
Looking at Old Tools in New Ways: Using Knockouts as Congenics to Study QTLs
A Novel Source of Congenics
It is safe to say that no one has ever entirely removed the flanking genes from an introgressed region by simple backcrossing — even 100 generations yields only an average introgressed segment length of 2 cM (Silver 1995). The problem of flanking regions has complicated analysis of introgressed genes for some time and is of particular concern for the analysis of complex traits using knockouts, (Gerlai 1996) so it is particularly satisfying to see this slightly vexing phenomenon turned to an advantage in QTL (Quantitative Trait Locus) analysis.
In this issue, Bolivar et al. (2001) take advantage of the fact that knockouts are typically generated using ES embryonic stem cells derived from 129 inbred strain and are often backcrossed to C57Bl6 (B6) animals when archived. This convention provides a convenient and growing source of available animals with a small region of 129 chromosome on an otherwise B6 background. The method demonstrated by Bolivar et al. compares the phenotype of B6 animals with the phenotype of B6 animals containing the introgressed 129 region and knockout (B6.129-KO). If there is a difference, the result is followed up with a simple two-part breeding scheme, described in the paper, that separates the effects of the knockout from the effects of the flanking region.
Coverage of the Genome in 129xB6 with Commercially Available Knockouts
One of the most intriguing aspects of the technique developed by Bolivar et al. is the commercial availability of a large number of B6.129-KO strains. Table 1gives names and chromosomal positions for 59 potentially useful knockout/congenics sold by the Jackson Laboratory. At least one knockout exists for every chromosome except 19.
B6.129-KO Strains Currently Commercially Available from the Jackson Laboratory (2001)
The average coverage of the genome implied by these animals depends on the number of backcross generations. We can, however, determine a range of possible coverage. The overly-conservative assumption that animals have all reached N20 (10 cM average flanking segment) gives a coverage of 400 cM (29%, assuming a 1500 cM genome), whereas assuming N10 (20cM average flanking segment) gives a coverage of 660 cM (44%). Figure 1 shows average coverage of the genome given the latter assumption. In either case, the easy availability of congenics to B6 that cover a substantial portion of the genome is quite intriguing.
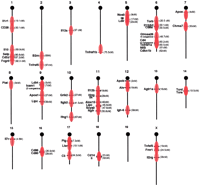
Genome coverage using B6.129-KO strains available from The Jackson Laboratory. Coverage of the genome is shown assuming 20cM introgressed segments (the average segment size for an N10 backcross). Actual segment size will differ considerably from 20cM; thus, lack of coverage in the diagram does not necessarily imply the absence of an introgressed segment. Heavier shading reflects the likely presence of multiple segments at that location due to the existence of multiple strains.
B6 x Other Strain Crosses
As Williams et al. (2001) note in their recent generation of consensus maps for the AXB, BXA, BXD, BXH, and CXB recombinant inbred (RI) sets, it will sometimes be the case that one strain (in this case B6) is a common progenitor in a number of crosses. If this progenitor differs phenotypically from the other strains in the crosses, it is quite possible to combine RI sets for a more powerful analysis. Williams et al. apply this reasoning to recombinant inbred (RI) mapping, but it applies as readily to the use of congenics. For example, if the B6 phenotype differs considerably from that of 129 and DBA, it seems reasonable to use the B6.129-KO flanking regions as congenics in further examination of QTLs initially identified in a B6xDBA cross.
It would not be cautious to depend on the B6.129-KO congenic for definitive confirmation of the original QTL's presence or absence, however, because there are numerous ways in which the 129 segment might yield unexpected results. (In the example cross, the phenotypic difference between B6 and 129 might result from loci entirely unrelated to the loci that differ between B6 and DBA.) However, there is also reason to suspect that examination of the 129 segment would yield a difference in behavior if the other strain also yields such a difference.
For example, in the region examined by Bolivar et al., (2001)emotionality-related QTLs had already been defined via crosses between B6 and Balb/cJ (Flint et al. 1995), C3H/HeJ (Caldarone et al. 1997), A/J (Gershenfeld et al. 1999), and DBA/2J (Owen et al. 1997; Wehner et al. 1997). Parsimony suggests that there is a particular B6 allele in the region that generates a difference in these related phenotypes. Even if some of the crosses are not detecting the same underlying genes, however, this is hardly a worst-case scenario. If there is a difference in phenotype between B6 and the B6.129-KO strain, examination of the 129 segment will still yield a well-mapped QTL.
Using B6.129-KO Animals in a Mapping Strategy
Assuming no overlap and knockout mice backcrossed to approximately N10 (20 cM average flanking intervals) it would take ∼75 strains to cover the entire genome. Realistically, of course, overlapping segments would increase this number considerably. It is also necessary to type multiple animals from each strain to detect subtle effects; Bolivar and coworkers do so comfortably with 12 animals of each strain. As a result of these two requirements, the number of animals required in order to use this method for a full genome scan rapidly increases beyond the 300–600 animals necessary for a moderately sensitive intercross- or backcross-based scan.
On the other hand, analysis of congenics provides a very definite position for the QTL and a very clear path to narrowing the QTL interval. Because only one locus differs between populations, analysis of congenics can also be very powerful for loci that operate independently of other loci involved in the phenotype, allowing identification and clear positioning of loci with very small effect sizes at any desired level of significance.
Regardless, the genome coverage from this set of congenics, although substantial, is not sufficient to eliminate the need for preliminary mapping using RI, F2, or N2 populations. In combination with some of the congenic and consomic panels described below, however, it might be possible to eliminate the initial mapping step in favor of congenic/consomic analysis. This would, however, have the significant drawback of eliminating detection of loci that act synthetically and would also eliminate information about epistasis and interactions with other loci. B6.129-KO animals will, however, be very useful in investigating particular regions of the genome nominated using other methods.
Additional Congenic Panels
The ability to use B6.129-KO animals as congenics is an extremely valuable development, even more so because the animals can be purchased easily. Several other notable panels have been generated over the past few years, however, as researchers have come to appreciate the potential use of congenic and consomic panels in QTL mapping, confirmation, and fine mapping. Panels that complement the B6.129-KO animals include the GTM (genome-tagged mice or congenic) animals developed by Iakoubova et al. (2001) and the CSS (chromosome substitution strain or consomic strain) animals developed by Nadeau et al. (2000b).
The GTM animals consist of two sets of 60 or more congenics. Each set spans essentially the entire genome with segments ranging between 20 and 40 cM. The sets consist of either DBA/2 or CAST/Ei segments introgressed onto a B6 background (B6.D2 or B6.CAST). The CSS animals, on the other hand, consist of introgressions of full chromosomes from one strain onto a different strain background. As of March 2000, Nadeau et al. reported the near completion of a B6 host strain with introgressed A/J donor chromosomes (B6.A) and further noted that they had begun construction of a A.B6, B6.129 and 129.B6 CSS lines.
Additionally, individual investigators have developed their own congenics for use in QTL and other investigations (for review of alcohol and drug-related QTL, see Bennet 2000). These congenics might prove useful to investigators interested in other traits as well. The existence of multiple sets of congenic and consomic lines greatly increases the chances of finding one or more congenic regions that can be used to further examine interesting phenotypes.
The Final Centimorgan
Identification of new QTLs or confirmation of existing QTLs using B6.129-KO congenics (or most other congenics discussed here) is unlikely to immediately reduce the mapping interval to the 1–2 cM range needed for fine mapping, because the congenics themselves average 10–20 cM. However, the use of this resource has the potential to considerably accelerate some fine mapping strategies in the 129xB6 and phenotypically similar backgrounds. It allows rapid confirmation of the QTL at high confidence levels and eliminates the need for multiple generations of backcrossing to generate an initial congenic. This, in turn, allows more time and resources to be devoted to narrowing the QTL interval.
Narrowing the interval can be accomplished by a variety of methods, including generation of congenics, typing of Advanced Intercross lines (AIL; Darvasi and Soller 1995), Recombinant Inbred Segregation Testing (RIST; Darvasi 1998), or by generating a much larger intercross or backcross. Each of these methods has its advantages and drawbacks, covered more fully in Darvasi (1998). For instance, AIL are very time-consuming to generate and unstable while RIST is relatively untried and requires RI strains. Congenic-based approaches such as ISCS (Darvasi 1998) are not a perfect answer either, but the availability of a variety of congenics certainly accelerates the process of confirmation and mapping and eliminates one of the major drawbacks of these approaches.
The challenge of sufficiently reducing the interval of a QTL has recently brought the future prospects for QTL analysis into question. Some researchers (e.g., Nadeau and Frankel 2000a) are doubtful that QTL cloning will become as routine and straightforward as broad QTL mapping has become in recent years. Others (e.g., Belknap et al. 2001) see the state of fine mapping today as analogous to the state of broad QTL mapping a decade ago and are confident that the tools and techniques necessary to go from phenotype to gene will soon be in place.
The virtual development of a large number of useful congenics by finding a new way to look at knockout strains distinctly highlights the importance of considering new breeding strategies in QTL analysis. Ultimately, I believe it will take a combination of novel viewpoints such as this one, increasingly fast automated genotyping technologies, (Buetow et al. 2001; Ranade 2001) and sequencing of many of the relevant strains (progress discussed in Marshall 2000) to uncover the sequence variations that inform complex traits. This is a considerable wish list, but enough of the pieces are already in progress that I am cautiously optimistic that it will be possible to routinely overcome these obstacles in future years.
Footnotes
-
EMAIL jlpeirce{at}princeton.edu; FAX (609) 258-0975.
-
Article and publication are athttp://www.genome.org/cgi/doi/10.1101/gr.206201.
- Cold Spring Harbor Laboratory Press








