatively spliced.
Features of Alu-Containing Alternatively Spliced Internal Exons
| EST/RNA confirming exon skip (1) | EST/RNA confirming exon insertion (2) | Exon len.(3) | No.sequences confirming exon skip (4) | No.sequences confirming exon insertion (5) | Place (6) | Effect on CDS (7) | Alu subfamily (8) | GenBank annotation (9) | |
| 1 | AB046854 | AF257238 | 75 | 1 | 1 | CDS | + | AluSc | Membrane-associated guanylate kinase |
| 2 | D86198 | BF223241 | 81 | 145 | 6 | CDS | + | AluJb | Dolichol-phosphate-mannose synthase |
| 3 | HSU76420 | HSU76421 | 120 | 3 | 9 | CDS | + | AluJb | dsRNA edenosine deaminase |
| 4 | AF161516 | AF152097 | 42 | 6 | 1 | CDS | + | AluSp | Similar to Rattus novergicus CDS5 activator binding |
| 5 | AB000459 | AB000460 | 123 | 10 | 1 | CDS | + | AluSq | Unknown protein product |
| 6 | Al791889 | HS4261062 | 102 | 1 | 3 | CDS | + | AluSp | Unknown protein product |
| 7 | AF013970 | AF069747 | 76 | 3 | 1 | CDS | alt n | AluJo | MTG8-like protein |
| 8 | AF042345 | H41675 | 98 | 2 | 7 | CDS | 3′t | AluJb | Ectopic viral integration site 5 |
| 9 | HSGPLP | BF207526 | 210 | 69 | 2 | CDS | 3′t | AluJb | Glutathione peroxidase-like |
| 10 | HSU64564 | HSU64570 | 138 | 15 | 2 | CDS | 3′t | AluJb | Myelin/oligodendrocyte glycoprotein |
| 11 | AF177862 | AA157902 | 95 | 139 | 1 | CDS | 3′t | AluJb | Nuclear protein of unknown function |
| 12 | AF086904 | AF217975 | 114 | 14 | 1 | CDS | 3′t | AluSq | Protein kinase Chk2 |
| 13 | HSM802141 | AK002113 | 138 | 8 | 2 | CDS | 3′t | FLAM_C | Strong similarity to rat exocyst complex protein Sec15 |
| 14 | AB032995 | BF087651 | 123 | 12 | 3 | CDS | 3′t | AluJo | Unknown protein product |
| 15 | HSM800948 | AA195214 | 126 | 1 | 1 | CDS | 3′t | AluJo | Unknown protein product |
| 16 | HSARSE | AA160312 | 286 | 2 | 1 | CDS | f/s | FLAM_C | Arylsulfatase E |
| 17 | HSU43746 | BE869603 | 126 | 2 | 1 | CDS | f/s | AluSx | Breast cancer susceptibility (BRCA2) |
| 18 | HSU15782 | BF247748 | 96 | 18 | 2 | CDS | f/s | AluJo | Cleavage stimulation factor 77kDa subunit |
| 19 | AF280109 | AF280111 | 121 | 4 | 1 | CDS | f/s | AluSg | Cytochrome P450 subfamily IIIA polypeptide 43 |
| 20 | AF121908 | AF065216 | 98 | 2 | 1 | CDS | f/s | AluSx | Cytosolic phospholipase A2 β |
| 21 | HSU06654 | AA071342 | 106 | 36 | 1 | CDS | f/s | AluJb | Differentiation antigen melan-A protein |
| 22 | HSU07707 | BE842355 | 101 | 4 | 1 | CDS | f/s | AluJb | Epidermal growth factor receptor substrate (eps15) |
| 23 | AF244135 | A1949382 | 61 | 17 | 3 | CDS | f/s | AluSg | Hepatocellular carcinoma-associated antigen 66 |
| 24 | HUMHRLFB | BE513181 | 151 | 23 | 3 | CDS | f/s | AluJo | hRlf β subunit (p102 protein) |
| 25 | HSICAM2 | BE261894 | 116 | 29 | 1 | CDS | f/s | AluJb | ICAM-2, cell adhesion ligand for LFA-1 |
| 26 | AB018010 | AW381165 | 132 | 53 | 4 | CDS | f/s | AluJb | Membrane glycoprotein 4F2 heavy chain |
| 27 | AF072247 | AA285195 | 128 | 25 | 2 | CDS | f/s | AluSg/x | Methyl-CpG binding domain-containing protein MBD3 |
| 28 | HUMMEVKIN | AF217536 | 118 | 12 | 2 | CDS | f/s | AluJb | Mevalonate kinase |
| 29 | AK001322 | AK022939 | 89 | 1 | 1 | CDS | f/s | AluJo | mRNA from NT2 neuronal precursor cells |
| 30 | D83735 | BE836938 | 122 | 54 | 3 | CDS | f/s | AluSx | Neutral calponin |
| 31 | AF010316 | AF217965 | 122 | 7 | 1 | CDS | f/s | AluJb | Microsomal glutathione transferase homolog |
| 32 | HSAJ4875 | AA225691 | 75 | 36 | 10 | CDS | f/s | AluSp | Putative glucosyltransferase |
| 33 | AF021819 | BE567765 | 93 | 198 | 1 | CDS | f/s | FLAM_C | RNA-binding protein regulatory subunit |
| 34 | AF095742 | BF038501 | 95 | 20 | 1 | CDS | f/s | AluSx | Serine protease ovasin |
| 35 | AF151858 | AA397587 | 71 | 34 | 4 | CDS | f/s | AluSc | Similar to putative t1/st2 receptor binding protein precursor |
| 36 | AF072810 | AW835499 | 82 | 6 | 1 | CDS | f/s | AluJo | Transcription factor WSTF |
| 37 | AK026835 | AA460397 | 77 | 13 | 1 | CDS | f/s | AluJb | Unknown protein product |
| 38 | HUMRSC765 | AU151565 | 91 | 33 | 1 | CDS | f/s | FLAM_A | Unknown protein product |
| 39 | BF513753 | AK000502 | 97 | 5 | 1 | CDS | f/s | AluSx | Unknown protein product |
| 40 | AK024815 | AL046389 | 101 | 1 | 1 | CDS | f/s | AluJo | Unknown protein product |
| 41 | AK001755 | AK023461 | 134 | 7 | 1 | CDS | f/s | AluSc | Unknown protein product |
| 42 | AB002315 | AL043085 | 151 | 3 | 1 | CDS | f/s | AluJb | Unknown protein product |
| 43 | AK022568 | BE898836 | 76 | 16 | 5 | CDS | f/s | AluJb | Weakly similar to Acyl-CoA dehydrogenase |
| 44 | AK022147 | AV714478 | 127 | 6 | 1 | CDS | f/s | AluSx | Weakly similar to the yeast GTPase-activating protein GYP7 |
| 45 | AF003924 | AW954573 | 122 | 6 | 3 | CDS | f/s | AluSg | Zinc finger protein ANC_2H01 |
| 46 | AF039918 | BE867770 | 117 | 2 | 1 | 5UTR | FRAM | CD39-like protein CD39L4 | |
| 47 | AF070674 | BF216095 | 130 | 7 | 2 | 5UTR | AluSx | Inhibitor of apoptosis protein-1 (MIHC) | |
| 48 | AF071107 | AF071108 | 84 | 8 | 2 | 5UTR | FLAM_A | SMAD5 | |
| 49 | AF130312 | BF184073 | 103 | 37 | 1 | 5UTR | AluSx | TATA box binding protein-related factor 2 | |
| 50 | AFO78864 | BE747669 | 131 | 21 | 1 | 5UTR | AluSx | TS58 | |
| 51 | BF086933 | AK002100 | 71 | 7 | 2 | 5UTR | AluSx | Unknown protein product | |
| 52 | AK001235 | BE788268 | 119 | 18 | 2 | 5UTR | FLAM_C | Unknown protein product | |
| 53 | AK001715 | BE740371 | 248 | 20 | 1 | 5UTR | AluJb | Unknown protein product | |
| 54 | HUMZFX | HSZFX3 | 128 | 2 | 1 | 5UTR | AluSx | Zinc finger protein X-linked | |
| 55 | BF306258 | AK024074 | 74 | 2 | 2 | N/A | AluSx | Moderately similar to zinc finger protein 91 | |
| 56 | AA435797 | HSU92992 | 122 | 8 | 1 | N/A | AluSg | mRNA from brain tissue, CAG repeat region | |
| 57 | HSM801006 | HSM800877 | 106 | 2 | 2 | N/A | AluJb | Similar to zinc finger helicase | |
| 58 | AK023856 | AA344993 | 98 | 16 | 2 | N/A | AluY | Unknown protein product | |
| 59 | AA210960 | AK021447 | 114 | 6 | 3 | N/A | AluSg | Unknown protein product | |
| 60 | T99367 | AB007962 | 118 | 2 | 1 | N/A | AluJb | Unknown protein product | |
| 61 | AK026653 | BF037972 | 147 | 8 | 1 | N/A | AluY | Unknown protein product |
[i] (1) One of the GenBank sequences (RNA or EST) showing the exon-skipping pattern. The name presented is the GenBank locus.
[ii] (2) One of the GenBank sequences (RNA or EST) confirming the existence of the Alu-containing exon. The name presented is the GenBank locus.
[iii] (3) The length of the Alu-containing exon.
[iv] (4) Number of expressed sequences (RNAs and ESTs) showing the exon-skipping pattern.
[v] (5) Number of expressed sequences (RNAs and ESTs) confirming the existence of the Alu-containing exon.
[vi] (6) The location of the Alu-containing exon along the mRNA is denoted as follows: (CDS) the exon is inserted within the protein-coding region; (5UTR) the exon is inserted within the 5′UTR; (N/A) missing or contradictory GenBank annotation.
[vii] (7) The effects of the insertion of the Alu-containing exon in the protein-coding region is denoted as follows: (+) the exon adds a domain, namely inserted in frame and do not contain an in-frame stop codon; (alt n) exon insertion causes the alteration of the amino terminus of the protein; (3′t) exon insertion contains an in-frame premature stop codon; (f/s) exon insertion causes a frame-shift.
[viii] (8) The subfamily of the Alu element, see Table 2. RepeatMasker (http://repeatmasker.genome.washington.edu/cgi-bin/RepeatMasker) was run on the DNA around each Alu-containing exon to determine the subfamily type.
[ix] (9) GenBank annotation of the locus.
We further analyzed the Alu-containing exons to check their influence on the transcripts they are inserted into. As a reference set of exons, we used a set of 62 alternatively spliced internal exons compiled by Hide et al. (2001) from 52 genes on chromosome 22. In their study, Hide et al. (2001) used a rigorous in silico method to scan the annotated genomic sequence of chromosome 22 to identify alternatively spliced internal exons that are skipped in some of the transcripts. We took the set of exons from chromosome 22 as a set representing the normal population of alternatively spliced internal exons, and compared it with the set of Alu-containing exons we found.
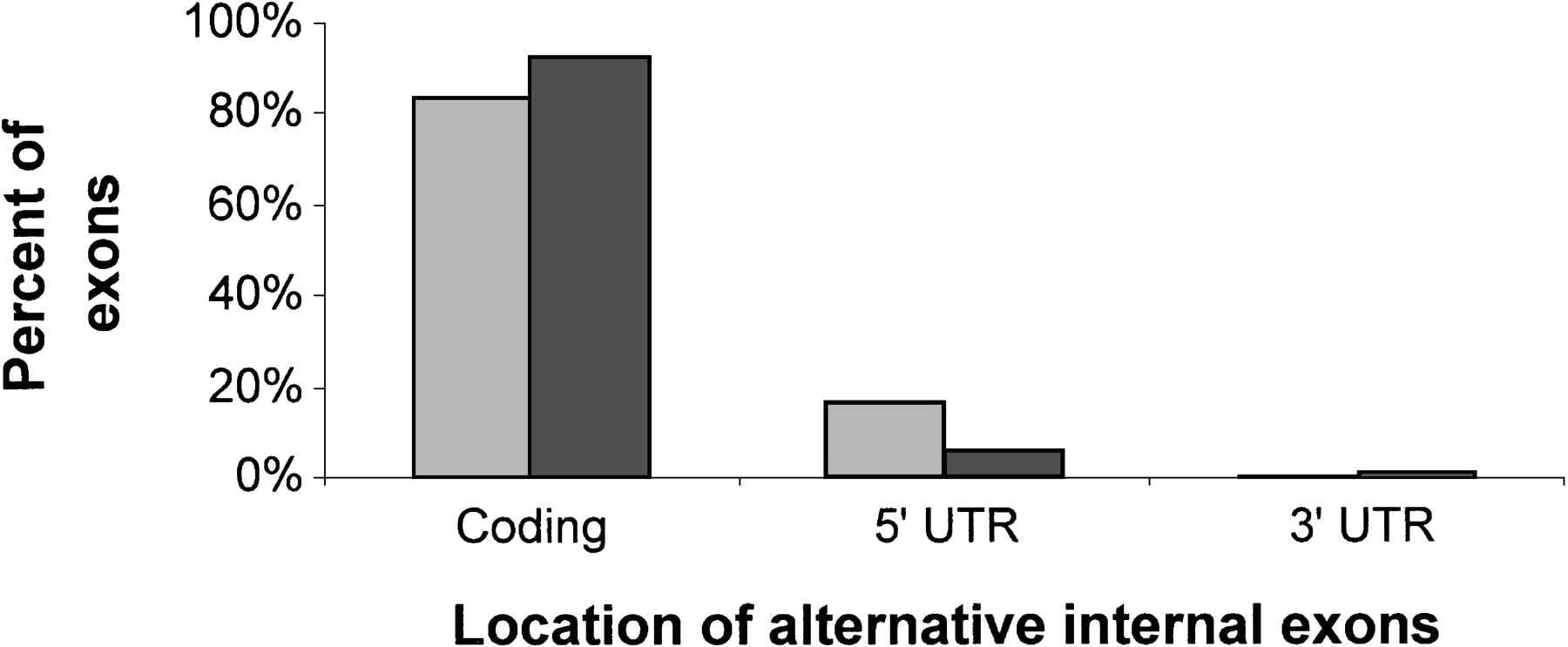
Of our 61 Alu-containing alternatively spliced internal exons, 54 had an unambiguous coding-region annotation in the GenBank cDNAs. Of these, 45 (83%) were located within the protein-coding region and 9 (17%) within the 5′ untranslated region (UTR). NoAlu-containing exons were found in the 3′ UTR. Although it is known that most expressed Alu sequences are found within the 3′ UTRs of mRNAs (Yulug et al. 1995), our finding is not surprising given that 3′ UTRs are mostly found in the terminal exon (Deutsch and Long 1999), whereas the exons in our study were internal ones. As seen in Figure 2, the distribution ofAlu-containing exons along the mRNA was similar to the distribution of alternatively spliced internal exons from chromosome 22. The slight bias of Alu-containing exons toward being found in the 5′ UTR of the mRNA was not statistically significant.
Location of alternatively spliced internal exons within the mRNA. Data for 54 Alu-containing exons, for which there was noncontradictory information in the GenBank annotation, is presented in lighter shaded bars. Data of 62 alternatively spliced internal exons from chromosome 22, compiled by Hide et al (2001) are presented as reference (darker shaded bars).
However, the influence of the Alu-containing exons on the coding region of the protein is significantly different from the influence of the alternatively spliced internal exons from chromosome 22 (Fig. 3). In 38 cases (84%) of 45Alu-containing exons that are located within the protein-coding region, the insertion of an Alu-containing exon results in a shortened protein, either through frameshift (30 cases, 66.6%) or through an in-frame stop codon within theAlu-containing exon itself (8 cases, 17.8%). In comparison, only 21 alternatively spliced internal exons (44%) from chromosome 22 set yielded a premature termination, 18 of them (38%) cause frameshift.
Effect of exon insertion on the protein-coding region. Data for 45 Alu-containing exons occurring within the protein-coding region are presented in lighter shaded bars. Data of 48 alternatively spliced internal exons from chromosome 22 (Hide et al. 2001), which occur in the protein-coding region, are presented as reference (darker shaded bars). Exons were considered as domain adding if their length was a multiple of three, and there was no in-frame stop codon within them. Exons were considered as causing a premature termination either when they caused a frame-shift or when they presented an in-frame stop codon. Data for alternatively spliced internal exons from chromosome 22 were calculated from Table 2 in Hide et al. (2001).
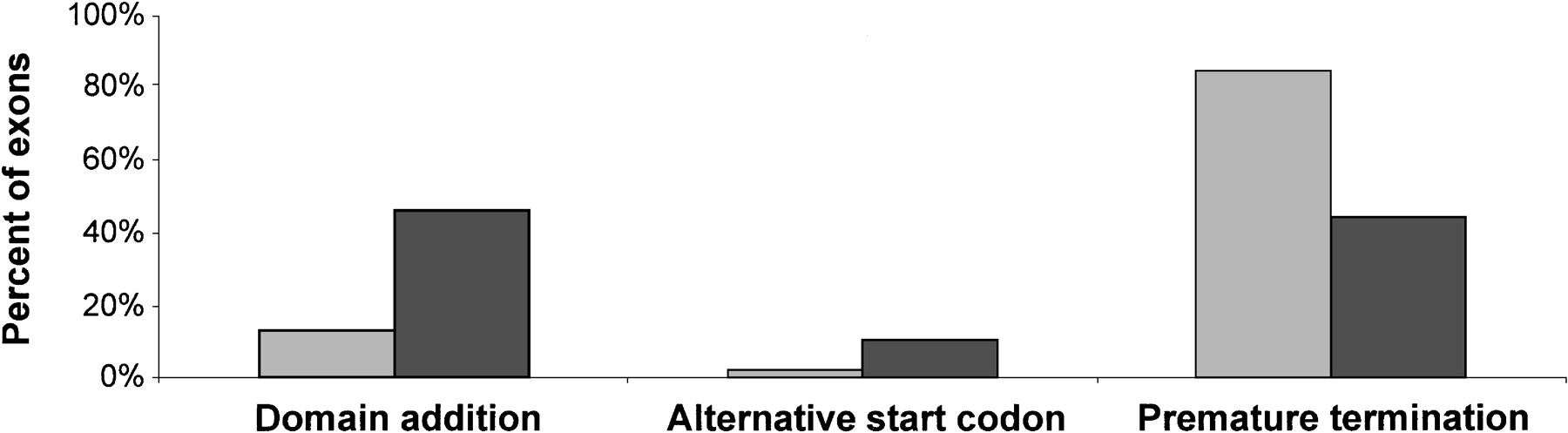
Only 6 (13%) Alu-containing exons neither contain stop codons nor affect the original termination codon. These exons can, therefore, be regarded as genuine domain donors. The lengths of these domains range between 15 and 42 amino acids, and their predicted isoelectric points vary from 3.4 to 11. The set of alternatively spliced internal exons from chromosome 22 behaves differently — 22 of the exons (46%) in this set are domain donors.
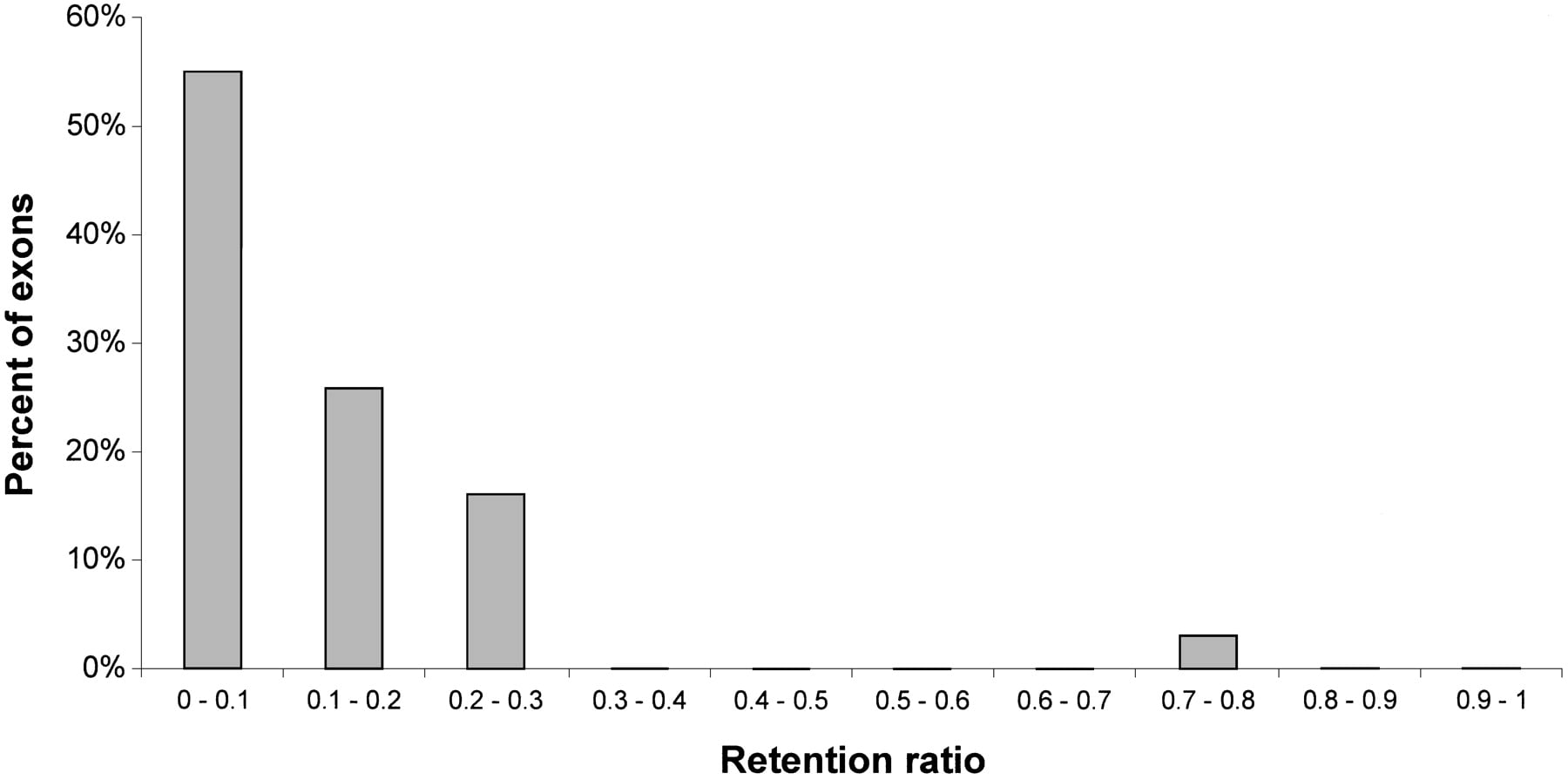
We suggest measuring the strength of the splice sites of an alternatively spliced internal exon by means of a retention ratio, which is calculated as the number of mRNA sequences that contain the alternatively spliced exon divided by the total number of mRNA sequences. In practice, the retention ratio for a gene or a locus was calculated as the observed number of expressed sequences that contain the alternatively spliced exon as well as the two flanking exons divided by the total number of expressed sequences aligned to the locus (see Table 1 for the number of sequences that confirmed each exon or skipped it). Most Alu-containing exons have a small retention ratio (average of 0.21), that is, they are only found in about one-fifth of all mRNA transcripts. This value is, of course, overestimated, because by necessity we took only loci in which there was at least one sequence showing an alternative internal exon. Loci with a small number of covering expressed sequences bias the ratio upward. Thus, the retention ratio for the 31 cases, in which the number of sequences is 10 or above, averages in 0.11 (Fig.4). In comparison, the average retention ratio of the 1115 alternatively spliced internal exons that do not contain Alu sequences is 0.41 (data not shown).
Retention ratios of highly covered Alu-containing exons. Retention ratio for each exon was calculated by the number of expressed sequences that contain the exon as well as both flanking exons, divided by the total number of sequences that contain both flanking exons. Only the 31 exons with 10 or more total sequences that contain both flanking exons were taken for this analysis. Therefore, every exon represents ∼3% of the exons dataset.
Following the convention in the literature, we define the poly(A)-containing Alu sequence as the plus strand and the complementary poly(T)-containing sequence as the minus strand. A total of 52 of the 61 Alu-containing exons (85%) involve the minus strand. The uneven distribution between the strands is probably due to the fact that the minus strand of the Alu consensus sequence contains more motifs that resemble splice sites than the plus strand (Makalowski et al.1994; Makalowski 2000). Table 3enumerates the splice sites utilized by theAlu-containing exons and the location of these splice sites along the consensus Alu sequence. There were seven sites in the minus strand of the Alu sequence that were utilized as 5′ splice sites (donors), of which three had not been reported previously (Makalowski 2000). Twelve sites in the minus strand of the Alusequence were utilized as 3′ splice sites (acceptors); all but one were not reported previously (Makalowski 2000). In the plus strand, we identified a single potential acceptor site and three potential donor sites — one of these was identified previously (Makalowski 2000).
Potential Splice Sites in the Alu Consensus Sequence that are Utilized byAlu-Containing Exons
| Alustrand | Type of potential splice site | Location inAlu consensus sequence[i] | Times utilized[ii] | Reported previously[iii] |
| Minus | 5′ splice site (donor) | 4 | 1 | No |
| 23 | 7 | Yes | ||
| 138 | 2 | Yes | ||
| 158 | 22 | Yes | ||
| 170 | 1 | Yes | ||
| 200 | 4 | No | ||
| 206 | 4 | No | ||
| Minus | 3′ splice site (acceptor) | 65 | 1 | No |
| 114 | 3 | No | ||
| 116 | 8 | No | ||
| 119 | 1 | No | ||
| 120 | 1 | No | ||
| 255 | 4 | No | ||
| 273 | 1 | No | ||
| 275 | 13 | No | ||
| 276 | 1 | No | ||
| 277 | 1 | No | ||
| 279 | 11 | Yes | ||
| 281 | 2 | No | ||
| Plus | 5′ splice site (donor) | 51 | 2 | No |
| 69 | 2 | Yes | ||
| 101 | 4 | No | ||
| Plus | 3′ splice site (acceptor) | 45 | 1 | No |
[i] Location of the potential splice sites in theAlu consensus sequence follows the numbering in Jurka and Milosavljevic (1991).
[ii] The number of Alu-containing alternatively spliced exons that utilize the splice site.
[iii] Compared with Makalowski (2000).
It has been proposed that Alu evolution proceeds through successive waves of fixation, in which each Alu subfamily is derived from a small number of source sequences belonging to an evolutionarily older subfamily (Jurka and Milosavljevic 1991; Batzer et al. 1996; Kapitonov and Jurka 1996). Key nucleotide positions are distinctive between Alu subfamilies (Jurka and Milosavljevic 1991; Batzer et al. 1996). We used a collection of 153,645 annotatedAlu elements mapped to the human genome (Stenger et al. 2001) to determine the frequency of each Alu subfamily in the human genome. RepeatMasker(http://repeatmasker.genome.washington.edu/cgi-bin/RepeatMasker) was run on the DNA around each Alu-containing exon to determine the borders of the Alu in the DNA and the subfamily type. We found that older subfamilies (such as Alu-J and Alumonomers) are significantly over-represented (P < 6.4 × 10−21) in the Alu-containing exons, whereas newer subfamilies (Alu-S and Alu-Y) are under-represented (Table2).
Distributions of Alu Subfamilies within the Genome andAlu-Containing Exons[i]
| Subfamily | Age (million years)[ii] | Distribution in the genome[iii] | Distribution in the set ofAlu-containing exons[iv] | ||||
| Occurrences | Percent | Occurrences | Percent | ||||
| Alumonomer | 112 | 1183 | 1% | 7 | 11% | ||
| Alu-J | 81 | 45156 | 29% | 26 | 43% | ||
| Alu-S | 48-31 | 88645 | 58% | 26 | 43% | ||
| Alu-Y | 19 | 15574 | 10% | 2 | 3% | ||
| Unknownfamily | — | 3087 | 2% | 0 | 0% | ||
[i] There is a statistically significant difference between two distributions (P < 6.4 × 10−21).
[ii] Age of Alu subfamilies from Kapitonov and Jurka (1996).
[iii] Distribution in the genome was calculated from a set of 153,645 human Alus compiled previously by Stenger et al. (2001), available at http://dir.niehs.nih.gov/ALU.
[iv] Subfamily types of the Alu sequences contained within alternatively spliced internal exons were determined usingRepeatMasker(http://repeatmasker.genome.washington.edu/cgi-bin/RepeatMasker).
The average length of an Alu-containing exon was 114 bases, with the longest exon being 286 bases, and the shortest 42 bases. As a typical Alu element contains 300 bases, the exons contain only a fraction of the Alu sequence. We usedRepeatMasker to determine the borders of the Aluelement on the genome. All Alu elements found within exons were extending into at least one of the flanking introns. We found no case of an Alu element totally contained within an exon, but this might be due to the fact that we limited our search to exons shorter than 400 bases, and an insertion of a full Alu element into an exon would result in a very long exon. We note that full-lengthAlu elements have been found previously in terminal exons. However, our study excluded terminal exons. These results indicate that all 61 Alu-containing exons found in our set resulted from exonization of part of an intronic Alu element, rather than directly inserted into pre-existing exons.
DISCUSSION
From our results, it is clear that constitutiveAlu-containing internal exons are either absent or very rare in the human transcriptome, whereas alternative Alu-containing internal exons appear frequently. Additionally, Alu-containing exons have a significantly lower average retention ratio than alternatively spliced internal exons that do not contain Alu. These findings imply that Alu splice-like sites that had evolved into strong constitutive splice sites were most probably selected against because of their interference with normal protein production. In contrast, mutational changes in Alu sequences resulting in the creation of weak splice sites are tolerated, especially if their retention ratio is low. There are several documented genetic diseases caused by a mutation that led to the creation of a strong splice site in an otherwise normal intronicAlu. For example, a G→C mutation in an Alu sequence within intron 3 of ornithine δ-aminotransferase (OAT), caused the creation of a strong donor site, consequently leading to the constitutive insertion of a novel Alu exon between exons 3 and 4. The insertion caused an in-frame stop codon, which led to OAT deficiency (Mitchell et al. 1991; Makalowski 2000). This is an example of the possible deleterious effect of Alu-containing exons that has become constitutively inserted within a transcript.
According to our data, older subfamilies (monomers and Alu-J) are over-represented in the set of Alu-containing exons compared with their distribution in the genome (Table 2). Since, by definition, members of older subfamilies were retroposed to the human genome earlier than members of newer subfamilies, they had more time to diverge from the Alu ancestor. Members of the Alu-J subfamilies show ∼86% identity to the Alu consensus sequence, whereas members of the Alu-S subfamilies show ∼92%–93% identity (Kapitonov and Jurka 1996). Therefore, the bias toward older subfamilies in the set of Alu-containing exons may reflect the number of substitutions needed to create a functional splice site within the retroposed Alu sequence to allow for its exonization.
Another possibility that would explain the fact that we did not find constitutive Alu-containing internal exons is that oldAlu-containing internal exons that became fixed show only a poor similarity to the consensus Alu sequence, and, therefore, could no longer be recognized by similarity searches as Aluderived.
We have chosen to focus on alternative splicing events of the exon-skipping type for two reasons. First, this type is the most frequent type of alternative splicing (Hide et al. 2001). Second, many unspliced ESTs found in the ESTs database (dbEST) represent sequenced introns (intron contamination) and contain Alu sequences, and, therefore, we preferred not to use unspliced expressed sequences as evidence for alternative splicing. In the exon-skipping type of alternative splicing, both variants are spliced — the skipping variant contains a large intron that skips the alternative internal exon, and the variant containing the exon has two introns, one on each of the alternatively spliced internal exon's sides.
Due to the strict nature of our search, not all alternatively spliced internal exons were retrieved, and, therefore, not all documentedAlu-containing exons appear in our database. We have taken only exons flanked by true introns on both sides. A true intron was defined as an intron abiding by the GT/AG, GC/AG, or AT/AC rules, without any of its nucleotides covered by an expressed sequence. Due to the large number of ESTs that represent intron contamination and align to places in the genome that are normally introns, many true exon-skipping cases were most probably disregarded in our study. In the same manner, our database of constitutively spliced internal exons is probably only a fraction of the complete set of constitutively spliced internal exons in the genome, because, in addition to the demand that the exon will be flanked by true introns, we have taken into account only exons covered by at least four sequences. Finally, we examined only genes for which the cDNA was deposited in GenBank, disregarding clusters made entirely of ESTs.
The literature describes numerous individual studies in whichAlu insertions were found within an mRNA. The vast majority of these cases are described as splice variants, with another splice variant that does not contain the Alu insertion in evidence. In the literature, we found two instances of internalAlu-containing exons that were reported to be found in all detected splice variants. Neither case appears in either our dataset of constitutive exons or in the alternative exons dataset. The reason for these exclusions was the alignment of intron-contaminated ESTs to these two loci. We have searched manually for ESTs matching these two loci. The human hematopoietic progenitor kinase (HPK1) contains anAlu-derived peptide in its carboxyl terminus. ThisAlu insertion was reported previously as fixed, that is, theAlu was present in all transcripts (Hu et al. 1996; Nekrutenko and Li 2001). We found 25 ESTs that skip the Alu-containing exon (exon 26), whereas only three sequences (two of them were mRNAs) contained the exon (data not shown). The zinc finger gene ZNF177 has been reported to contain both an Alu and an L1 fragment in the constitutively spliced exon 4 (Baban et al. 1996; Landry et al. 2001). Apart from the two mRNAs reported by (Baban et al. 1996), we failed to find a single EST that may be used to determine whether or not this exon is really constitutive. However, we predict that splice variants that do not contain this exon will be discovered in the future.
We have shown that exonized Alu elements are alternatively spliced. Thus, Alu elements have the evolutionary potential to enhance the coding capacity and regulatory versatility of the genome without compromising its integrity.
METHODS
The Gencarta Database and its LEADS output was licensed from Compugen Ltd. (http://www.cgen.com). Briefly, theLEADS output was created as follows. ESTs and cDNAs from GenBank version 121 were cleaned from terminal vector sequences, and low-complexity stretches and repeats in the expressed sequences were masked. Sequences with internal vector contamination and sequences identified as immunoglobulins or T-cell receptors were discarded. In the next stage, expressed sequences were heuristically compared with the genome to find likely high-quality hits. They were then aligned to the genome by use of a spliced alignment model that allows long gaps. Only sequences having >94% identity to a stretch in the genome were used in further stages. Sequences having hits to more than one locus in the genome were analyzed to choose the correct locus, taking into account percent identity and intron content (to differentiate between genes and processed pseudogenes). Sequences mapping to two or more chromosomes, or sequences in which the inferred introns were longer than 400,000 were discarded as suspected chimeras. Low-quality sequence ends that disagreed with the DNA were trimmed. In the clustering and assembly stage, overlapping expressed sequences and corresponding genomic sequences were multiply aligned. Positions on the genomic sequence in which there is at least one sequence that opens or closes a long gap were considered splice sites. Where possible, long gaps begin with a GT or GC dinucleotide and end with an AG dinucleotide. The resulting multiple alignment is represented as a directed graph, in which each vertex represents the multiple alignment of sequences between two detected splice sites. An edge exists between two vertices if at least one sequence continues from the first multiple alignment to the second. Every sequence has a hyperedge consisting of the vertices through which it passes.
The 13,097 clusters that contained at least 4 expressed sequences, of which at least 1 was a cDNA sequence, were selected for the internal-exon search. An intron was defined as a vertex containing only the genomic sequence, and a true intron as an intron abiding by the GT/AG, GC/AG, or AT/AC rules. An exon was defined as a vertex containing at least one expressed sequence, and an internal exon was defined as an exon embedded between two true introns. Substructures of the cluster containing three exons separated by two introns, in which the second exon is an internal exon, were searched. An internal exon was classified as an alternatively spliced internal if there was at least one sequence that contained the three exons, and one sequence that contained both flanking exons, but skipped the middle one. A constitutively spliced internal exon was defined as an internal exon covered by at least four sequences, for which no alternative splicing was observed. The search was limited to exons shorter than 400 bases.
Constitutive and the alternative exons were searched using thePERL programs GetConstitutiveExons.pl and GetAlternativeCassetteExons.pl, respectively (http://www.kimura.tau.ac.il/∼rotem/ALU/). Packages used by these programs for parsing the LEADS output, compiled for SUN architecture, can be downloaded from http://www.cgen.com/parse_LEADS. Exons datasets can be downloaded fromhttp://www.kimura.tau.ac.il/∼rotem/ALU. Both exons datasets were compared with the NCBI Alu database (ftp://ncbi.nlm.nih.gov/pub/jmc/alu/, (Claverie and Makalowski 1994)) using the BLASTn program with default parameters. Genomic sequences near the Alu-containing exons were extracted from LEADS clusters using theGetAlternativeCassetteExons.pl program. Exon-intron structures of genes containing Alu exon were double checked using the Sim4 Program for spliced alignment (Florea et al. 1998). Isoelectric point was predicted using the Expasy online servicehttp://www.expasy.ch/tools/pi_tool.html. Location in mRNA and influence on protein-coding regions were inferred manually from GenBank annotations. Data for alternatively spliced internal exons from chromosome 22 were calculated from Table 2 in Hide et al. (2001).Alu subfamilies, orientation, and borders on the genomic sequence were determined using RepeatMasker(http://repeatmasker.genome.washington.edu/cgi-bin/RepeatMasker). Subfamilies of genomic Alu sequences were inferred fromhttp://dir.niehs.nih.gov/ALU/map (Stenger et al. 2001).
WEB SITE REFERENCES
ftp://ncbi.nlm.nih.gov/pub/jmc/alu/; The NCBI Alu database.
http://dir.niehs.nih.gov/ALU/map; Database of Alu elements in the human genome from Stenger et al. (2001).
http://repeatmasker.genome.washington.edu/cgi-bin/RepeatMasker; TheRepeatMasker program by Smit and Green.
http://www.cgen.com; Compugen home page.
http://www.expasy.ch/tools/pi_tool.html; A tool that computes isoelectric point (pI) and molecular weight (Mw).
http://www.kimura.tau.ac.il/∼rotem/ALU/; Supplementary material from corresponding author.
We thank Dr. Galit Rotman for valuable review and discussion. We also thank the Compugen LEADS team for help in various productions.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Notes
[17] Corresponding author.
Notes
[18] E-MAIL [email protected]; FAX +972-3-6409403.
[19] Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.229302.
REFERENCES
- ↵S. BabanJ.D. FreemanD.L. Mager(1996) Transcripts from a novel human KRAB zinc finger gene contain spliced Alu and endogenous retroviral segments. Genomics 33:463–472.
- ↵T.R. BarnettL. DrakeW. Pickle(1993) Human biliary glycoprotein gene: Characterization of a family of novel alternatively spliced RNAs and their expressed proteins. Mol. Cell. Biol. 13:1273–1282.
- ↵M.A. BatzerP.L. DeiningerU. Hellmann-BlumbergJ. JurkaD. LabudaC.M. RubinC.W. SchmidE. ZietkiewiczE. Zuckerkandl(1996) Standardized nomenclature for Alu repeats. J. Mol. Evol. 42:3–6.
- ↵I.W. CarasM.A. DavitzL. RheeG. WeddellD.W. Martin Jr.V. Nussenzweig(1987) Cloning of decay-accelerating factor suggests novel use of splicing to generate two proteins. Nature 325:545–549.
- ↵J.M. ClaverieW. Makalowski(1994) Alu alert. Nature 371:752.
- ↵M. DeutschM. Long(1999) Intron-exon structures of eukaryotic model organisms. Nucleic Acids Res. 27:3219–3228.
- ↵L. FloreaG. HartzellZ. ZhangG.M. RubinW. Miller(1998) A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 8:967–974.
- ↵H. HamdiH. NishioR. ZielinskiA. Dugaiczyk(1999) Origin and phylogenetic distribution of Alu DNA repeats: Irreversible events in the evolution of primates. J. Mol. Biol. 289:861–871.
- ↵W.A. HideV.N. BabenkoP.A. van HeusdenC. SeoigheJ.F. Kelso(2001) The contribution of exon-skipping events on chromosome 22 to protein coding diversity. Genome Res. 11:1848–1853.
- ↵M.C. HuW.R. QiuX. WangC.F. MeyerT.H. Tan(1996) Human HPK1, a novel human hematopoietic progenitor kinase that activates the JNK/SAPK kinase cascade. Genes & Dev. 10:2251–2264.
- ↵International Human Genome Sequencing Consortium(2001) Initial sequencing and analysis of the human genome. Nature 409:860–921.
- ↵J. JurkaA. Milosavljevic(1991) Reconstruction and analysis of human Alu genes. J. Mol. Evol. 32:105–121.
- ↵V. KapitonovJ. Jurka(1996) The age of Alu subfamilies. J. Mol. Evol. 42:59–65.
- ↵J.R. LandryP. MedstrandD.L. Mager(2001) Repetitive elements in the 5′ untranslated region of a human zinc- finger gene modulate transcription and translation efficiency. Genomics 76:110–116.
- ↵W.H. LiZ. GuH. WangA. Nekrutenko(2001) Evolutionary analyses of the human genome. Nature 409:847–849.
- ↵W. Makalowski(2000) Genomic scrap yard: How genomes utilize all that junk. Gene 259:61–67.
- ↵W. MakalowskiG.A. MitchellD. Labuda(1994) Alu sequences in the coding regions of mRNA: A source of protein variability. Trends Genet. 10:188–193.
- ↵A.J. MighellA.F. MarkhamP.A. Robinson(1997) Alu sequences. FEBS Lett. 417:1–5.
- ↵G.A. MitchellD. LabudaG. FontaineJ.M. SaudubrayJ.P. BonnefontS. LyonnetL.C. BrodyG. SteelC ObieD. Valle(1991) Splice-mediated insertion of an Alu sequence inactivates ornithine δ-aminotransferase: A role for Alu elements in human mutation. Proc. Natl. Acad. Sci. 88:815–819.
- ↵A. NekrutenkoW.H. Li(2001) Transposable elements are found in a large number of human protein-coding genes. Trends Genet. 17:619–621.
- ↵C.W. Schmid(1996) Alu: Structure, origin, evolution, significance and function of one- tenth of human DNA. Prog. Nucleic Acid Res. Mol. Biol. 53:283–319.
- ↵A. ShoshanV. GrebinskiyA. MagenA. ScolnicovE. FinkD. LehaviA. Wasserman(2001) Designing oligo libraries taking alternative splicing into account. in Microarrays: Optical Technologies and Informatics, Proc SPIE, eds M.L. BittnerY. ChenA.N. DorselE.D. Dougherty(SPIE, Bellingham, WA), 4266:86–95.
- ↵J.E. StengerK.S. LobachevD. GordeninT.A. DardenJ. JurkaM.A. Resnick(2001) Biased distribution of inverted and direct Alus in the human genome: Implications for insertion, exclusion, and genome stability. Genome Res. 11:12–27.
- ↵M.N. SzmulewiczG.E. NovickR.J. Herrera(1998) Effects of Alu insertions on gene function. Electrophoresis 19:1260–1264.
- ↵I.G. YulugA. YulugE.M. Fisher(1995) The frequency and position of Alu repeats in cDNAs, as determined by database searching. Genomics 27:544–548.