
Genome Evolution at the Genus Level: Comparison of Three Complete Genomes of Hyperthermophilic Archaea
Abstract
We have compared three complete genomes of closely related hyperthermophilic species of Archaea belonging to thePyrococcus genus: Pyrococcus abyssi, Pyrococcus horikoshii, and Pyrococcus furiosus. At the genomic level, the comparison reveals a differential conservation among four regions of the Pyrococcus chromosomes correlated with the location of genetic elements mediating DNA reorganization. This discloses the relative contribution of the major mechanisms that promote genomic plasticity in these Archaea, namely rearrangements linked to the replication terminus, insertion sequence-mediated recombinations, and DNA integration within tRNA genes. The combination of these mechanisms leads to a high level of genomic plasticity in these hyperthermophilic Archaea, at least comparable to the plasticity observed between closely related bacteria. At the proteomic level, the comparison of the threePyrococcus species sheds light on specific selection pressures acting both on their coding capacities and evolutionary rates. Indeed, thanks to two independent methods, the “reciprocal best hits“ approach and a new distance ratio analysis, we detect the false orthology relationships within the Pyrococcus lineage. This reveals a high amount of differential gains and losses of genes since the divergence of the three closely related species. The resulting polymorphism is probably linked to an adaptation of these free-living organisms to differential environmental constraints. As a corollary, we delineate the set of orthologous genes shared by the three species, that is, the genes that may characterize the Pyrococcus genus. In this conserved core, the amino acid substitution rate is equal between P. abyssi and P. horikoshii for most of their shared proteins, even for fast-evolving ones. In contrast, strong discrepancies exist among the substitution rates observed in P. furiosus relative to the two other species, which is in disagreement with the molecular clock hypothesis.
The complete genome projects span the major branches of the archaeal and eubacterial phylogenetic trees and many eukaryotic genomes will be soon available. This genomic revolution has provided a considerable amount of data and enables comparative studies between distant organisms at the comprehensive and integrative level of genomes. They have revealed a remarkable genomic plasticity because dynamic rearrangements occurred so frequently, not only at large evolutionary distances but also between related species such asEscherichia coli and Haemophilus influenza, that gene order conservation is restricted to a few operons (Koonin and Galperin 1997; Siefert et al. 1997; Smith et al. 1997; Watanabe et al. 1997; de Rosa and Labedan 1998). Genomic comparisons have also highlighted the high variability of gene content, leading to a very small set of universal proteins mainly restricted to informational families (proteins involved in replication, transcription, and translation) (Huynen and Bork 1998; Kyrpides et al. 1999; Koonin et al. 2000). This underlines the considerable plasticity in biochemical pathways, with several solutions being independently invented in the course of evolution to achieve essential functions. Even at smaller evolutionary intervals, many individual genes show tree topologies in fundamental disagreement with the organismal phylogeny. Such mosaic phylogenies have revealed the unforeseen importance of lineage-specific losses and acquisition by horizontal transfer in the course of evolution. The relative extent of horizontal gene transfer versus lineage-specific gene losses has been hotly debated, in particular between Eubacteria and Archaea (Aravind et al. 1998; Kyrpides and Olsen 1999). The importance of DNA exchange between very distant prokaryotes belonging to distinct domains questions the commonly accepted scenario of the emergence of life and the universal phylogenetic tree (Koonin et al. 1997; Gupta 1998; Forterre and Philippe 1999). Nevertheless, all of these studies have emphasized that identification of truly orthologous relationships between genomes is a prerequisite to performing confident comparative genomic analysis. In fact, orthologous genes evolved from a common ancestral gene by speciation, whereas paralogous genes resulted from a duplication event (Fitch 1970). However, intricate relationships are hidden behind these definitions and the identification of true orthologs is not trivial in practice.
As a consequence, the interpretation of genomic comparisons between distant lineages is a challenging task. Comparisons of closely related species constitute a complementary approach crucial to the understanding of the forces at work in genome evolution. At the genomic level, they provide a unique opportunity to understand the mechanisms that determine chromosomal organization and evolution. At the proteomic level, this is a powerful strategy to assess the genuine extent of gene losses and gains that lead to the observed divergence of coding capacity. Until now, within- and between-species comparisons have been performed only on pathogenic Eubacteria (Himmelreich et al. 1997;Herrmann and Reiner 1998; Alm et al. 1999; Kalman et al. 1999; Read et al. 2000). They provided new insights into molecular evolution at the genome scale in Eubacteria and permitted the correlation of specific genes with phenotypic properties. In contrast, little is known about the evolution of closely related archaeal genomes, which are of particular interest because they show an eubacterial form with an eukaryotic content (Keeling et al. 1994). Indeed, most of the proteins involved in cell division or in metabolic pathways are eubacterial-like, whereas the informational genes are eukaryotic-like (Brown and Doolittle 1997; Koonin and Galperin 1997; Doolittle and Logsdon 1998).
Here we present the detailed genome-scale comparison of three closely related species of free-living Archaea: Pyrococcus abyssi, Pyrococcus horikoshii (Kawarabayasi et al. 1998), andPyrococcus furiosus (Maeder et al. 1999). This was made possible by the recent sequencing of P. abyssi whose annotations and phylogenetic relationships with nonpyrococcal species will be discussed elsewhere (O. Poch, in prep.). The three species are hyperthermophilic Euryarchaea belonging to the Thermococcales order (Fiala and Stetter 1986; Erauso et al. 1993; Gonzalez et al. 1998). We have compared these three genomes at different levels: chromosomal organization, evolutionary distances, and gene content.
RESULTS
General Features of the Pyrococcus Genomes
The P. abyssi sequence consists of a 1,765,118-bp chromosome (44.7% GC) and a 3444-bp multicopy plasmid (Erauso et al. 1996). In the chromosomal sequence, 1765 open reading frames (ORFs) were identified and annotated with the integrated GScope program (R. Ripp, in prep.). Biological roles were assigned to 51% of them (14% are informational proteins and 37% operational ones). Several genome features of P. abyssi (http://www.genoscope.cns.fr/Pab/),P. horikoshii (Kawarabayasi et al. 1998;http://www.bio.nite.go.jp/ot3db_index.html), and P. furiosus(Maeder et al. 1999; http://www.genome.utah.edu/sequence.html;http://www.ornl.gov/hgmis/publicat/99santa/157.html) affirmed the close relationship between the three species, including similar GC content and RNA elements in the three species (Table1). Two types of long clusters of tandem repeats (LCTRs) are common to the three genomes. Eight inteins are located at the same insertion site in the three Pyrococcus, reflecting a strong conservation of these mobile genetic elements.P. furiosus differs from the two others by a larger genome size and the presence of insertion sequences (ISs). The P. furiosus genome also shows a significantly larger amount of paralogous proteins. These differences are in agreement with the ribosomal RNA phylogenetic analyses (Gonzalez et al. 1998) indicating that P. abyssi and P. horikoshii have diverged after the speciation of P. furiosus. Among the paralogous proteins encoded by all the three Pyrococcus genomes, we identified a new extended family with a rare ATP-binding motif (GxRRxGK[S,T]). The multiple alignment analysis of these proteins (data not shown) leads us to divide them into two subfamilies and reveals that several of these proteins show authentic frameshifts in the three species. Counterparts were found in Archaea (Thermococcus sp., Methanococcus jannaschii, Methanobacterium thermoautotrophicum, Sulfolobus solfataricus) and in the hyperthermophilic bacterium, Thermotoga maritima, but no duplication is observed in these species.
General Comparative Features of thePyrococcus Genomes
Genome Comparison of the Three Pyrococcus Species
Pairwise comparison of the three genomes reveals a higher nucleotide conservation between P. abyssi and P. horikoshii(1122 kb in common) than between P. abyssi and P. furiosus (847 kb) or between P. horikoshii and P. furiosus (898 kb). Analysis at 2-kb resolution of inversion and/or transposition events permits the delineation of major collinear segments between each pair of genomes (Fig.1A). The preserved segments between P. abyssi and P. horikoshii are longer (up to 300 kb) than those observed between P. furiosus and either of these two species. This indicates a large amount of chromosomal rearrangements since the divergence of P. furiosus from the common ancestor of P. abyssi and P. horikoshii. Nevertheless, even between these two latter close species, 17 major inversions or transpositions are observed.
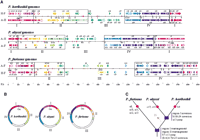
Comparison of the chromosomal organization of the threePyrococcus genomes. (A) Each genome is represented by three lines. The medium horizontal black line symbolizes the genome. A black vertical line represents the replication origin (Ori). The black vertical ticks indicate the positions of the tRNA genes; orange and red vertical ticks represent the insertion sequences, and the genes encoding the Pyrococcus-specific ATP-binding proteins, respectively. The upper and lower lines of colored arrows illustrate the oppositions of colinear segments between the considered genome and each of the other two genomes as obtained by three pairwise nucleotide comparisons (see Methods). A-H is the comparison between P. abyssi and P. horikoshii(arrows labeled from a1 to a17). A-F is the comparison between P. abyssi and P. furiosus (labeled from b1 to b25). H-F is the comparison between P. horikoshii and P. furiosus(labeled from c1 to c27). The arrows have been labeled arbitrarily, oriented, and colored according to the P. abyssi genome (arrows a1–a17 and arrows b1–b25), and the P. horikoshiigenome (arrows c1–c27). For representation convenience, only segments >10 kb are plotted. White boxes inside arrows specify indel areas >2 kb. The P. abyssi genome has been reverse complemented to facilitate the analysis. Only the tRNAs located at the boundaries (2 kb resolution) of colinear segments or indels areas are labeled according to the amino acid one-letter code. The regions of differential conservation (I, II, III, and IV) are delimitated by black boxes. The scale is indicated at the bottom of the figure. (B) Circular representation of the three chromosomes showing the four regions (I, II, III, and IV) according to the colors defined above and the origin of replication Ori. (C) Schematic diagram of the phylogenetic relationships among the three Pyrococcus species. Red circles illustrate the absence of segments (named on the side) in one of the three species. Blue boxes represent major events that occurred withinPyrococcus lineage (see text). The arrow labeled IS symbolizes the putative loss of IS in the common ancestor of P. abyssiand P. horikoshii.
Four main regions are distinguished in the Pyrococcus genomes according to their conservation pattern (regions I–IV in Fig. 1A,B), and excluding the b7/c13 and b12/c19 transpositions, no DNA fragment exchange occurred between these regions. The region I, containing the replication origin (Myllykallio et al. 2000), and the region IV, containing the ribosomal operon, are the most conserved in terms of gene organization and content even though the synteny is less well preserved in region I (Fig. 1A,B). In region II, the gene order and content are roughly maintained between P. abyssi and P. horikoshii, whereas numerous rearrangements occur in the third species. Region III, containing the termination origin, is a hotspot of translocation and indel (insertion-deletion) events in the threePyrococcus genomes and is significantly larger in P. furiosus. Comparing the P. abyssi and P. horikoshii genomes, the most remarkable event is the inversion of region I across the origin (Fig. 1B). Inversions of the region containing the replication or termination origins have been widely established (Segall et al. 1988; Mahan and Roth 1991). To determine in which of the two species this inversion occurred, we precisely compared the orientation of region I segments in the three genomes. However, there are so many disruptions of segment order in the P. furiosus region I that no clear answer could be provided.
An in-depth analysis has allowed us to highlight numerous additional rearrangements. This analysis takes advantage of the close relationship among the three species to deduce recombination scenarios according to the parsimony hypothesis. Numerous chromosomal features common toP. abyssi and P. horikoshii are different in P. furiosus, inferring that major events occurred before the divergence of P. abyssi and P. horikoshii (Fig. 1C). Besides the extensive rearrangements in regions I and II mentioned above, we notice the segment b7/c13 transposition between regions II and III, the large DNA inversion in region IV (segment (b16-b17)/c23), and the absence of numerous segments in the plasticity zone (a8, a10, a11, a12, and a13). All these rearrangements and indels must have contributed to the specificity of the P. furiosus evolution. We can also infer some events that occurred during or after the speciation of P. abyssi and P. horikoshii. In addition to the region I inversion, we notice the segment a16/b18 inversion in region IV and the segments a11 and a13 translocation in region III. In this latter region, namely the zone of plasticity, the segments c15 and c17 common to P. horikoshii and P. furiosus are absent in the P. abyssi genome, suggesting loss of these large collinear regions (35 kb and 11 kb, respectively inP. horikoshii) in P. abyssi. Similarly, the segment b9 present only in P. abyssi and P. furiosus is likely to have been lost in P. horikoshii since the divergence of P. abyssi and P. horikoshii (Fig. 1C).
We then looked for dispersed genetic elements that may promote the observed intergenomic disruption synteny. Putative targets for homologous recombination in Archaea are LCTRs detected previously in several archaeal chromosomes (Charlebois et al. 1998). Such LCTRs are indeed present in the three Pyrococcus genomes, but we found no clear correlation between their location and segment boundaries. In contrast, a close inspection of pairwise comparison results reveals that 15 of the 46 tRNA genes of P. horikoshii (nine in P. abyssi and eight in P. furiosus) are located exactly at the segment extremities, and 14 tRNA genes in P. horikoshii(16 in P. abyssi and 12 in P. furiosus) define the boundaries of indel areas (Fig 1A). This suggests that tRNAs may represent favorite targets for recombination and indel events within the Pyrococcus lineage. We find two P. horikoshii-specific regions that support directly the hypothesis of DNA integration within tRNA genes. These two regions (4 kb and 21.6 kb, respectively) are flanked by a perfect direct repeat (45 bp and 48 bp, respectively) that is strictly identical to the 3′ end of tRNAVal and tRNAAla genes, respectively. In each case, one of the repeats constitutes the 3′ end of the tRNA gene, and the embedded region contains a gene encoding a protein (PHO1864 and PHO1200, respectively), weakly similar to the Sulfolobusviruslike particle, SSV1-encoded integrase, which has been shown to mediate the integration of the SSV1 virus within a tRNAArggene of its host, the Crenarchaea, Sulfolobus shibatae (Reiter et al. 1989; Palm et al. 1991; Muskhelishvili et al. 1993). The consequences of DNA integration within tRNA genes on the chromosomal organization is well exemplified in region IV whose overall synteny allows us to deduce a recombination scenario linked unambiguously to tRNAAsp, tRNAVal, and tRNASer. Regardless of the real chronology, if we start from the P. furiosusgenome, we observe that a recombination event occurred between the tRNASer and tRNAAsp, leading to the inversion of the c23 segment in the P. horikoshii genome. Another recombination event involving the tRNAAsp and tRNAVal, located at the two extremities of the a16/c24 segment, leads to the inversion of segment a16 in the P. abyssi genome. The two tRNAAsp and tRNAValtherefore are separated only by 1051 bp.
On the other hand, when examining the genome of P. furiosus, we detected some correlation between the disruption pattern and the 24 transposons-associated, IS-like elements. Their distribution is clearly nonrandom because ISs are highly concentrated in regions II and III, which are the most shuffled parts of the P. furiosus genome. Furthermore, nine are located on collinear segment boundaries, eight are found within large indel regions, and five are in completely scrambled regions. These elements, which promote rearrangements by direct transposition or homologous recombination, are not found inP. horikoshii and P. abyssi genomes. This suggests an invasion of the P. furiosus genome by IS, which may participate to the differentiation of the P. furiosus genome, or by a loss of IS by the P. abyssi and P. horikoshiicommon ancestor, leading to the conservation of larger segments observed between these two species. The existence of vestigial IS in both P. abyssi (position 761 435 to 761 646) and P. horikoshii (368 535 to 368 779) genomes strongly supports the latter hypothesis.
Finally, our analysis of dispersed genetic elements has highlighted an intriguing feature concerning the location of the numerous genes that contain an atypical ATP-binding motif belonging to thePyrococcus family mentioned above. These genes are always positioned in indel areas and are, with one exception, all located within regions II and III (Fig. 1A).
Proteome Comparison
The number of predicted ORFs is quite different among the threePyrococcus species, especially when considering P. abyssi and P. horikoshii (1765 and 2061 ORFs, respectively) whose genome sizes are comparable (Table 1). These differences may be linked to both discrepancies of annotation and to the variable number of ORFs with no identifiable homolog in the databases (Fig. 2). Therefore, in the subsequent analysis, we consider only those ORFs with at least one homolog (1723, 1651, and 1947 ORFs in P. abyssi, P. horikoshii, and P. furiosus, respectively). The average amino acid identity between the closest homologs of P. abyssiand P. horikoshii is 77%. This average amino acid identity is lower between P. furiosus and P. abyssi counterparts (72%) and between P. furiosus and P. horikoshiicounterparts (73%). This confirms the closer relationship betweenP. abyssi and P. horikoshii relative to P. furiosus. As observed previously in proteomic comparisons (Kalman et al. 1999), proteins of different functional classes have evolved at different rates after divergence of the three species. In particular, the hypothetical and operational proteins show, on average, higher substitution rates than informational ones.
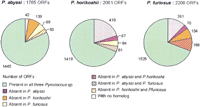
Homology relationship distribution within the Pyrococcusgenus. The figure shows, for each species, the fractions of genes common to the three Pyrococcus, shared by twoPyrococcus, unique to one Pyrococcus, and with no homolog in the current databases.
Three Species' Differential Gains or Losses of Genes
To establish the list of genes which, unambiguously, are not shared by the three Pyrococcus species, we chose a low percent identity cutoff (20%) based on multiple alignment of complete sequences (see Methods). These genes represent differential losses or gains of functions within the Pyrococcus lineage, regardless of nonorthologous gene displacement (Koonin et al. 1996). The fraction of genes absent in at least one genome is relatively important (278 genes in P. abyssi, 232 in P. horikoshii, and 422 inP. furiosus) and reveals extensive differential gains or losses among the Pyrococcus species (Fig. 2). This fraction is extended particularly in P. furiosus and includes 198 genes unique to P. furiosus, which is in agreement with its larger genome size. The number of genes common to P. abyssi andP. furiosus, but absent in P. horikoshii (139 inP. abyssi and 154 in P. furiosus), is very high compared to the fraction of genes shared by P. horikoshii andP. furiosus, but missing in P. abyssi (67 in P. horikoshii and 70 in P. furiosus) (Fig. 2). This suggests important losses in the P. horikoshii genome after the divergence of P. abyssi and P. horikoshii from their common ancestor. In term of functions, some differential losses or gains of well-characterized operons have been reported previously between P. furiosus and P. horikoshii (Maeder et al. 1999). With the exception of the his operon, all the amino acid biosynthetic operons of P. furiosus missing in P. horikoshii are present in P. abyssi (Table2). The maltose and phosphate operons are also shared by P. abyssi and P. furiosus, but absent in P. horikoshii. This confirms a substantial loss of complete biosynthetic pathways in P. horikoshii. Concerning the chemotaxis-related genes reported to be absent in P. furiosus(Maeder et al. 1999), they are present in both P. abyssi andP. horikoshii. Compared to the two others, the P. abyssi genome contains three clustered eubacterial-like restriction/modification enzymes, which are located at the junction between regions I and IV.
Main Characterized Operons Involved in Differential Losses or Gains within Pyrococcus Lineage
Triangular Distance Relationships Within the Common Set
Excluding the genes without any homolog and those involved in differential losses or gains, we obtain for each species the set of common proteins. These common sets consist of 1445, 1419, and 1525 genes in P. abyssi, P. horikoshii, and P. furiosus, respectively (Fig. 2). The differences between these numbers reflect the variable extent of paralogous genes in each genome: 557 of 1445 (39%) in P. abyssi, 537 of 1419 (38%) in P. horikoshii, and 687 of 1525 (45%) in P. furiosus.
To obtain an overall understanding of the homologous relationships among the three compared proteomes, we use the multiple alignments of complete sequences to calculate the distances between each protein of a given genome and all counterpart(s) present in the alignment (see Methods). A pyrococcal gene trio is then defined as one gene from one pyrococcal genome and its two best homologs in the otherPyrococcus genomes. As proteins of different functional classes evolved at very different rates, we studied the distance ratios (see Methods) within each triangular homologous relationship rather than absolute distances. This provides a new tool to assess the relative substitution rates of proteins. The ΔHF/ΔAH and ΔAF/ΔAH ratio distributions have the same overall form with a majority of values >1 (Fig. 3A,B), confirming the proximity of P. abyssi and P. horikoshii relative to P. furiosus. Nevertheless, the two distributions show a large dispersion of values, reflecting either nonorthologous relationships or a strong difference in the evolution rate of the genes after the divergence of the three species. In contrast, the ΔAF/ΔHF ratios are, surprisingly, quite homogeneous and highly concentrated around 1 (Fig. 3C). This experimental observation shows the existence of anisoceles-triangular distance relationship for most of the proteins of the three Pyrococcusspecies. In other words, whatever the variability of a considered gene trio, the distance observed between P. abyssi and P. furiosus proteins is equal to the distance observed between P. horikoshii and P. furiosus proteins. This infers either a very recent divergence of P. abyssi and P. horikoshiiand/or an overall similar rate of evolution in these two species.
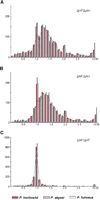
Distance ratios within trios of homologous genes from the threePyrococcus species. The figures represent the distributions of (A) ΔHF/ΔAH ratios, (B) ΔAF/ΔAH ratios, and (C) ΔAF/ΔHF ratios. Intervals include the upper bound ([0.45, 0.55] for instance), and the last interval of the distributions embrace all values >3.05. Histograms show distance ratio distributions for all trios of homologous genes and calculated independently in each species: P. horikoshii in red, P. abyssi in blue, and P. furiosus in yellow. The black curves represent the distance ratio distributions of the minimal calculated set of true orthologs common to the three species. Abbreviations are ΔAF for distance between P. abyssi andP. furiosus homologs, ΔAH for distance between P. abyssi and P. horikoshii homologs, and ΔHF for distance between P. horikoshii and P. furiosus homologs.
Orthologous and Nonorthologous Relationships
The analysis of triangular homology relationships has revealed that the common sets are probably composed of both orthologs and nonorthologs. Therefore, we tried to isolate nonorthologous trios of genes. To achieve this goal, we combined two independent approaches. The first approach is based on the commonly used reciprocal best hits method (Tatusov et al. 1997; Tekaia et al. 1999; Snel et al. 1999). The second method takes advantage of the isosceles triangular relationships existing among the Pyrococcus species and isolates all trios of genes showing a biased ΔAF/ΔHF ratio (see Methods). Such trios are likely to be composed of genes with false orthologous relationships or with an unusual evolution rate ratio between P. abyssi andP. horikoshii.
Table 3 shows the total number of trios with questionable orthology relationships detected independently in each Pyrococcus genome (228 in P. abyssi, 202 inP. horikoshii, and 297 in P. furiosus). The nonreciprocal relationships of homology are overrepresented in theP. furiosus genome. This is probably linked to the high number of paralogs in this species, which induces numerous one-to-many and many-to-many homologous relationships. The number of putative true orthologs shared by the three genomes has been calculated independently in each genome and is equivalent (1217, 1217, and 1228 genes in P. abyssi, P. horikoshii, and P. furiosus, respectively), corroborating the relevance of our analysis. The distribution of the distance ratios between these putative true orthologs are represented in Figure 3. ΔHF/ΔAH and ΔAF/ΔAH distributions still show a large dispersion of values, revealing that differences among distance ratios are not because of false orthology relationships, but should reflect substitution rate discrepancies between P. furiosus and the two other species.
Detection of Spurious Triangular Relationships in thePyrococcus Genomes
Some false orthologs have been identified by the two methods (55 inP. abyssi, 45 in P. horikoshii, and 54 in P. furiosus), highlighting the consistency of our combined approach in the detection of spurious orthology. Roughly 100 gene trios could only be detected by the existence of biased triangular relationship. A manual inspection of such trios shows that this new approach is powerful for distinguishing suspicious orthologs among both distant and close homologs. As an example, the asparagine synthetase of P. abyssi (PAB1605) is distantly related (phylogenetic distances of 73 and 71) to an asparagine synthetase of P. horikoshii and ofP. furiosus. Within this trio of genes, the ΔAF/ΔHF ratio is 2.37. Regarding the domain organization and the phylogenetic tree of this family (data not shown), PAB1605 is not orthologous to the other two, although reciprocal best hits were observed.
Spatial Clustering of the Predicted False Orthologs
Strikingly, the chromosomal localization of questionable orthologs is not random, because most of them (77% in P. abyssi and 81% in P. horikoshii and P. furiosus) are either clustered with each other or with genes missing in at least onePyrococcus genome (Table 4). In addition, when available, the functions of the proteins encoded by such clustered genes are frequently related and can concern metabolic pathways as well as processes linked to informational proteins. This is exemplified by seven clustered genes conserved in P. furiosusand P. abyssi genomes (PAB2176 to PAB0185) that are probably involved in glycerolipid metabolism. Among them, two false orthologs are detected in the P. horikoshii genome and five genes are clearly absent, suggesting that the entire cluster has been lost inP. horikoshii. This loss is probably associated with a recombination event, because the cluster is located precisely at a breakpoint. Similarly, we have identified a long cluster (>30 kb) of 21 genes in the P. abyssi genome (PAB1411–PAB1389). Among these genes, 16 show biased triangular relationships and five are absent in both P. furiosus and P. horikoshii genomes. The cluster includes lipopolysaccharid biosynthesis-related proteins, some dehydrogenases, and a hydrogenase operon composed of six proteins. The hydrogenase operon is eubacterial-like and closely related to the hydrogenase-4 of E. coli, suggesting that an operon gain has occurred in this plasticity zone after the speciation of P. abyssi. Finally, we noted another cluster conserved between P. horikoshii and P. furiosus. This cluster encompasses eight genes with biased triangular relationships: P. horikoshii andP. furiosus homologs are closer than P. horikoshiiand P. abyssi homologs. Most of these genes encode hypothetical proteins but two of them encode putative helicases.
Distribution of Genes with Spurious Orthology Relationship in the ThreePyrococcus Genomes
Thus, at the genomic level, the clustering of both false orthologs and genes involved in gains/losses events allowed us to extend plasticity regions previously limited to gene losses, but also to reveal hidden zones of plasticity. At the evolutionary level, these regions may correspond to (1) stretches of genes differentially lost or acquired since the divergence of the three species, or (2) sets of genes involved in related functions that have evolved under differential selection pressure.
DISCUSSION
Genomic Plasticity between Closely Related Archaea
Our comparative genomic analysis confirms the close proximity and evolutionary tree topology of the three Pyrococcus species deduced from ribosomal RNA phylogenetic studies (Gonzalez et al. 1998). At the comprehensive level of genomes, the shared evolutionary history of the three hyperthermophilic archaeons is reflected by the conservation of RNA elements, the similarity in GC contents, and the degree of sequence conservation. The phylogenetic proximity of the three Archaea is further attested by the existence of extended collinear segments between the genomes, that is, regions with a conserved gene order regardless of indel areas. The closer proximity ofP. abyssi and P. horikoshii is affirmed by their average amino acid identity (77%) and their chromosomal organization. Nevertheless, the evolutionary distance between P. abyssi andP. horikoshii is not negligible relative to P. furiosus because the average amino acid identities are also high between P. furiosus and the two other species (72% withP. abyssi and 73% with P. horikoshii).
Because our analysis is the first comparison between three complete genomes of Archaea at the genus level, we have no data on the chromosomal organization conservation at small evolutionary distances in this domain. In contrast, several within- and between-species comparisons of pathogenic Eubacteria are available. Given the discrepancies in methods and in genome size, the comparison of the relative genomic plasticity existing in the two domains is uncertain. Nevertheless, the rearrangements within the Pyrococcus lineage appear far more numerous than the six major breakpoints reported between the two intracellular parasitic Eubacteria, Mycoplasma pneumoniae and Mycoplama genitalium (Himmelreich et al. 1997; Herrmann and Reiner 1998). Regarding dot plots of gene similarities, the level of DNA reorganization observed betweenChamydia trachomatis and C. pneumoniae (Kalman et al. 1999; Read et al. 2000) is of the same range as that between P. abyssi and P. horikoshii. In contrast, the shuffling observed between P. furiosus and the two otherPyrococcus species is more important than in theChlamydia taxon. Thus, in the first approximation, archaeal species show at least as much genomic plasticity as eubacterial species, despite their eukaryoticlike replication and repair machinery (Brown and Doolittle 1997; DiRuggiero et al. 1999).
The conservation of gene order can be considered as an indicator of the genome evolution rate, and some studies have attempted to build phylogenies based on gene order (Hannenhalli et al. 1995; Sankoff and Blanchette 1999). Nevertheless, as reported previously, the relation between genome rearrangements and protein identity is not linear (Huynen and Bork 1998). In our analysis, the DNA shuffling observed between the P. furiosus genome and the two others clearly overestimates the divergence time of P. furiosus from the common ancestor of P. abyssi and P. horikoshii. More generally, when comparing the different studies performed at the genus level, the extent of chromosomal rearrangements between closely related species appears to be independent of sequence conservation. This is exemplified by the remarkably high level of amino acid conservation within the Pyrococcus lineage compared to the average amino acid identity (67%) within the Mycoplasma, whereas the overall synteny is more preserved in this latter taxon.
Mechanisms Involved in Chromosomal Reorganization in thePyrococcus Genus
The absence of linear correlation between the chromosomal rearrangement rate and the evolutionary distances raises the question of the mechanisms at work in the pyrococcal genomic plasticity. Our detailed genome-to-genome comparisons shed light on the existence of four regions in the Pyrococcus chromosomes and allows us to isolate some of the broad mechanisms that may shape the evolutionary dynamic of DNA in the Pyrococcus genus: site-specific integration within tRNA genes, rearrangement linked to replication arrest, and IS-mediated recombination.
In the three Pyrococcus genomes, the tRNA genes are frequently located at the boundaries of synteny blocks along the whole chromosome, strongly suggesting that site-specific recombinations within tRNA genes have occurred many times during the Pyrococcus divergent evolution and have concerned all the regions of the genomes. In P. horikoshii, two inserted fragments contain a predicted gene weakly homologous with an integrase involved in the integration of the SSV1 virus within a tRNAArg gene of its host, the Crenarchaea,Sulfolobus shibatae (Reiter et al. 1989; Palm et al. 1991;Muskhelishvili et al. 1993). This supports the existence of a common mode of DNA integration within tRNA genes between the Crenarchaea and the Euryarchaea. In Eubacteria, some tRNA genes can constitute the integration site of plasmids and phages (Reiter et al. 1989; Dupont et al. 1995) and may also be the target site of recombination of pathogenicity islands (Hou 1999). Thus, the involvement of tRNA genes in site-specific recombination appears as a widespread mechanism able to promote integration of various autonomous genetic elements. InPyrococcus, the origin of the acquired DNA remains mysterious because no virus has been isolated yet in these taxon.
Our analysis has stressed the predominant role of IS-mediated recombination and rearrangement linked to replication arrest in DNA shuffling within Pyrococcus genomes. The implication of these mechanisms has been reported previously in some Eubacteria and Archaea (Hackett et al. 1994; Louarn et al. 1994; Bierne et al. 1997;Myllykallio et al. 2000). Our genome-scale comparison gives us a unique opportunity to estimate their relative contribution in DNA reorganization. Indeed, the differential conservation pattern existing among the four regions of the Pyrococcus chromosomes is directly correlated with the location of the replication terminus and the differential presence of IS. The regions II and III are particularly shuffled in P. furiosus and are the main location of the IS unique to this species. In region III, which contains the replication terminus, the extensive shuffling observed in the P. furiosus genome may result from the cumulative effects of these two mechanisms. In contrast, in region II, the real impact of IS-mediated recombinations is clearly illustrated by the numerous chromosomal rearrangements in the P. furiosus genome, compared to the synteny observed between the two other species. In corollary, the effects of the rearrangements linked to replication arrest are observable directly in region III of P. abyssi and P. horikoshii genomes because there is no IS to disrupt the gene order. It is interesting to note that almost all the genes containing the Pyrococcus-specific ATP-binding motif are also concentrated in the same two regions. They are always located at the boundaries or in indel regions and thus could be involved in recombination events associated with deletion or insertion, but the precise mechanism remains to be elucidated.
These results raise the question of the quasi-exclusion of both these genes and the IS from regions I and IV. Region I contains the replication origin and the rRNA operon, whereas region IV contains the ribosomal protein operon. It is then tempting to speculate that stabilizing forces maintain a relative synteny in these regions to ensure an efficient expression of such informational genes that have crucial effects on the fitness of the cell. Similarly, the presence of vestigial IS in P. abyssi and P. horikoshii genomes suggests that IS was present in the common ancestor of the three species and was subsequently lost in the lineage leading to P. abyssi and P. horikoshii. Thus, the loss of IS may have resulted from a negative selection in the common ancestor of P. abyssi and P. horikoshii.
Testing the Molecular Clock Hypothesis at the Genome Scale
The extended genomic plasticity observed within thePyrococcus lineage raises the question of the evolution of proteomes in the three species in both terms of evolutionary rate and coding capacity. Our distance ratio analysis has revealed the existence of an isoceles-triangular relationship among most of the trios of homologous genes. In other words, for a given trio of genes, the distance between P. furiosus and P. abyssicounterparts is equal to the distance between P. furiosus andP. horikoshii homologs. This equality could reflect a negligible evolutionary distance between P. abyssi and P. horikoshii relative to P. furiosus, but the average identity observed between the three Pyrococcus genomes denies this hypothesis. Thus, the equality of distances may result from the equality of the amino acid substitution rates in P. abyssi and in P. horikoshii, even for fast-evolving proteins. In contrast, the distance between P. furiosus proteins and their orthologs in P. abyssi or in P. horikoshii is not proportional to the distance observed between P. abyssi andP. horikoshii orthologs (Fig. 3A,B). This reveals that the equality of amino acid substitution rate among P. furiosus and the two other lineages is not verified for all proteins. On small subsets of genes, several attempts to test the equality of evolutionary rates among two or several related species have shown that the results depend on the considered genes (Muse and Weir 1992; Takezaki et al. 1995; Akashi 1996; Robinson et al. 1998; Ballard 2000). At the genome scale, a recent analysis of the mitochondrial genomes of mammalian species has revealed that the global molecular clock was clearly violated for both the amino acid and nucleotide data (Yoder and Yang 2000). Another study among distant species suggests that a generalized version of the molecular clock hypothesis may be valid on the genome scale (Grishin et al. 2000). These conflicting results emphasize the importance of the evolutionary intervals considered, because a comparison between distant groups of species may omit the subtle but significant differences existing between closely related species. In our analysis, the molecular clock hypothesis is not verified by thePyrococcus lineage. Some of the P. furiosus genes may have evolved at an accelerated rate in response to specific selection pressure. This could be linked to environmental constraints becauseP. furiosus was isolated from a marine solfatara in the south of Italy (Fiala and Stetter 1986) whereas P. abyssi andP. horikoshii were found in hydrothermal vent sites in the Pacific Ocean (Erauso et al. 1993; Gonzalez et al. 1998).
Divergence of Gene Content within Pyrococcus Lineage
Gene content comparisons require the identification of shared orthologous genes. Given the complexity of homologous relationships, this constitutes a challenging task in comparative genomics (Tatusov et al. 1997; Koonin et al. 2000). A commonly used method relies on reciprocal best hits (Tatusov et al. 1997; Snel et al. 1999; Tekaia et al. 1999). This method is efficient in many cases but may be insufficient to detect differential losses of paralogs of an ancestral gene (Snel et al. 1999). We thus used a complementary approach that takes advantage of the isoceles triangular relationships existing among the Pyrococcus species. An atypical distance relationship may reflect nonorthologous relations. This assumption is greatly supported by the spatial clustering of the detected false-positive orthologs with each other or with genes missing in at least one Pyrococcus. In such clusters, genes are frequently implicated in related functions or in the same biosynthetic pathways. Thus, a meticulous analysis of nonorthologous relationships permits the delineation of extended zone of diversity and complete cascades of genes that have been acquired or lost since the divergence of the three species.
In a corollary, the obtained set of likely orthologous genes should correspond to the conserved core of the Pyrococcus genus, that is, to the vertically inherited and stable genes shared by the three species. This includes genes belonging to conserved archaeal families (Makarova et al. 1999; Graham et al. 2000), but alsoPyrococcus-specific genes of unknown function. Given the close proximity of the three species, the conserved core is surprisingly small because it represents roughly two-thirds of the proteomes of each species. This highlights the extreme polymorphism of the coding capacity in the Pyrococcus taxon and is likely to reflect an adaptation of the metabolism to specific environmental constraints.
At the functional level, even informational genes encoding restriction/modification enzymes and helicases are concerned by differential losses or gains. Numerous genes of P. furiosushave been reported previously to be absent in P. horikoshii(Maeder et al. 1999), including operons involved in maltose and trehalose transport, phosphate uptake, TCA cycle, and amino acid biosynthesis. Our analysis reveals that most of these operons are in fact present in P. abyssi. The same tendency is also observable at the entire proteome level because the fraction of genes shared only by P. abyssi and P. horikoshii is less important than the fraction of genes common only to P. abyssiand P. furiosus. This is unexpected given the phylogenetic relationships of the three species and suggests massive or numerous losses in the P. horikoshii lineage since the P. abyssi and P. horikoshii divergence. Genome phylogenies based on shared orthologs have been proposed recently and suggest that gene content carries a strong phylogenetic signature (Snel et al. 1999;Tekaia et al. 1999). Such phylogenies are powerful in the delineation of major lineages, but our analysis of three closely related species reveals that they could be sensitive to the frequency of independent gains and losses at small evolutionary intervals. This raises the question of the biological signification of the apparently random gains and losses. Until now, genome-scale comparisons of closely related species have been restricted to pathogenic Eubacteria (Himmelreich et al. 1997; Read et al. 2000). Because these Eubacteria are obligate intracellular parasites, differential losses can be interpreted as an adaptation to parasitic life. Such reductive evolution has been reported in Richettsia prowasecki, which shows a high fraction of noncoding DNA and of pseudogenes (Andersson and Andersson 1999). The three Pyrococcus are also engaged in extensive DNA traffic since their divergence, although they are free-living Archaea faced with extreme environmental conditions. Thus, losses and gains are not restricted to parasitic Eubacteria but may constitute a recurrent phenomenon in the evolution of prokaryotes even at small evolutionary intervals. Our study has revealed that the mechanisms promoting genomic plasticity are similar in the deeply-branched EuryarchaeaPyrococcus and in Eubacteria. In this context, the origin of the acquired DNA detected in these hyperthermophilic organisms (work in progress) would provide new insights intothe complex picture offered by genome sequences. More generally, future comparative analysis of closely related free-living organisms would undoubtedly enhance our understanding of the evolutionary forces that determine genomic plasticity.
METHODS
Complete Genome Sequences
The complete genome sequence of P. abyssi has been determined at Genoscope and annotated at the Structural Biology and Genomic Laboratory (IGBMC). Sequence and annotations are available athttp://www.genoscope.cns.fr/Pab/. The nucleotide sequence of the whole genome of P. abyssi was submitted to EMBL database under accession no. EMBL: AL096836. The P. abyssi genome sequence was analyzed and annotated using GScope (R. Ripp, in prep.), which is an integrated program written in Tcl/Tk, specially designed for the visualisation and analysis of prokaryotic genomes. TheP. horikoshii and P. furiosus complete genome sequences and predicted ORFs were retrieved athttp://www.bio.nite.go.jp/ot3db_index.html andhttp://www.genome.utah.edu/sequence.html, respectively.
Genome Comparison
To analyze the similarity and collinearity between the threePyroccoccus genomes, we performed pairwise BLASTNcomparisons (Altschul et al. 1997) among the three complete sequences. The P. abyssi sequence has been reverse-complemented before the analysis for representation convenience. BLASTNparameters were chosen to extend High-Scoring Pairs (HSPs): nucleotide mismatch penalty −1, no filter, gap opening penalty 6, and gap extension penalty 1. Only the HSPs longer than 2000 bp and with a percent identity >60 were considered in the subsequent analysis. Two overlapping HSPs were detected and manually removed. The sum of the resulting HSPs reflect the overall homology degree maintained between a pair of genomes in terms of nucleotide conservation.
The HSPs were then parsed by a Tcl/Tk script to determine extended conserved regions, missing regions, and breakpoints (inversions and/or transpositions) between a pair of genomes. Major collinear segments between a pair of genomes (arrows in Fig. 1) are defined as one or several consecutive HSPs in the same orientation in the two genomes. Gaps longer than 2000 bp within these colinear segments are represented by white boxes inside arrows in Figure 1. The number of arrows thus indicates the amount of major genomic rearrangements other than indel events between two genomes.
Proteome Comparison
All predicted proteins of P. abyssi, P. horikoshii, and P. furiosus were searched against protein public databases using BLASTP (Altschul et al. 1997). At most, 70 sequences among all homologs detected by theBLASTP search with an expectation value <10–3were used to construct Multiple Alignments of Complete Sequences using the new program DbCLUSTAL, specially designed for genome-scale studies (Thompson et al. 2000). Tests performed on the alignments used in our analysis revealed that 98% of the alignments were reliable (Thompson et al. 2000). Two genes were considered to be putative homologs if they showed >20% identity in the Multiple Alignment of Complete Sequences. A gene with no homolog in a genome was considered to be absent from this genome. Two homologous genes found in the same genome were considered to be paralogs.
The distances between homologous genes were calculated according to the BioNJ method (Gascuel 1997). ΔAF, ΔAH, and ΔHF denote the distances between P. abyssi and P. furiosus, P. abyssi and P. horikoshii, P. horikoshii andP. furiosus homologs, respectively. For a particularPyrococcus genome, we defined a trio as a gene in the considered genome and its two best homologs in the otherPyrococcus genomes. For each trio, we calculated three distances ratios: ΔAF/ΔAH, ΔHF/ΔAH, and ΔAF/ΔHF. We used two methods to detect suspicious orthologous relationships within a trio of genes. The first method is based on the definition of structural homologs proposed by Tekaia and coworkers (1999): Two genes are considered as false orthologs if they are not the best homolog of each other in the considered species. In the second method, we assumed that an extreme ΔAF/ΔHF ratio within a trio of genes denote a spurious orthologous relationship. To define extreme values, we calculated the upper and lower quartiles and the interquartile range (difference between the two quartiles) from the ΔAF/ΔHF ratio distribution. The ΔAF/ΔHF ratio is considered as an extreme value only if the ΔAF/ΔHF value is greater than (upper quartile + inter quartile range * 4) or lower than (lower quartile – inter quartile range * 4).
WWW REFERENCES
http://www.bio.nite.go.jp/ot3db_index.html (Pyrococcus horikoshii genome and proteome)
http://www.genome.utah.edu/sequence.html (Pyrococcus furiosus genome and proteome)
http://www.genoscope.cns.fr/Pab/ (Pyrococcus abyssi genome and proteome)
http://www.ornl.gov/hgmis/publicat/99santa/157.html (description of thePyrococcus furiosus genome)
Acknowledgments
We thank the Utah Genome Center (Dept. of Human Genetics, University of Utah) for access to sequence data on P. furiosus. We acknowledge Jean Weissenbach and William Saurin for the P. abyssi sequencing and their constant support. We thank Julie Thompson, Frederic Plewniak, and Luc Moulinier for helpful discussions and critical reading of the manuscript, Jean-Louis Mandel for useful advice, and Dino Moras for his continuous encouragement during this work. Special thanks are due to Patrick Forterre for enlightening discussions. We also thank Serge Uge for computer system facilities. This work was supported by institute funds from CNRS, INSERM, the French Genome project, and the Fond de Recherche Hoechst Marion Roussel.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
↵4 Corresponding author.
-
(This article had been held until publication of the P. furiosus sequence.)
-
E-MAIL poch{at}igbmc.u-strasbg.fr; FAX 33 3 88 65 32 76.
-
Article published on-line before print: Genome Res.,10.1101/gr.165301.
-
Article and publication are at www.genome.org/cgi/doi/10.1101/gr.165301.
-
- Received September 18, 2000.
- Accepted December 21, 2000.
- Cold Spring Harbor Laboratory Press








