
Integration of Cot Analysis, DNA Cloning, and High-Throughput Sequencing Facilitates Genome Characterization and Gene Discovery
Abstract
Cot-based sequence discovery represents a powerful means by which both low-copy and repetitive sequences can be selectively and efficiently fractionated, cloned, and characterized. Based upon the results of a Cot analysis, hydroxyapatite chromatography was used to fractionate sorghum (Sorghum bicolor) genomic DNA into highly repetitive (HR), moderately repetitive (MR), and single/low-copy (SL) sequence components that were consequently cloned to produce HRCot, MRCot, and SLCot genomic libraries. Filter hybridization (blotting) and sequence analysis both show that the HRCot library is enriched in sequences traditionally found in high-copy number (e.g., retroelements, rDNA, centromeric repeats), the SLCot library is enriched in low-copy sequences (e.g., genes and “nonrepetitive ESTs”), and the MRCot library contains sequences of moderate redundancy. The Cot analysis suggests that the sorghum genome is approximately 700 Mb (in agreement with previous estimates) and that HR, MR, and SL components comprise 15%, 41%, and 24% of sorghum DNA, respectively. Unlike previously described techniques to sequence the low-copy components of genomes, sequencing of Cot components is independent of expression and methylation patterns that vary widely among DNA elements, developmental stages, and taxa. High-throughput sequencing of Cot clones may be a means of “capturing” the sequence complexity of eukaryotic genomes at unprecedented efficiency.
[Online supplementary material is available at www.genome.org. The sequence data described in this paper have been submitted to the GenBank under accession nos.AZ921847-AZ923007. Reagents, samples, and unpublished information freely provided by H. Ma and J. Messing.]
When a solution of denatured genomic DNA is placed in an environment conducive to renaturation, the rate at which a particular sequence reassociates is proportional to the number of times it is found in the genome. This principle forms the basis of DNA renaturation kinetics (also called Cot analysis), a technique by which the redundant nature of eukaryotic genomes was first demonstrated (Britten and Kohne 1968; see Fig. A in the online supplement to this article for a review of Cot analysis,www.genome.org). In a typical renaturation kinetics study, samples of sheared genomic DNA are heat-denatured and allowed to reassociate to different Cot values [Cot value = the product of nucleotide concentration in moles per liter (C0 or Co), reassociation time in seconds (t), and, if applicable, a factor based upon the cation concentration of the buffer; for review, see Britten et al. 1974]. For each sample, renatured DNA is separated from single-stranded DNA using hydroxyapatite (HAP) chromatography, and the percentage of the sample that has not reassociated (%ssDNA) is determined. The logarithm of a sample's Cot value is plotted against its corresponding %ssDNA to yield a Cot point, and a graph of Cot points ranging from little or no reassociation until reassociation approaches completion is called a Cot curve (Peterson et al. 1998). Mathematical analysis of a Cot curve permits estimation of genome size, the proportion of the genome contained in the single-copy and repetitive DNA components, and the kinetic complexity of each component. Interspecific comparison of Cot data has provided considerable insight into the structure and evolution of eukaryotic genomes (e.g., Britten and Kohne 1968; Davidson et al. 1975; Goldberg et al. 1975; Galau et al. 1976; Hake and Walbot 1980; Geever et al. 1989).
With the advent of molecular cloning techniques, most genome researchers abandoned Cot analysis. However, the principles of nucleic acid hybridization developed through Cot research form the basis of many molecular biology techniques, and information generated in Cot studies remains central to current knowledge of genome structure (for review, see Goldberg 2001).
Repetitive DNA has proven a particularly difficult problem in the investigation of eukaryotic genomes, especially in plants where large genomes are common. In some plants (e.g., maize, SanMiguel and Bennetzen 1998), recent retroelement amplification has made BAC end sequencing and assembly of shotgun-sequenced clones almost impossible. Consequently, substantial efforts are taken to isolate DNA regions that do not contain repetitive sequences. One means of obtaining single/low-copy sequences is to prepare cDNA libraries. However, the representation of genes in a given cDNA library is only indicative of gene expression in the source tissue(s), and gene copy number is not accurately reflected in cDNA libraries even if “normalization” techniques (e.g., Ko 1990; Soares et al. 1994; Neto et al. 1997;Poustka et al. 1999) are employed. Repetitive DNA is often more highly methylated than low-copy DNA, and consequently some researchers have used methylation-sensitive restriction enzymes (e.g., McCouch et al. 1988) or bacterial host strains that preferentially restrict methylated DNA (Rabinowicz et al. 1999) to produce genomic libraries enriched in low-copy (ostensibly genic) DNA. However, cloning strategies involving the preferential exclusion of hypermethylated DNA may result in the loss of important/interesting genes because the pattern and significance of DNA methylation can differ markedly between species (e.g., Simmen et al. 1999), genes within an organism (e.g., Lois et al. 1990; Wölfl et al. 1991), developmental stages (for review, seeHeslop-Harrison 2000), and different regions of the same gene (e.g., Li et al. 1993; Riesewijk et al. 1996). Clearly, alternative strategies for isolating and sequencing the unique elements of genomes are needed.
The results of a Cot analysis provide the information needed to isolate the major kinetic components of a genome in a manner independent of sequence expression (Britten and Kohne 1968) and/or methylation (Burtseva et al. 1979). However, to our knowledge DNA fractionated via Cot/HAP techniques has not previously been used in the construction of genomic libraries. Here we describe the production and characterization of genomic libraries derived from the three major kinetic components of sorghum (Sorghum bicolor) DNA. Sorghum was chosen for this study because (to our knowledge) it has not been the subject of a Cot analysis, it has a 4000–6000 year history of cultivation (Kimber 2000), it is one of the most agronomically important plant species in the world (Smith 2000), and its relatively small genome is a valuable “window” into the low-copy sequence diversity of closely related, large-genome crops such as maize and sugarcane (see Draye et al. 2001).
Our results suggest that cloning of isolated kinetic components is a useful and powerful means to clone genomic sequences based upon their relative iteration and to efficiently discover new DNA sequences in a manner independent of expression and/or methylation patterns. The combination of Cot-based cloning and high-throughput sequencing of Cot libraries [Cot-based cloning and sequencing (CBCS)] represents a means by which the sequence complexity of large genomes can be “captured” at a fraction of the cost of shotgun sequencing.
RESULTS
Melting Temperatures and GC Content of Sorghum DNA
Melting curves were generated for sheared sorghum DNA in 0.03, 0.12, and 0.5 M sodium phosphate buffer (SPB), and melting temperatures (Tm) for DNA in each buffer were determined using first-derivative analysis. The melting temperatures for sorghum DNA in 0.03, 0.12, and 0.5 M SPB are 75.1°, 84.1°, and 93.1°C, respectively.
For DNA dissolved in buffers with a monovalent cation concentration (Mmvc) between 0.01 and 0.2 M, the GC content of the DNA can be calculated using the formula %GC = 2.44 (Tm − 81.5 − 16.6 log Mmvc) (Mandel and Marmur 1968). Consequently, the sorghum DNA samples in 0.03 M SPB (Na+ = 0.045 M) and 0.12 M SPB (Na+ = 0.18 M) result in %GC estimates of 38.9% and 36.5%, respectively. The average of these two values is 37.7%.
Cot Analysis
A Cot curve for Sorghum bicolor was prepared according toPeterson et al. (1998) and analyzed using the computer program ofPearson et al. (1977). The analysis providing the lowest RMS (root mean square deviation) and goodness of fit values (0.02554 and 0.02712, respectively) is a three-component fit with no constrained variables.
In all Cot analyses, a certain fraction of the DNA forms duplexes even at Cot values approaching zero. Such early renaturation is thought to be due to base pairing between complementary sequences on the same DNA molecule (i.e., foldback DNA; Britten et al. 1974). As shown (Fig. 1), approximately 16% of sorghum DNA had reassociated by the earliest Cot point (10−5 M•sec) and consequently was not included in the curve. No detectable reassociation was observed until a Cot value of about 0.02 M•sec.
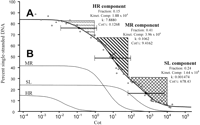
Sorghum cot analysis. (A) Complete Cot curve, data analysis, and component isolation. A least-squares curve (thick black line) was fitted through the data points (open circles) using the computer program of Pearson et al. (1977). The curve consists of highly repetitive (HR), moderately repetitive (MR), and single/low-copy (SL) components characterized by fast, intermediate, and slow reassociation, respectively. For each component, the following values have been placed to the right of the component's general location: Fraction = the proportion of the genome found in that component, Kinet. Comp. (kinetic complexity) = the length in nucleotide pairs of the longest nonrepeating sequence calculated from the Cot data, k = the observed reassociation rate in M−1•s−1, and Cot½ = the value on the abscissa of the complete Cot curve at which half the DNA in the component has reassociated. Black diamonds mark the positions on the complete Cot curve of the Cot½ values for HR, MR, and SL components. For a Cot component, 80% of the sequences in that component will renature in the “two Cot decade region” (TCDR) flanking the component's Cot½ value (brackets centered at Cot½ markers; Britten and Davidson 1985). We utilized this principle in isolating HR, MR, and SL Cot components for Cot library construction. In brief, all double-stranded DNA within a component's TCDR was isolated except for areas that overlap the TCDRs of other components (regions marked by upward vertical dashes, diagonal stripes, and crosshatching delimit areas of the curve used in HRCot, MRCot, and SLCot library construction, respectively). Note that the area isolated for use in constructing the SLCot library extends a short way past the right end of the TCDR for the SL component; this is presumably not a problem as any double-stranded DNA in the region to the right of the SL component TCDR is likely to be single-copy. (B) The predicted individual renaturation profiles of the HR component, MR component, and SL component are shown.
Four percent of the sorghum DNA did not reassociate by the highest Cot value (20,000 M•sec). DNA that does not reassociate by such a high Cot value is thought to be damaged and incapable of binding to HAP (e.g., Kiper and Herzfeld 1978).
The sorghum Cot curve consists of a fast, an intermediate, and a slow reassociating component. The complete Cot curve, renaturation profiles of the three Cot components, and the reassociation rate (k), Cot½ value, kinetic complexity, and genome fraction of each component are presented in Figure 1. In diploid organisms, the slowest reassociating component of a Cot curve generally represents single-copy DNA sequences. In such cases, genome size can be estimated by comparing the k value of the slow reassociating component to E. coli's rate constant (k = 0.22 M−1•sec−1) and DNA content (Zimmerman and Goldberg 1977). The genome of E. coli(strain K12, substrain MG1655) is 4,639,221 bp (Blattner et al. 1997). Assuming that the sorghum slow reassociating component (k = 0.001474 M−1•sec−1) is composed of single-copy DNA, the estimated 1C genome size of sorghum would be G = (4,639,221 bp × 0.22 M−1•sec−1) ÷ 0.001474 M−1•sec−1 = 6.92 × 108 bp or 692 Mbp. While this value is slightly lower than the reported values based on Feulgen densitometry (753–837 Mbp, Laurie and Bennett 1985) and flow cytometry (748–772 Mbp, Arumuganathan and Earle 1991), there is only a 7.5%–17% difference between the Cot-based genome size and these previous estimates. Consequently, it is likely that the slow reassociating component is primarily single-copy DNA, and thus we refer to it as the single/low-copy (SL) component.
Assuming that the SL component has a repetition frequency of 1, the average repetition frequency of the DNA in the other components can be estimated by dividing their k values by the k value of the SL component (Hood et al. 1975). The predicted repetition frequencies of sequences in the fast reassociating component and in the intermediate reassociating component are 7.8864/0.001474 (5350.3) and 0.1062/ 0.001474 (72.1), respectively. In light of their relative repetitiveness, the fast and intermediate reassociating components are hereafter referred to as the highly repetitive (HR) and moderately repetitive (MR) components.
Library Construction, Blot Analysis, and Sequencing
HRCot, MRCot, and SLCot libraries were generated from isolated Cot components (see Methods for details on component isolation and cloning). The relative iteration of the insert DNA in the three Cot libraries was examined by comparing the intensity with which Cot clone probes hybridized to replica Southern blots of sorghum genomic DNA. The average intensity of hybridization to blots incubated with radiolabeled HRCot sequences was 43,067 cpm (± 6248) while the average values for the MRCot and SLCot blots were 3783 cpm (± 1419) and 1377 cpm (± 253), respectively.
We sequenced a total of 384 HRCot, 480 MRCot, and 576 SLCot clones of which 253, 409, and 499 (respectively) met our sequence quality criteria (i.e., Ph/Pr value >16 over 300 continuous bases, high-quality insert sequence ≥50 bp). The (253 + 409 + 499 = 1161) “quality clones” were BLASTed against the GenBank Nr (nonredundant), GenBank EST, and SUCEST Sugarcane EST (http://sucest.lbi.dcc.unicamp.br/en/) databases. For each quality clone, only bit scores (S‘) of 55.44 or greater were deemed significant and used in characterizing the clone. Unlike E values (E) commonly used to compare the quality of hits, bit scores provide a means of comparing the significance of database hits independent of database and query size (see www.ncbi.nlm.nih.gov for details). For a database of 3.5 billion nucleotides (slightly larger than the effective size of the GenBank Nr and EST databases at the time of sequence analysis) and an effective query length of 159 nt, a bit score of 55.44 is roughly equivalent to an E value of 1 × 10−5. For a given quality clone, the term “primary hit” was used to indicate the database sequence (if any) showing the highest significant homology to that clone.
Categorization of Cot Clones
Each Cot clone was placed into a single descriptive category (“BLAST category”) based upon the scheme shown in Figure 2. Because of the difficulty associated with evaluating EST hits (see Limitations of the EST Data and Table A in the online supplement to this article,www.genome.org), GenBank Nr database hits were given priority in the classification scheme with EST hits used only to categorize clones without significant Nr hits or with Nr hits to genomic sequences of unknown character. Results of category assignment and a list of characterized gene and repeat sequences recognized by various Cot clones are given in Table 1. An overview of the data is shown in Figure 3.
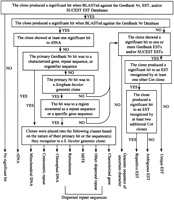
Classification of Cot clones based on sequence analysis. For comparative purposes, each of the sequenced Cot clones meeting the minimum sequence quality requirements (see Methods section) was assigned to a single descriptive “BLAST category” based upon its significant database hits according to the classification scheme shown.
BLAST-Based Categorization of HRCot, MRCot, and SLCot Clonesu
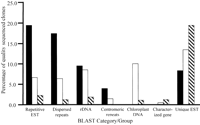
Sequence composition of different Cot libraries. Black bars represent HRCot clones, white bars represent MRCot clones, and diagonally striped bars represent SLCot clones. The BLAST group “Dispersed repeat sequences” is composed of the Retroelement, MITE, and Other dispersed repeat BLAST categories (see Fig. 2 and Table 1).
All three libraries possessed more clones in the no significant hit BLAST category than any other. Roughly 70% of the SLCot clones showed no significant database hits, whereas about 50% of the MRCot clones and 35% of the HRCot clones fell into this category.
HRCot hits were primarily to plant repetitive DNA sequences (Lapitan 1992; Bennetzen et al. 1998; Heslop-Harrison 2000) including retrotransposons and other dispersed repeat sequences, rDNA sequences, and sorghum centromeric repeat sequences. The relative percentage of clones showing homology to repetitive ESTs was considerably higher for the HRCot library (19.4%) than the other two libraries (6.6% for MRCot, 2.2% for SLCot). None of the HRCot clones produced a significant hit to a characterized gene sequence, and the percentage of unique EST clones in the HRCot library was much lower than corresponding values for the MRCot and SLCot libraries (Table 1, Fig. 3).
Among the three Cot libraries, the SLCot library showed the highest percentage of hits to characterized gene sequences andunique ESTs. No centromeric sequences were detected, and only 6.2% of the SLCot clones fell into any of the repeat sequence categories.
The MRCot library showed intermediate levels of repeat sequences and unique sequences. With regard to low-copy sequences, the percentage of MRCot sequences in the unique EST category was roughly the mean of the corresponding values for the HRCot and SLCot libraries (Fig. 3). Of the characterized gene sequences detected in the Cot libraries, 25% were found in the MRCot library (the remaining 75% were in the SLCot library). Although the HRCot library had the greatest fraction of clones with homology to known repeats (Table 1), some repeat sequences (presumably of moderate iteration) were more abundant in the MRCot library. For example, clones with homology to the retroelement Leviathan were three times more common in the MRCot library than the HRCot library. Likewise, sequences with homology to retrotransposon genes/pseudogenes in the GenBank Nr database were limited to MRCot clones (Table 1). Of particular note, 10% of the MRCot sequences correspond to chloroplast DNA, presumably a contaminant in the nuclear DNA isolation process (Fig. 3). However, chloroplast sequences were detected in less than one percent of the SLCot clones and none of the HRCot clones. While chloroplast DNA was not a desired end product of Cot library construction, the observation that chloroplast sequences are almost exclusively limited to MRCot clones neatly illustrates the “two Cot decade” principle used in the isolation of individual Cot components; that is, 80% of the copies of a given DNA sequence are contained within a span of two Cot decades (Fig. 1; Britten and Davidson 1985). Based on the Cot curve, the MR component constitutes 41% of the genome, but if a tenth of this component is actually chloroplast DNA, the percentage of the genome found in the MR component may be closer to 37%.
Of the 1161 Cot clone sequences used in sequence analysis, only one clone showed a significant primary hit to a mitochondrial DNA sequence. This clone (SLCot4G05) appears to contain a portion of the Sorghum bicolor F0-F1 ATPase alpha subunit gene (GenBank AJ278690).
Retrosor-6
One of the largest continuous sorghum DNA sequences in the GenBank Nr database is a 126 kb BAC clone containing the 22 kD kafirin cluster (GenBank AF061282; V. Llaca, A. Lou, and J. Messing, unpubl.). A total of 15.2% of the HRCot clones, 2.4% of the MRCot clones, and 1.0% of the SLCot clones showed primary hits to this BAC. Interestingly, 34 of 39 (87.1%) of the HRCot and 7 of 7 (100%) of the MRCot primary hits to the kafirin cluster BAC are localized within a 7377 bp sequence found only once in the BAC (bases 127,895–135,271). None of the SLCot hits to the kafirin cluster BAC recognize the 7377 bp sequence. Although the 7377 bp sequence represents only 4.5% of the bases in the kafirin cluster BAC, it accounts for 13.4% of all primary HRCot hits, making it the most frequently recognized S. bicolor sequence.
In their annotation of the kafirin cluster BAC, (GenBank AF061282) V. Llaca, A. Lou, and J. Messing have deemed the 7377 bp sequence a “retroelement”. Although they have named five other sorghum retroelements (Retrosor-1, Retrosor-2, Retrosor-3, Retrosor-4, and Retrosor-5), they did not name the 7377 bp retroelement sequence. Our study of the sequence likewise suggests that it is a retroelement (see Fig.4A), and with the support of J. Messing (pers. comm.), we have named the sequence Retrosor-6. Retrosor-6 possesses no large open reading frames (ORFs) although nucleotide-protein BLAST (blastx) results indicate that it shares limited homology to an ORF1 polyprotein (S’ = 43.9 bits) of the gypsy-type retroelement Athila(Pélissier et al. 1995) and a putative Arabidopsis polprotein (S' = 42.4 bits). The apparent absence of gag andenv genes and the limited homology to known polsequences suggest that the copy of the retroelement found in the kafirin cluster is no longer capable of autonomous replication.
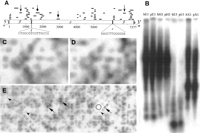
Retrosor-6. (A) The structure of Retrosor-6and the distribution of the 41 sorghum Cot clones with primary homology to Retrosor-6. Retroelement features of Retrosor-6include (α) duplicated target site sequences flanking both ends of the sequence, (β) long terminal repeats (LTRs) (bases 1–1279 and 6098–7377) with the canonical LTR start/end nucleotides 5′-TG…CA-3′, (χ) a primer binding site complementary to the plant tRNA for asparagine (bases 1286–1301), (δ) an internal sequence region with homology to ORF1 of the Arabidopsisgypsy-type retroelement Athila, and (ε) a polypurine tract (bases 6083–6095) (see Murphy et al. 1995 for review of retroelement structure). A scale showing distance in base pairs has been positioned underneath the diagram of the retroelement. For each Cot clone recognizing Retrosor-6, a thin black line has been placed above the retroelement marking the relative position(s) and length of the sequence shared by that clone and Retrosor-6. Because the LTRs have almost identical sequences (99.5% sequence identity), all of the clones with homology to one LTR have a similar/identical degree of homology to the other LTR. For these clones, lines have been positioned above both LTRs. (B) Hybridization of a Retrosor-6probe (diamond-headed arrow in A) to a Southern blot. The labels at the head of each lane indicate the source of DNA in that lane and the restriction enzyme with which the DNA was digested. Specifically, b = S. bicolor, p = S. propinquum, E1 = EcoRI, H3 = HindIII, E5 = EcoRV, and X1 = XbaI. The two species show essentially identical hybridization patterns and intensities. (C) S. bicolor grid probed with a sequence from the Retrosor-6LTR (chevron-headed arrow in A). (D) A grid identical to that in ‘C’ probed with a Retrosor-6 partial internal sequence (triangular-head arrow in A). The hybridization patterns observed for grids probed with the internalRetrosor-6 sequence are virtually identical to those produced by the LTR sequence probe for both S. bicolor (C andD) and S. propinquum (data not shown). (E) A section of the S. propinquum BAC grid probed with part of the LTR sequence of Retrosor-6. The number of copies ofRetrosor-6 in the S. propinquum and S. bicolor genomes were estimated as described in Table B (available in the online supplement to this article, www.genome.org) and the Methods section. The region on the grid used as “background” is enclosed within a circle. Examples of clones showing relatively intense hybridization signals are marked by arrows (triangular-heads) whereas clones with relatively weak but interpretable hybridization signals are marked by arrowheads (chevrons).
To examine the abundance and dispersal pattern of Retrosor-6in the genome of S. bicolor and to check for its presence in the wild species S. propinquum, a Cot clone containing 190 bp of the Retrosor-6 sequence was radiolabeled and used to probe a Southern blot containing restriction-digested S. bicolor andS. propinquum DNA. As shown in Figure 4B, theRetrosor-6 hybridization pattern for both sorghum species is essentially the same, consisting of a few dark bands within a smear of hybridization signal.
While most of the Retrosor-6 retroelement shows numerous Cot clone hits (Fig. 4A), the region between bases 2000 and 4000 has only two hits. To explore whether this region has diverged more rapidly than other parts of Retrosor-6, high-density BAC grids from both sorghum species were probed with a Cot clone containing part of theRetrosor-6 LTR sequence (e.g., Fig. 4C), and duplicate copies of the grids were probed with a sequence from the 2–4 kb region of the retroelement (Fig. 4D). When the autoradiograms for the LTR region and the 2–4 kb region were digitally aligned and compared, only minimal differences could be detected in the hybridization patterns for a particular species (e.g., Fig. 4C and D).
To estimate the copy number of Retrosor-6 in the genomes ofS. bicolor and S. propinquum, the BAC grids probed with the Retrosor-6 LTR sequence were analyzed using a densitometer (see Fig. 4E). The grid densitometry results suggest that there are approximately 6275 copies of Retrosor-6 in theS. bicolor genome and 6748 copies in the S. propinquum genome (see Table B in the online supplement to this article, www.genome.org). Assuming an average size for the retroelement of 7377 bp, Retrosor-6 accounts for approximately 6.0% and 6.3% of genomic DNA in S. bicolor and S. propinquum, respectively.
Of note, two of the randomly selected HRCot clones hybridized to Southern blots (see Library Construction, Blot Analysis, and Sequencing above) were later shown to contain portions ofRetrosor-6. One clone (HRCot2G11) containing part of theRetrosor-6 LTR produced the highest level of hybridization of any of the randomly selected Cot clones with a specific activity of 10,000 cpm. The second clone (HRCot3B01), carrying part of the internal sequence of Retrosor-6, resulted in a hybridization intensity of 5000 cpm, that is, half that of the clone containing the LTR sequence.
Molecular Genetic Markers, BAC End Sequences, and Cot Clones
Cot clones were BLASTed against approximately 1500 molecular markers (see the section Molecular Markers in the online supplement to this article, www.genome.org) from a high-density sorghum molecular map based on RFLP segregation in the progeny of a cross between S. bicolor and S. propinquum(Chittenden et al. 1994; Draye et al. 2001). Fourteen Cot clones contained inserts with significant homology (S' ≥76.28) to a total of nine markers on the molecular map (see Table C in the online supplement).
The Cot clone sequences also were compared to 116 sorghum BAC end sequences (H. Ma, J. Bowers, and A. Paterson, unpubl.). None of the BAC ends showed significant homology to SLCot clones. However, 12 BAC ends possessed significant homology to HRCot clones, six recognized MRCot clones, and two recognized both HRCot and MRCot clones. In total, 20 of the 116 BAC ends (17%) exhibited significant homology to at least one of the 1161 sorghum Cot clone sequences. Assuming that the Cot libraries are representative of the sequence complexities of the components from which they were prepared, a 15% probability that any randomly selected sorghum genomic sequence will share significant sequence identity with one or more of the 1161 quality Cot clones (see the section Probability of Significant BAC End/Cot Clone Homology in the online supplement) would be predicted. The observed percentage of BAC ends with homology to the Cot clones (17%) and predicted percentage (15%) are not significantly different (seeTest of Significance of a Binomial Proportion in the online supplement), suggesting that the Cot libraries are reflective of their respective Cot components.
DISCUSSION
Although renaturation kinetics has long been used to characterize genomes (Britten and Kohne 1968), to our knowledge the present study is the first report in which Cot components isolated from genomic DNA have been cloned and sequenced. Our results indicate that (1) the Cot libraries differ with regard to sequence iteration and composition in a predictable manner, (2) most of the highly repetitive DNA in the sorghum genome is found within the sequenced HRCot quality clones, (3) a previously unnamed sorghum retroelement is a major component (and perhaps the most abundant sequence) in both the S. bicolor andS. propinquum genomes, (4) Cot clones can be used to augment the information content of both molecular and physical maps, and (5) sequencing of clones from Cot libraries may represent a means by which the diversity of sequences found in a genome can be efficiently “captured”.
Effectiveness of Cot-Based Cloning
The three Cot libraries differ in relative sequence iteration and composition in a manner reflecting the nature of the components from which they were derived; that is, construction of repetition-based DNA libraries using Cot techniques is effective. When Southern blots of sorghum genomic DNA were probed with randomly selected, radiolabeled Cot clone inserts, those blots hybridized with HRCot sequences exhibited a mean labeling intensity (cpm) >10 times that of MRCot-probed blots and >30 times that of SLCot-probed blots. Detailed sequence analysis of 250–500 Cot clone inserts from each of the three Cot libraries revealed that the HRCot library is rich in sequences traditionally found in high-copy numbers (e.g., retrotransposons, rDNA, centromeric repeats), the SLCot library is enriched in sequences with homology to characterized genes and unique ESTs, and the MRCot library possesses its own subset of repeat sequences as well as exhibiting some overlap with the HRCot and SLCot libraries (Table 1, Fig. 3). Additionally, the observed percentage (17%) of random sorghum BAC end sequences recognizing one or more of the 1161 sorghum Cot clone sequences was found to be statistically indistinguishable from the percentage expected if the Cot libraries are representative of their respective Cot components (15%). Because the methods employed do not rely upon differential expression and/or methylation of sequences, Cot-based cloning provides a means by which any genomic sequence (including genes expressed at low levels or during short developmental timeframes) can be isolated and cloned based upon its relative iteration.
Retrosor-6
The previously discovered sequence exhibiting primary homology to the greatest number of Cot clones is a 7377 bp retroelement found in the sorghum kafirin cluster sequence (GenBank AF061282; V. Llaca, A. Lou, and J. Messing, unpubl.). This retroelement (now calledRetrosor-6) is highly reiterated in both S. bicolorand S. propinquum (Fig. 4B). These two species possess the same chromosome number and can be crossed (e.g., Chittenden et al. 1994), but the resulting progeny exhibit the aberrant segregation and partial sterility that typifies interspecific hybrids. Two lines of evidence suggest that most copies of Retrosor-6 in both sorghum species are similar to the copy of the retroelement found in the kafirin gene cluster. First, S. bicolor and S. propinquum BAC grids probed with part of the Retrosor-6LTR showed hybridization patterns nearly identical to those observed for duplicate blots probed with part of the internal region of the retroelement (Fig. 4C,D). Second, a Southern blot probed with a portion of the Retrosor-6 LTR exhibited hybridization signal about twice that of a duplicate blot probed with an internal sequence of similar length—an observation suggesting that there are roughly two copies of the LTR for each copy of the internal sequence. Based on the assumption that most copies of Retrosor-6 are similar to the kafirin cluster copy of the retroelement, densitometric analysis of BAC grids indicates that Retrosor-6 accounts for approximately 6% of the DNA in both sorghum species (see Table B in the online supplement for this article, www.genome.org). Because S. bicolor and S. propinquum have similar genome sizes and possess roughly the same number of copies of Retrosor-6, the retroelement may have been introduced into a common ancestor of the two species rather than into the species separately. However, on the assumption that Retrosor-6 provides no selective advantage to the genome and hence can undergo mutation without influencing fitness, the preponderance of apparently intact copies of Retrosor-6and the relatively high level of shared sequence identity between the LTRs of the kafirin cluster copy of Retrosor-6 (615/618 bp matches, S' = 1171 bits) suggest that the retroelement may be fairly new to the Sorghum genus.
Cot Clones and the Sorghum Molecular Map
Cot clone sequences were compared with the sequences of markers on the sorghum molecular map (Bowers et al. 2000). Three of the nine molecular markers recognized by Cot clones appear to be rDNA sequences. The three “rDNA molecular markers” are found at essentially the same locus on S. bicolor linkage group C (see Table Cin the online supplement to this article, www.genome.org). The 18S-5.8S-26S rDNA locus has been localized by fluorescence in situ hybridization to the longest S. bicolor mitotic metaphase chromosome (Sang and Liang 2000). Likewise, we recently demonstrated that the longest S. bicolor pachytene chromosome is the nucleolus organizer chromosome (Draye et al. 2001). Consequently, it appears that S. bicolor mitotic metaphase chromosome1, meiotic chromosome 1, and linkage group C are the same entity, the first instance in which a sorghum linkage group has been assigned to a cytologically distinguishable chromosome.
Potential Bias in the Sorghum Cot Libraries
E. coli possesses three endonuclease systems that preferentially restrict methylated DNA; McrA, McrBC, and Mrr. These restriction systems do not cleave DNA that has been methylated by the bacterium's endogenous methylase systems (Redaschi and Bickle 1996). In preparing the sorghum Cot libraries, we used the Promega pGEM-T Easy cloning kit and the accompanying host strain JM109. While JM109 lacks functional McrA and Mrr restriction systems (it ismcrA −, mrr −), it does possess a functional McrBC protein (mcrBC +). The McrBC protein cleaves DNA sequences with the following configuration: 5′-PumCN40–80PumC-3′ (Pieper et al. 1997). Consequently, it is possible that certain methylated (presumably highly repetitive) sequences from sorghum are underrepresented in one or more of the Cot libraries due to preferential restriction by the McrBC system. However, it is likely that the relatively small size of the Cot clone inserts (∼100–400 bp) and the relatively large size of McrBC recognition sites (≥44 bp) substantially decreased possible effects of McrBC during cloning. The limited effect of the McrBC genotype on sorghum Cot library construction is suggested by the observation that the highest proportion of HRCot clones showing significant hits to the GenBank Nr database contain sequences that are frequently methylated in plants, that is, retrotransposons (Rabinowicz et al. 1999) and centromeric sequences (Moore et al. 1993) (Table 1, Fig. 3). Regardless, to construct a Cot library that is truly representative of a particular Cot component, one should use a host strain with a genotype that is insensitive to DNA methylation patterns.
Continued Use of Sorghum Cot Libraries
Now that the feasibility of Cot-based cloning has been demonstrated, we have begun to use the sorghum Cot libraries as a means to augment the information content of the rapidly-growing S. bicolor andS. propinquum physical maps (Bowers et al. 2001; Draye et al. 2001). For example, Cot clones with homology to Retrosor-6 are being used to determine the genetic and physical distribution of this element by evaluating colocalization of Retrosor-6 and genetically mapped RFLPs on S. bicolor and S. propinquum BACs. This basic principle will likely be used to physically map other repeat sequences. Cot clone insert sequences with homology to characterized plant genes (see Table 1) will be used to find sorghum homologs/orthologs in BAC clones and position these sequences on the physical maps.
Comparison of BAC ends with sorghum Cot clone sequences provides a means to identify BAC ends that contain repetitive DNA sequences. As described in the Results, 17% of sorghum BAC ends (n = 116) show homology to HRCot/MRCot sequences. These BAC ends likely contain repetitive elements and thus may be of limited use in contig assembly.
Experimental Modifications and Applications
While the goals of the present study were to investigate the feasibility/usefulness of cloning isolated Cot components and further characterize the sorghum genome, Cot clones could be employed in other ways. Additionally, many of the experimental parameters utilized in this project could be altered to meet different research needs. For example:
- (1)
- In our research, only double-stranded DNA resulting from reassociation was used in preparing Cot libraries. However, HAP-fractionated single-stranded DNA can be used in Cot-based cloning as well. In this regard, we have taken single-stranded Cot DNA, generated complementary strands via the random primer method (Mackey et al. 1995), and used TA-cloning techniques (Kawata et al. 1998) to produce ssDNA-derived Cot clones (D. Peterson, A. Nagel, S. Wessler, and A. Paterson, unpubl.). The use of ssDNA fractions in cloning would be advantageous in instances where the quantity of genomic DNA is limited. Additionally, fewer base pair mismatches would be expected if primer extension techniques rather than strand renaturation were used to generate duplexes for cloning and sequencing.
- (2)
- Foldback sequences could be cloned to produce a “foldback Cot” (FBCot) library. Although most foldback DNA is probably repetitive in nature (Davidson et al. 1971), some foldback sequences may be single/low-copy DNA; likewise the foldback fraction may contain some sequences not represented in the HR, MR, and/or SL components. Consequently, FBCot libraries may be a source of useful sequence information. We have used random primer/TA-cloning techniques to produce FBCot clones for S. bicolor, although these clones have not yet been sequenced (D. Peterson, A. Nagel, S. Wessler, and A. Paterson, unpubl.).
- (3)
- If the DNA fragments in a component are of a length optimal for automated sequencing (about 500–1000 bp), the fragments can be cloned using standard techniques. If the DNA fragments in an isolated component are relatively short (e.g., 200 bp as in the present research), prior to cloning the fragments can be joined together using DNA linkers with highly recognizable sequences under reaction conditions that result in concatemers with mean lengths in the optimal size range for sequencing. The generation and cloning of concatemers as described above is similar to the SAGE (serial analysis of gene expression) technique (Velculescu et al. 1995).
- (4)
- By using renaturation kinetics to further purify/characterize isolated Cot components into subcomponents (i.e., minicot analysis; see Britten et al. 1974; Goldberg 1978; Kiper and Herzfeld 1978), the resolution of Cot analysis (and subsequently Cot-based cloning) could be increased.
- (5)
- In species where methylation is known to be associated with repetitive DNA (e.g., Rabinowicz et al. 1999), cloning of isolated SL sequences intomcrBC +/mcrA +/mrr +bacterial strains may further decrease contamination of the resulting library with repeat sequences.
- (6)
- EST/cDNA and genomic libraries could be screened with isolated Cot fractions to identify populations of clones containing probable unique and/or repetitive sequences.
- (7)
- The possibility of affordably automating (and thus standardizing) many of the Cot analysis/HAP fractionation procedures is well within modern capabilities.
Capture of Sequence Complexity Using Cot-Based Cloning and Sequencing (CBCS)
While analysis of complete genome sequences is the ultimate means by which the genomes of different species can be compared, genome sequencing may not be an affordable, realistic, and/or desirable option for species with large, highly repetitive genomes. An alternative to genome sequencing is the “capture” (isolation, cloning, and sequencing) of an organism's sequence complexity (Britten et al. 1974); that is, the combined length in nucleotide pairs of the different DNA sequences that comprise a genome (Britten et al. 1974). Because most prokaryotic genomes are relatively devoid of repetition, the sequence complexity of a bacterial genome is roughly the same as its genome size (Britten and Kohne 1968). In contrast, the sequence complexity of a eukaryotic genome is the combined length of all of its single-copy DNA sequences plus one copy of each repeat sequence (e.g., a genome composed of 100,000 copies of sequence A, 9000 copies of sequence B, 3400 copies of sequence C, two copies of sequence D, and one copy each of sequencesE–Z would have a sequence complexity of A +B + C + D + E + F…+Z bp). Cot analysis provides an accurate means of estimating the sequence complexity of kinetic components (Britten et al. 1974). To distinguish between the exact sequence complexity of a component/genome (presumably only determinable by complete component/genome sequencing) and an estimate of its sequence complexity based on a Cot analysis, the term “kinetic complexity” is used to identify the latter (Britten et al. 1974). However, this convention does not mean that kinetic complexity values do not accurately reflect sequence complexity—as an analogy, the exact genome size of an organism cannot really be determined except by complete genome sequencing, although genome size can be accurately estimated using Feulgen densitometry, flow cytometry, Cot analysis, and other methodologies. Because each repeat sequence is counted only once in determination of a genome's sequence complexity, the contribution of repeat sequences to sequence complexity is generally quite small. In contrast, single/low-copy sequences account for the vast majority of a genome's sequence complexity (e.g., 98% of the combined kinetic complexity of the sorghum HR, MR, and SL Cot components is found in the SL component; Fig. 1).
The use of bacterial strains sensitive to DNA methylation has been proposed as a means to capture the low-copy sequences that comprise most of a genome's sequence complexity (i.e., “methyl filtration”,Rabinowicz et al. 1999). However, methyl filtration and similar approaches such as PstI cloning are based on the assumption that hypermethylated sequences represent DNA that is nongenic whereas hypomethylated sequences represent low-copy DNA. In most (if not all) instances, using cloning/sequencing techniques centered on differential sequence methylation will result in the loss of many important and interesting genes: (1) it is common knowledge that methylation is one of the primary means by which genes are regulated, and that the methylation status of genes (or portions of genes) differs markedly between tissues and/or developmental stages (Siegfried and Cedar 1997;Heslop-Harrison 2000), (2) some genes are normally active when hypermethylated (Lois et al. 1990; Wölfl et al. 1991; Heslop-Harrison 2000) and may not function if they are demethylated (Li et al. 1993), (3) in some genes methylation at one site enhances transcription whereas methylation at another site reduces transcription (Li et al. 1993; Riesewijk et al. 1996), and (4) some species normally possess hypermethylated genes and hypomethylated repeat sequences (Simmen et al. 1999).
We propose “Cot-based cloning and sequencing” (CBCS) as a means to capture the sequence complexity of a genome in a manner independent of methylation. In CBCS, isolated kinetic components are cloned to produce Cot libraries, and clones from each library are sequenced using high-throughput methods. To obtain comparable sequence complexity coverage for different Cot components, Cot clones from each Cot library are sequenced in proportion to the kinetic complexity of the component from which they were derived.
The usefulness of CBCS is best demonstrated when compared with shotgun sequencing (the sequencing of randomly selected clones
from a genomic library), the primary means by which genomes are currently sequenced. In shotgun sequencing, the number of
different clones (n) that need to be sequenced in order to have 99% confidence that all genomic elements have been sequenced
at least once (i.e., that the sequence complexity of the genome has been captured) can be calculated using the following formula: where Z = mean insert size in bp and G = 1C genome size in bp (Paterson 1996). For a standard sorghum (1C = 760 Mb) genomic library containing 600 bp inserts, 99% confidence can be obtained by sequencing
5.8 × 106 randomly selected genomic clones. In Cot library construction, genomic DNA is separated into kinetic/sequence complexity-based
components prior to cloning and sequencing. Consequently, for a Cot library the probability of sequencing 99% of the DNA elements
in the component used to construct that library is a function of the component's kinetic complexity (γ) rather than genome
size:
where Z = mean insert size in bp and G = 1C genome size in bp (Paterson 1996). For a standard sorghum (1C = 760 Mb) genomic library containing 600 bp inserts, 99% confidence can be obtained by sequencing
5.8 × 106 randomly selected genomic clones. In Cot library construction, genomic DNA is separated into kinetic/sequence complexity-based
components prior to cloning and sequencing. Consequently, for a Cot library the probability of sequencing 99% of the DNA elements
in the component used to construct that library is a function of the component's kinetic complexity (γ) rather than genome
size:
Assuming one had sorghum HRCot, MRCot, and SLCot libraries with 600 bp inserts, 99% confidence could be obtained for (1) the HR component by sequencing 142 HRCot clones, (2) the MR component by sequencing 3.1 × 104 MRCot clones, and (3) the SL component by sequencing 1.3 × 106 SLCot clones. The total number of Cot clones that would need to be sequenced to have 99% confidence that all HR, MR, and SL component elements had been sequenced would be (142 + 3.1×104 + 1.3×106 =) 1.33×106clones. However, the HR, MR, and SL components comprise only 80% of sorghum DNA; the remaining 20% is divided between foldback DNA (16%) and damaged (unannealable) sequences (4%). With regard to the latter sequence category, it represents a small proportion of the genome and presumably contains no sequences that are not found in one or more of the other fractions. Assuming that damage is a random event that would affect all portions of the genome in a manner proportional to their relative fractions, less than one-quarter of the 4% unannealable DNA (i.e., <7.6 million base pairs or 1.0% of the entire genome) would be high sequence complexity (single/low-copy) DNA. Consequently, the unannealable DNA can essentially be ignored. The same is not true of the foldback fraction (see Experimental Modifications and Applications above). To be fairly secure of retrieving the useful sequence information from the foldback fraction, it can be assigned a “kinetic complexity” equal to the number of base pairs it contains. In sorghum, the foldback fraction contains (0.16 × 760 Mbp =) 1.2 × 108 bp of DNA, and thus using Equation and a mean insert size of 600 bp, sequencing of 9.2 × 105 “foldback Cot” (FBCot) clones would give 99% confidence that all sequences in the foldback fraction had been sequenced at least once. Consequently, using CBCS and the highly conservative assumption that the foldback fraction is largely single-copy DNA, the sequence complexity of the entire sorghum genome could be captured (∼99% confidence) by sequencing a total of (1.33 × 106 + 9.2 × 105 =) 2.3 × 106 Cot clones. Undoubtedly sequencing of 2.3 million clones would be a significant undertaking. However, capturing the sequence complexity of the sorghum genome using the shotgun approach would require sequencing of (5.8 × 106 ÷ 2.3 × 106 =) 2.5 times as many clones. The relative advantage of CBCS over shotgun sequencing is even more pronounced for species possessing genomes with higher proportions of repetitive DNA—for some plants and animal species, CBCS allows genome sequence complexity capture using less than one-tenth the number of clones that would be required using shotgun sequencing (D. Peterson, S. Wessler, and A. Paterson, in prep.). In all cases, the minimum number of clones needed to attain a specific level of sequence complexity coverage can be calculated in advance of initiating sequencing.
Sorghum has a relatively high percentage of foldback DNA compared to many species for which Cot analyses have been performed. Several strategies employed prior to FBCot sequencing (e.g., screening high-density grids of the FBCot library with randomly selected FBCot clones) could be used to identify highly redundant FBCot sequences and subsequently reduce the number of FBCot clones that would need to be sequenced to attain a desired level of sequence complexity coverage.
CBCS may not provide information on small variations in individual members of repetitive DNA families; such information is important in the disambiguation and assembly of complete genomic sequences. This limitation of CBCS might be remedied by coupling it with various techniques designed to detect small variations in related sequences (i.e., DNA resequencing techniques; see Nickerson et al. 1997;Hacia 1999; Kurg et al. 2000; Xiao and Oefner 2001). Regardless, the ability to capture the sequence complexity of a higher organism with far less investment than is required by shotgun sequencing may greatly accelerate the timetable for genome-wide study of many of the world's biota.
METHODS
Plant Material
Sorghum bicolor (L.) Moench (breeding line BTx623) DNA was used for Cot analysis, Cot library construction, and as a source of DNA in blotting experiments. For comparative purposes, Southern blots and colony blots containing DNA from Sorghum propinquum Kunth, a noncultivated sorghum species crossed with BTx623 to make a detailed genetic map (Bowers et al. 2000; A. Paterson and J. Bowers, in prep.) were probed with BTx623 DNA probes (see below).
Melting Curves and Cot Analysis
DNA isolation, preparation, and melting analyses were performed as described (Peterson et al. 1997, 1998). Cot analysis was performed according to Peterson et al. (1998) except that 0.5 M SPB was used to elute double-stranded DNA from HAP columns rather than 0.48 M SPB. A least squares analysis of the Cot data was performed using the computer program of Pearson et al. (1977).
Cloning of Cot Components
Highly repetitive (HR), moderately repetitive (MR), and single/low-copy (SL) DNA components of the Cot curve were prepared for cloning as outlined in Figure 5. The sections of the Cot curve used for cloning (i.e., roughly the two Cot decade regions flanking the Cot½ value of each component) are shown in Figure 1. DNA sample concentrations were determined using KOH-denaturation and spectrophotometry as described by Peterson et al. (1998).
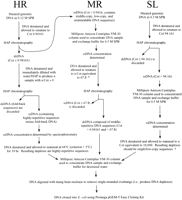
Overview of the steps involved in cloning HR, MR, and SL Cot components. DNA was denatured by heating samples in boiling water for 5–10 min. For samples in a particular sodium phosphate buffer (SPB), renaturation was allowed to occur at the criterion (Tm − 25°C) unless noted otherwise (see Britten et al. 1974 for details). Single-stranded DNA (ssDNA) and double-stranded (dsDNA) were separated using hydroxyapatite (HAP) chromatography. To attain the equivalent of a specific Cot value when starting with an isolated Cot fraction, the desired Cot value should be multiplied by the fraction of the genome remaining single-stranded at the Cot value of the starting material (see Hood et al. 1975). This principle was employed once in the isolation of the MR component (*) and once in the SL component isolation (†): * From the Cot curve, 0.67 of the genome is single-stranded at a Cot of 0.94161. To achieve renaturation equivalent to Cot 67.8 with whole genomic DNA, the Cot >0.94161 DNA was renatured to a Cot of (67.8 × 0.67 =) 45.4.† From the Cot curve, 0.28 of the genome is single-stranded at a Cot of 94.161. To achieve renaturation equivalent to Cot 10,000 with whole genomic DNA, the Cot >94.161 DNA was renatured to a Cot of (10,000 × 0.28 =) 2800.
Isolated Cot components were digested with mung bean nuclease (Promega) to remove single-stranded DNA overhangs (see manufacturer's instructions), and the resulting blunt-ended molecules were cloned intoE. coli (JM109) using the Promega pGEM®-T Easy cloning kit (cat. no. A1380). The HRCot, MRCot, and SLCot libraries were plated onto selective media, and positive clones were transferred via sterile toothpicks into freezing medium in 96-well microtiter plates. In total, four plates of HRCot, five plates of MRCot, and six plates of SLCot clones were obtained. Cot libraries were replicated using a hand-held 96-pin replicator and stored at −80°C (seePeterson et al. 2000 for details). Each clone was named based upon the library, plate, row, and column in which it was found (e.g., HRCot3A10 = HRCot library, plate 3, row A, column 10).
Sequencing
Plasmids were isolated from Cot clones using an alkaline lysis method with modifications made for the 96-well plate format (Marra et al. 1997). Cycle sequencing reactions were performed using the BigDye Terminator Cycle Sequencing Kit Version 2 (Applied Biosystems, Foster City, CA) and an MJ Research (Watertown, PA) PTC-100 thermocycler. Finished cycle sequencing reactions were filtered through Sephadex filter plates (Krakowski et al. 1995) directly into Perkin-Elmer MicroAmp Optical 96-well reaction plates. Sequencing was performed using an ABI 3700 automated DNA Analyzer. ABI sequencer trace data was evaluated using the programs PHRED,CROSSMATCH, and PHRAP (see www.phrap.org for additional information). Only clones with a Ph/Pr value >16 over 300 continuous base pairs and insert sequences ≥50 bp in length were used in sequence analyses.
Sequence Analysis
The sequence of each Cot clone was compared to sequences in the GenBank Nr and EST databases (http://www.ncbi.nlm.nih.gov), and the SUCEST Sugarcane EST database (http://sucest.lbi.dcc.unicamp.br/en/) using standard BLAST (blastn) protocols (Altschul et al. 1997). Based on the nature of the hits (if any), each Cot clone insert sequence was placed into a single descriptive “BLASTcategory” according to the scheme shown in Figure 2.
The Retrosor-6 sequence (bases 127,895–135,271 of GenBankAF061282) was compared to data in the GenBank Nr database using standard blastn (nucleotide query – nucleotide database) and blastx (nucleotide query – protein database) programs (Altschul et al. 1997).
Southern Blots
Southern blots containing S. bicolor and S. propinquum DNA were prepared and probed as described by Chittenden et al. (1994). For simple determination of hybridization intensity, 15 clones from each Cot library were randomly selected as sources of probes. Clone inserts were preferentially amplified by PCR and labeled with 32P-dCTP using nick translation. Each blot was hybridized with 1.8 ng/mL (= 20 μCi/mL) of radiolabeled probe DNA in hybridization buffer for 16 h at 65°C. Excess solution was drained from blots, and blots were given three successive 20 min washes (65°C) in 0.25× SSPE (aqueous 0.75 M NaCl, 50 mM NaH2P04·H20, 6.3 mM EDTA, pH 7.4) containing 0.25% SDS (1.0 L per wash with agitation). Membranes were blotted dry with paper towels and wrapped in plastic wrap. A Geiger-Muller counter was used to measure the relative amount of hybridization (cpm) of each probe to its corresponding blot.
After sequence analysis, one of the S. bicolor/S. propinquum Southern blots was probed with radiolabeled insert from a Cot clone with substantial sequence identity to Retrosor-6(HRCot3E04). Hybridization conditions were identical to those described above. An autoradiogram of the blot was obtained using standard protocols.
Colony Blotting
High-density grids containing 18,432 double-spotted clones were prepared from the S. bicolor BAC library BTx623 (D. Begum, unpubl.) and the S. propinquum library SP/YRL (Lin et al. 1999) as described by Choi and Wing (1999). For each BAC library, two identical BAC grids (i.e., two grids containing the same clones in the same order) were selected for analysis. One S. bicolor grid (SB1) and one S. propinquum grid (SP1) were each probed with part of the long terminal repeat (LTR) sequence (clone MRCot2B04) of Retrosor-6, whereas the duplicate filters (SB2 and SP2) were probed with a sequence found in the central region ofRetrosor-6 (clone HRCot3C12) (Choi and Wing 1999). Autoradiogram images were digitally captured using an Alpha Innotech (San Leandro, CA) AlphaImager 2200 image capture/analysis system. The two SB images were aligned, superimposed, and compared using Adobe Photoshop 6.0. SP images were likewise compared and analyzed.
To estimate the Retrosor-6 copy number in the genomes ofS. bicolor and S. propinquum, the AlphaImagerSpot Densitometry application (AlphaImager 2200 v. 5.1) was used to analyze one section (i.e., one-sixth) of BAC grid SB1 and one section of grid SP1 (see Fig. 4E). For a section, a region within the section containing no visible probe hybridization was selected and set as “background.” The “Integrated Density Value” [IDV = Σ (each pixel value − background)] for the entire section was then determined. Because BAC clones are double-spotted on grids, the IDV of the section was divided by two to yield the “Section IDV.” Using a circular sampling tool with a fixed diameter slightly smaller than a clone, IDV readings were taken for 50 different clones ranging from the lowest detectable hybridization signal to the highest hybridization intensity (Fig. 4E). Clones were selected from all areas of a grid section. The mean density value of the five clones with the lowest IDVs (LowIDV) and the mean value of the five clones with the highest IDVs (HighIDV) were determined. For both S. bicolor and S. propinquum, comparison of the LowIDV and HighIDV indicate an approximately fourfold difference in clone hybridization intensity. It was assumed that the LowIDV represents clones with one copy ofRetrosor-6, and therefore inferred that the HighIDV represents clones with four copies of Retrosor-6. To determine the mean number of clones per section, the SectionIDV was divided by the LowIDV. The resulting value was used to estimate the Retrosor-6 copy number per genome and the percentage of the genome composed ofRetrosor-6 DNA (see Table B in the online supplement to this article, www.genome.org).
Comparison of Cot Clones With Sorghum Molecular Markers and BAC End Sequences
Cot clone sequences were compared to roughly 1500 molecular markers (see the section Molecular Markers in the online supplement for GenBank accession numbers) on the sorghum molecular genetic map using standard BLAST (blastn) procedures (Altschul et al. 1997). The chromosomal positions of Cot clones containing sequences with high sequence similarity to molecular genetic markers (S' ≥76.28) are shown in Table C of the online supplement. BAC end sequences (n = 116) obtained from H.-M. Ma wereBLASTed against the GenBank dbGSS database (which contains the sorghum Cot clone sequences). Significant hits to Cot clones (S' ≥76.28) were noted.
WEBSITE REFERENCES
http://sucest.lbi.dcc.unicamp.br/en/; SUCEST: The Sugarcane EST Project.
http://www.genome.org; Genome Research website.
http://www.ncbi.nlm.nih.gov; National Center for Biotechnology Information (home of GenBank).
http://www.phrap.org; The Phred/Phrap/Consed System home page.
Acknowledgments
We thank Glenn Galau, William Pearson, and Stephen Stack for advice. This project was supported in part by USDA-NRICGP award 99–35300–7819 to D.G.P.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵3 These authors contributed equally to the research. They are listed in alphabetical order.
-
↵4 Corresponding author.
-
E-MAIL dgp{at}arches.uga.edu; FAX (706) 583-0160.
-
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.226102. Article published online before print in April 2002.
-
- Received November 28, 2001.
- Accepted March 6, 2002.
- Cold Spring Harbor Laboratory Press








